

Abstract
C‐X‐C motif chemokine ligand 14 (CXCL14) is a novel gene that is expressed in many normal cells but is absent from or expressed at very low levels in cancerous tissues such as head and neck squamous cell carcinoma (HNSCC), prostate cancer, and pancreatic cancer. However, the relationship between CXCL14 and hepatocellular carcinoma (HCC) remains unclear. Therefore, the exact function of CXCL14, which may modulate antitumor immune responses in certain cancers, was evaluated. CXCL14 was downregulated in HCC tissues compared to adjacent normal tissues. Moreover, overexpression of CXCL14 had an inhibitory effect on cell proliferation, induced apoptosis and inhibited the invasion of HCC cells in vitro. Upregulation of CXCL14 by lentivirus also significantly suppressed the growth of subcutaneous tumors in nude mice in vivo. We further demonstrated that the loss of CXCL14 expression was regulated by promoter hypermethylation. CXCL14 induced tumor cell apoptosis through both the mitochondrial and nuclear apoptosis pathways. CXCL14 suppressed tumor cell proliferation through regulation of the cell cycle by downregulation of cyclins and cyclin‐dependent kinases. In conclusion, CXCL14 plays a pivotal role as a potential tumor suppressor in HCC. The re‐expression or upregulation of this gene may provide a novel strategy in HCC therapy in the future.
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third most common cause of cancer‐related deaths worldwide. Eighty‐two percent of cases (and deaths) are in developing countries and the death rate is the second highest in China because of the high prevalence of chronic hepatitis B virus infection and liver cirrhosis.1, 2, 3 Japan, unlike other Asian countries, has a high proportion of HCC caused by hepatitis C virus (HCV) infection accounting for 80–90% of all cases.4 Although there have been many advances in HCC therapy, such as recent target therapies, liver transplantation, proton therapy and interventional radiological treatment, the overall patient outcome has not been improved substantially. The 5‐year survival rate is limited to 25–39% after surgery.5, 6, 7, 8, 9 Therefore, novel treatments for liver cancer are urgently needed.10, 11 New treatments such as immunotherapy and potential novel gene therapy show potential for the treatment of HCC.12, 13
Recent studies have suggested important functions of chemokines in various aspects of tumor growth.14 Chemokines contribute to leukocyte infiltration in tumors, and some chemokines, such as IL‐8, CXCL1–3 and CXCL5, also have direct proangiogenic effects. In addition, chemokines, especially the CXC family, are potentially important factors in tumor growth, immunity, invasion, metastasis and potent mediators of neoangiogenesis.15, 16, 17
The chemokine C‐X‐C motif chemokine ligand 14 (CXCL14), was initially named BRAK because of its isolation from the human breast and kidney‐derived cells; however, its function and receptor remain unclear.18, 19, 20 CXCL14 was reported to induce B cells, monocytes, and dendritic cell (DC) infiltration into normal and tumor tissues and to inhibit angiogenesis.21, 22, 23 CXCL14 is expressed universally and abundantly in normal tissues but is absent from or expressed minimally in cancerous tissues such as head and neck squamous cell carcinoma (HNSCC) and breast cancer cells, but heightened CXCL14 expression has been reported in adenocarcinomas such as prostate cancer and lung cancer. 24, 25, 26, 27, 28 CXCL14 was reported to be expressed in normal liver tissues, but the expression and role of CXCL14 in liver cancer remain unknown.18 The only report regarding CXCL14 and liver cancer is on intronic polymorphism rs2237062 in the CXCL14 gene, which influences HBV‐related HCC progression.29 Therefore, in the present study we evaluated the expression, potential function and mechanism of CXCL14 in regulating HCC biology.
Materials and Methods
Patients and specimen collection
This study included 126 HCC patients who had undergone hepatectomy between 2008 and 2011 at our hospital (First Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang, China). The study was approved by the local ethics committee, and informed consent was obtained from all patients. HCC was diagnosed in all patients before or after hepatectomy and was confirmed by histopathological examination. None of the patients received pre‐surgical chemotherapy or radiation therapy.
Cell lines and cell culture
Seven HCC cell lines (HepG2, Hep3B, HuH7, HCCLM3, SK‐Hep1, SMMC‐7721, and Bel‐7402) and one normal liver cell line (L‐02), which are all maintained at our institution, were used in this study. All cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C and were passaged using standard cell culture techniques.
Bisulfite treatment of DNA, methylation‐specific PCR and combined bisulfite restriction analysis
DNA samples were modified using EZ DNA Methylation Golden Kit (Zymo Research, Irvine, CA, USA). The bisulfite‐modified DNA was amplified by methylation‐specific PCR (MSP) using the primer pairs shown in Table S1. Combined bisulfite restriction analysis (COBRA) was performed to semi‐quantitate the methylated and unmethylated DNA after bisulfite modification.
Bisulfite sequencing PCR
To determine the methylation pattern of 5′ CpG islands of the CXCL14 gene promoter in liver cancer cells and liver tissues, bisulfite‐treated DNA was subjected to bisulfite sequencing PCR (BSP). The primers shown in Table S1 were used to amplify CpG islands in the promoter region with an expected product 358 bp in length. Twenty‐nine CpG sites spanning the −147 to +211 bp region were evaluated.
Quantitative real‐time PCR analysis
Total RNA was extracted from cell lines or tissues and cDNA was synthesised (Bio‐Rad, Hercules, CA, USA). Real‐time PCR was performed according to the manufacturer's instructions. The primer sequences were shown in Table S1.
5‐aza and TSA treatment
Hepatocellular carcinoma cells were seeded at a density of 1 × 106/mL in 10‐cm dishes. After overnight culture, cells were treated with 10 μM of the DNA demethylating agent 5‐aza‐2′deoxycytidine (5‐aza; Sigma‐Aldrich, St. Louis, MO, USA) for 96 h. Some cell lines were further treated with 300 nM of the histone deacetylase inhibitor trichostatin A (TSA; Sigma‐Aldrich) for an additional 24 h. After treatment, the cells were harvested for RNA extraction.
Western blot analysis
Approximately 50 μg of tissue or 30 μg of cellular protein lysates were used for separation by electrophoresis in 12% PAGE gels (Invitrogen, Carlsbad, CA, USA). Blots were immunostained with primary antibody and secondary antibody, respectively. The primary antibodies against CXCL14 (ab46010, Abcam, Cambridge, UK), and β‐actin (Sigma‐Aldrich) were used in this experiment.
Immunohistochemistry
Immunohistochemical analysis was performed using paired paraffin‐embedded tissues from HCCs and adjacent benign liver tissues using primary antibodies against CXCL14 (1:200; ab46010, Abcam). The extent of CXCL14 staining was scored by assigning the percentage of positive tumor cells (0, none; 1, <20% of positive staining cells; 2, 20–50% of positive staining cells; 3, >50% of positive staining cells).
Lentivirus infection and stable transfections
Replication‐defective lentivirus encoding the complete CXCL14 open reading frame (LV‐CXCL14) and a lentivirus vector encoding green fluorescent protein (LV‐GFP), which was used as the control, were constructed by Invitrogen. HepG2 and SMMC‐7721 cells were treated with lentivirus at a multiplicity of infection (MOI) of 20 pfu per cell in 2% FBS medium with 8 μg/mL Polybrene (Sigma‐Aldrich). Five microliters/mL Blasticidin S HCl (Invitrogen) was added to the medium to select stable transformants.
Cell viability assay
Cell viability assays were performed using Cell Counting Kit‐8 (Dojindo Laboratories, Kumamoto, Japan). We read the absorbance at 450 nm to determine the cell viability in each well.
Colony formation assay
For colony formation assays, the cells with LV‐GFP or LV‐CXCL14 were plated (3000 cells/dish) in 10‐cm dishes. The surviving colonies (>50 cells) were stained with crystal violet and counted after 10 days of culture.
Annexin V apoptosis assay
Cells undergoing early and late apoptosis were quantified using a flow cytometer (CYTOMICS FC 500; Beckman Coulter, Miami, FL, USA) following staining with Annexin V‐APC and propidium iodide (Tianjin Sungene Biotech Co. Ltd, Tianjin, China).
Flow cytometry analysis of cell cycle distribution
Cells for cell cycle assays were fixed in 70% ethanol, stored at 4°C for more than 24 h, and stained with DNA PREP stain (Beckman Coulter, Brea, CA, USA) before flow cytometry.
Analysis of cell invasion and migration
Cell invasion and migration assays were performed using 24‐well transwell plates (Millipore, Billerica, MA, USA). Filters for invasion assays were coated with Matrigel (BD Biosciences, San Jose, CA, USA) in the upper compartment before cell seeding. Then, 4 × 104 cells were seeded in the filters and the lower compartment was filled with cell culture medium supplemented with 20% FBS. Invasive and migratory cells on the bottom surface were stained with crystal violet and counted after 24 h of culture. The invading or migrating cells were examined, counted, and photographed using digital microscopy.
In vivo animal study
Four‐week‐old male BALB/c nude mice were divided randomly into two groups of 10 mice each. The HCC cells of stable transfections were injected subcutaneously into the armpit of mice in a total volume of 100 μL (2 × 106 cells in PBS). Approximately 10 days after cell inoculation, tumor volumes for each mouse were monitored with a caliper every 2 days by measuring in two directions (length and width). The volume was calculated as length × (width)2/2. Then mice were killed. Tumors were removed and weighed. The experiments were performed according to the institutional ethics guidelines.
Statistical analysis
Data are displayed as means ± standard deviation (SD). anova and Student's t‐test were used to determine the statistical significance of differences between experimental groups in vitro and in vivo. The relationship between CXCL14 expression and clinicopathological variables was assessed by the χ2 test. Additionally, all statistical analyses were performed using the spss 19.0 program for Windows (SPSS Inc., Chicago, IL, USA). A value of P < 0.05 was taken to indicate statistical significance. Graphs were created using GraphPad Prism 5.
Results
CXCL14 downregulated or silenced by promoter methylation in HCC cell lines
As shown in Figure 1(a), CXCL14 transcription was reduced or silenced in all seven HCC cell lines examined compared to the normal L‐02 liver cells (100%), inferring aberrant gene silencing of CXCL14 in HCC.
Figure 1.
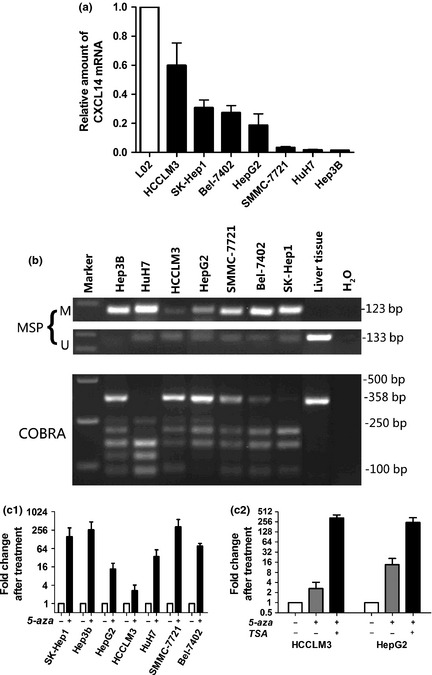
(a) C‐X‐C motif chemokine ligand 14 (CXCL14) was silenced or downregulated in all hepatocellular carcinoma (HCC) cell lines, as determined by quantitative reverse transcription‐polymerase chain reaction (RT‐PCR). (b) Methylation of CXCL14 was determined by methylation‐specific PCR (MSP) and combined bisulfite restriction analysis (COBRA). (c) CXCL14 mRNA expression was restored after treatment with the demethylation reagent 5‐aza alone (c1) or in combination with TSA (c2).
Aberrant promoter CpG methylation is known to be related to gene silencing. We next explored CXCL14 methylation status by MSP and COBRA. Partial methylation was detected in seven HCC cell lines (Fig. 1b). The detailed methylation status of individual CpG sites was examined by BSP (Fig. S1A). The BSP results confirmed those of the MSP and COBRA analyses (Fig.S1B). The HCC cell lines with lower expression of CXCL14 (Hep3B, HuH7, and SMMC‐7721) had a predominantly higher frequency of methylated CpG sites.
Pharmacological demethylation restores CXCL14 expression
All seven HCC cell lines were treated with the DNA methyltransferase inhibitor 5‐aza. Re‐expression of CXCL14 was observed in all cell lines examined (Fig. 1c1). As treatment of 5‐aza resulted in a lower reactivation level of CXCL14 in HCCLM3 and HepG2 cells, these two cell lines were further treated with TSA. The restored expression of CXCL14 was observed more in HCCLM3 and HepG2 treated with 5‐aza and TSA than in with 5‐aza only (Fig. 1c2).
CXCL14 expression is decreased in liver tumor tissues
Immunohistochemistry was performed to evaluate CXCL14 protein expression in 42 paired HCCs and their adjacent non‐tumor tissues (Fig. 2a). CXCL14 protein was expressed predominantly in the cytoplasm in adjacent non‐tumor tissues but was significantly downregulated in HCC tissues (P < 0.001; Fig. 2b). The analysis of CXCL14 protein level by western blot confirmed the immunohistochemistry results (Fig. 2c).
Figure 2.
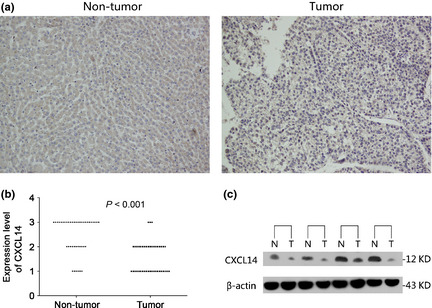
(a) Representative images of C‐X‐C motif chemokine ligand 14 (CXCL14) protein expression in hepatocellular carcinoma (HCC) and their adjacent non‐tumor tissues determined by immunohistochemistry, original magnifications, ×100. (b) CXCL14 protein was downregulated significantly in primary tumor specimens compared with the adjacent non‐tumor tissues by immunohistochemistry (P < 0.001, n = 42). Immunohistochemistry scoring was performed according to the percentage of positive tumor cells (1, < 20%; 2, 20–50%; 3, >50%). (c) The downregulated protein of CXCL14 was confirmed by western blot analysis. N, adjacent normal tissues; T, hepatic tumor tissues.
Increased expression of CXCL14 mediated by lentivirus vector in liver cancer cell lines and its effect on cancer cell proliferation
Re‐expression of CXCL14 in the stably transfected HepG2 and SMMC‐7721 cells was confirmed by quantitative RT‐PCR (Fig. 3a1) and western blotting (Fig. 3a2), which indicated that LV‐CXCL14 infection resulted in efficient overexpression of CXCL14. Ectopic expression of CXCL14 caused a significant decrease in the viability of HepG2 (P < 0.001) and SMMC‐7721 (P < 0.001, Fig. 3b) cells. To further validate the inhibitory effect of CXCL14 on cell proliferation, a colony formation assay was performed (Fig. 3c). The group with CXCL14 overexpression exhibited fewer surviving colonies (P < 0.01) compared with the LV‐GFP group.
Figure 3.
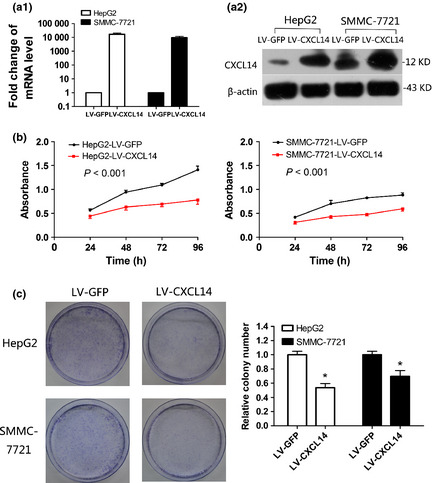
Effect of ectopic C‐X‐C motif chemokine ligand 14 (CXCL14) expression on tumor proliferation. (a) Ectopic expression of CXCL14 mRNA (a1) and protein (a2) in HepG2 and SMMC‐7721 cell lines was confirmed by reverse transcription‐polymerase chain reaction (RT‐PCR) or western blot analysis, respectively. (b) CXCL14 significantly inhibited viability of HepG2 (P < 0.001) and SMMC‐7721 cells (P < 0.001). (c) The effect of CXCL14 on cancer cell growth was confirmed by colony formation assay. The left panel shows representative images of colony formation by hepatocellular carcinoma (HCC) cells with stable transfection. Quantitative analysis of colony numbers is shown in the right panel. Data are means ± SD; *P < 0.01.
Overexpression of CXCL14 induces cell apoptosis and regulates the cell cycle
Suppression of cell growth in tumor cells is usually associated with concomitant activation of cell death pathways. Therefore, we examined the contribution of apoptosis to the observed growth inhibition of CXCL14‐transfected cells using flow cytometry with Annexin V‐APC and propidium iodide double staining (Fig. 4a1). Our results showed a significantly higher percentage of apoptosis (Q2 + Q4) in the HepG2 cells with LV‐CXCL14 compared to those with with LV‐GFP (Fig. 4a2, P < 0.001). This effect was also observed in SMMC‐7721 cells with LV‐CXCL14 (P < 0.001).
Figure 4.
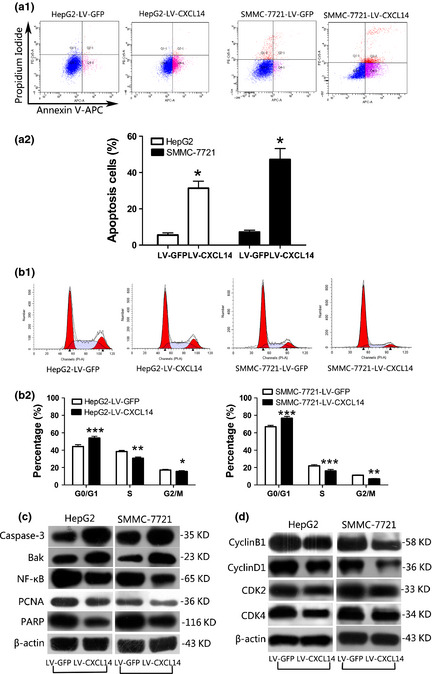
C‐X‐C motif chemokine ligand 14 (CXCL14) induced apoptosis and regulated the cell cycle in hepatocellular carcinoma (HCC) cell lines. (a) CXCL14 induced apoptosis in HepG2 and SMMC‐7721 cells as shown by flow cytometry following Annexin V‐APC and propidium iodide double staining. (a1) Representative fluorescence‐activated cell sorting (FACS) images of HCC cells with LV‐GFP or LV‐CXCL14. (a2) Quantitative analyses of apoptotic cells (Q2 + Q4). The experiment was repeated in triplicate. Data are means ± SD, *P < 0.001. (b) CXCL14 regulated the cell cycle in HepG2 and SMMC‐7721 cells, as shown by cytometry. (b1) Representative FACS images of HCC cells with LV‐GFP or LV‐CXCL14. (b2) The alteration of cell cycle distribution was analyzed. The experiment was repeated in triplicate. Data are means ± SD, *P < 0.001, **P < 0.05, ***P < 0.01. (c) Effect of CXCL14 on protein expression of pro‐apoptosis regulators and anti‐apoptosis mediators in HepG2 and SMMC‐7721 cells by western blotting. (d) Effect of CXCL14 on protein expression of cyclins and cyclin‐dependent kinase (CDKs) by western blotting.
Furthermore, we analyzed the cell cycle alternations by flow cytometry (Fig. 4b1). We observed a significant increase in G0/G1 phase arrest in LV‐CXCL14‐infected HepG2 cells (Fig. 4b2; P < 0.01) accompanied by a significant decrease in S phase (P < 0.05) and G2/M phase (P < 0.001). A similar result was obtained in SMMC‐7721 cells with LV‐CXCL14 (P < 0.01; P < 0.01; P < 0.05, respectively).
To elucidate the molecular basis of apoptosis we examined pro‐apoptosis mediators, including caspase‐3 and Bak, anti‐apoptosis regulators, including nuclear factor kappa‐B (NF‐κB), proliferating cell nuclear antigen (PCNA) and nuclear enzyme poly (ADP‐ribose) polymerase (PARP) in stably transfected HepG2 and SMMC‐7721 cells (Fig. 4c). To explain the cell cycle alteration we examined cyclin B1, cyclin D1, cyclin‐dependent kinase 2 (CDK2) and cyclin‐dependent kinase 4 (CDK4) (Fig. 4d).
Ectopic expression of CXCL14 significantly increased the protein levels of caspase‐3, Bak and degradation of PARP, suggesting that CXCL14 induced apoptosis through caspase‐dependent pathways. Decreased PCNA and NF‐κB indicated that CXCL14 may promote apoptosis through nuclear pathways. In addition, decreased cyclin B1 and D1, CDK2 and CDK4 may explain the suppression of proliferation effect of CXCL14.
CXCL14 promotes cells migration but has inhibitory effect on invasion
The effect of CXCL14 on the migration and invasiveness of HCC cells was analyzed using the Matrigel model (Fig. 5a). The average number of migratory cells with LV‐CXCL14 was increased significantly compared to cells with LV‐GFP (Fig. 5b; P < 0.001). Conversely, CXCL14 had a significant inhibitory effect on invasion (P < 0.01). To further evaluate this, we assessed the effect of CXCL14 on secretion of MMPs, which are key mediators of extracellular matrix degradation. CXCL14 significantly decreased the protein levels of MMP‐1 and MMP‐9 (Fig. 5c), but no statistically significant differences were observed for MMP‐2 (data not shown).
Figure 5.
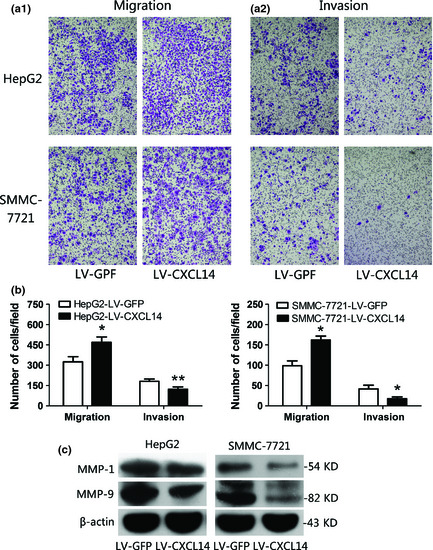
C‐X‐C motif chemokine ligand 14 (CXCL14) induced alteration of migration and invasion in hepatocellular carcinoma (HCC) cell lines. (a) Representative images of migration (a1) and invasion (a2) of HepG2 cells and SMMC‐7721 cells using the Matrigel model (original magnification, ×100). (b) The alteration of migration and invasion was analyzed. CXCL14 significantly promoted cell migration but had an inhibitory effect on invasion. The experiment was repeated in triplicate. Data are means ± SD, *P < 0.001, **P < 0.01. (c) Effect of CXCL14 on protein expression of matrix metalloproteinases (MMPs) according to western blot analysis.
CXCL14 inhibits tumor growth in nude mice
The tumor growth curves of SMMC‐7721 and HepG2 stably transfected with LV‐CXCL14 or LV‐GFP in nude mice are shown in Figure 6(a1). The tumor volume was significantly smaller in LV‐CXCL14‐transfected nude mice than LV‐GFP control mice (P < 0.01, P < 0.05, respectively). At the end of the experiment, the nude mice were killed and the xenograft tumor was isolated. The mean tumor weight was significantly lower in LV‐CXCL14‐transfected nude mice than LV‐GFP control mice (P < 0.01; Fig. 6 a2), indicating that CXCL14 acts as a tumor suppressor in HCC. Finally, to confirm the ectopic expression of CXCL14, tumor sections were assessed for CXCL14 expression by western blotting (Fig. 6b).
Figure 6.
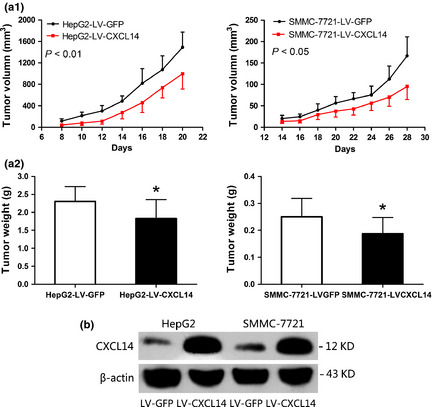
C‐X‐C motif chemokine ligand 14 (CXCL14) inhibited growth of tumors derived from HepG2 and SMMC‐7721 in vivo. (a1) Tumor growth curve of LV‐CXCL14 cells in nude mice compared with LV‐GFP cells. Data are means ± SD (n = 10/group). (a2) Histogram showing the mean tumor weights of the LV‐CXCL14 and LV‐GFP groups. *P < 0.01. (b) Tumor sections were excised and analyzed for CXCL14 expression by western blotting.
CXCL14 is associated with hepatic cirrhosis and frequent CXCL14 methylation in primary HCCs
To determine whether CXCL14 expression was associated with the clinical and pathological features of HCC, we performed quantitative PCR analysis of CXCL14 in paired primary cancerous and adjacent non‐cancerous tissues from 126 HCC patients. One hundred and eleven cases (88.1%) exhibited significant downregulation of CXCL14 in cancerous tissues (Fig. 7a). In all of the 126 tissue pairs, CXCL14 expression was significantly lower in tumors compared to adjacent non‐cancerous tissues (P < 0.001; Fig. 7b). The relationship with clinical characteristics was analyzed and the results are shown in Table 1. A positive correlation was found between CXCL14 expression and hepatic cirrhosis, but not between CXCL14 expression and portal vein tumor thrombus (PVTT) or preoperative Alpha‐fetoprotein level status. We further investigated CXCL14 methylation status in 20 primary HCCs by COBRA. Frequent methylation was detected in HCCs (12/20 = 60%), but no methylation was detected in 10 normal liver tissue specimens (Fig. 7c), indicating that CXCL14 methylation is tumor‐specific. A negative correlation was found between CXCL14 expression and methylation status in the 20 primary HCCs (P < 0.05, Table S2). Detailed BSP analyses confirmed the findings by COBRA that the CXCL14 promoter was frequently methylated in primary HCC but not in normal liver tissue (Fig. S1C).
Figure 7.
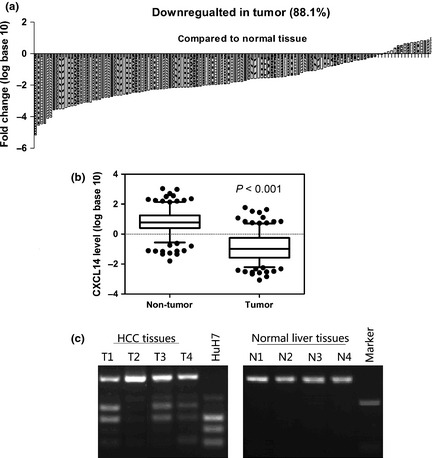
Relative C‐X‐C motif chemokine ligand 14 (CXCL14) expression levels in hepatocellular carcinoma (HCC) tissues. (a) In the vast majority of cancerous tissues (88.1%), the mRNA level of CXCL14 was reduced. (b) In all 126 tissue pairs, the downregulation of CXCL14 expression was significant in tumors compared to adjacent non‐cancerous tissues. (c) Promoter methylation of CXCL14 in primary HCC and normal liver tissues determined by combined bisulfite restriction analysis (COBRA).
Table 1.
Clinicopathological correlation of C‐X‐C motif chemokine ligand 14 (CXCL14) expression in human hepatocellular carcinoma (HCC)
| Variables | CXCL14 expression in HCC | P‐value | ||
|---|---|---|---|---|
| Total n = 126 | Low (n = 63) | High (n = 63) | ||
| Age | ||||
| <50 years | 32 | 19 | 13 | 0.219 |
| ≥50 years | 94 | 44 | 50 | |
| Gender | ||||
| Female | 20 | 13 | 7 | 0.144 |
| Male | 106 | 50 | 56 | |
| Hepatitis B | ||||
| Absent | 32 | 15 | 17 | 0.682 |
| Present | 94 | 48 | 46 | |
| Hepatitis C | ||||
| Absent | 125 | 62 | 63 | 1.000 |
| Present | 1 | 1 | 0 | |
| Liver cirrhosis | ||||
| Absent | 43 | 27 | 16 | 0.039a |
| Present | 83 | 36 | 47 | |
| Preoperative AFP level | ||||
| <400 ng/mL | 80 | 38 | 42 | 0.459 |
| ≥400 ng/mL | 46 | 25 | 21 | |
| Histopathologic grading | ||||
| Well + moderately | 58 | 25 | 33 | 0.153 |
| Poorly | 68 | 38 | 30 | |
| Tumor size | ||||
| <5 cm | 46 | 23 | 23 | 1.000 |
| ≥5 cm | 80 | 40 | 40 | |
| Tumor number | ||||
| Single | 94 | 45 | 49 | 0.413 |
| Multiple | 32 | 18 | 14 | |
| PVTT | ||||
| Absent | 96 | 49 | 47 | 0.676 |
| Present | 30 | 14 | 16 | |
P < 0.05. The significance of the difference between groups in the table was assessed by the Student's t‐test and χ2 tests (Fisher's exact test).
AFP, alpha‐fetoprotein; PVTT, portal vein tumor thrombus.
Discussion
In this study we showed for the first time that CXCL14 is expressed in normal liver tissues but was absent or downregulated in all seven HCC cell lines and tissues investigated. Reportedly, the silence of CXCL14 is attributable to the promoter methylation status in prostate cancer and lung cancer.20, 28 Bisulfite sequencing of the promoter region of the CXCL14 gene showed dense methylation in the HCC cell lines. The silencing of CXCL14 can be reversed by pharmacological demethylation, inferring that methylation is the predominant mechanism for the inactivation of CXCL14 in HCC. As 5‐aza results in only low‐level reactivation of CXCL14 in HCCLM3 and HepG2 cell lines, we tested whether histone modification mediates CXCL14 silencing in these cells using the histone deacetylase inhibitor TSA. Further restored expression of CXCL14 was observed in HCCLM3 and HepG2 treated with 5‐aza together with TSA compared with 5‐aza only (Fig. 1c2), suggesting that histone modifications also play a role in the transcriptional silence of CXCL14.30 We also demonstrated that the protein expression of CXCL14 was significantly decreased in primary HCC tissues compared with their adjacent non‐tumor tissues by immunohistochemistry and western blotting. COBRA and BSP results showed the promoter of CXCL14 was partly methylated in primary HCC tissues, whereas there was no methylation in normal liver tissue. These results suggest CXCL14 to be a potential tumor suppressor and that its downregulation could affect the development of HCC.
The presumptive tumor suppressor function of CXCL14 in human HCC was further investigated by both in vitro and in vivo assays. Ectopic expression of CXCL14 in the HepG2 and SMMC‐7721 cell lines showed significant growth‐suppressing effect by inhibition of cell viability and colony formation. Moreover, CXCL14 enhanced apoptosis and resulted in cell cycle arrest in the G0/G1 phases in HepG2 and SMMC‐7721 cells as determined by flow cytometry. The suppression of tumor growth in CXCL14 re‐expressed cells was confirmed in nude mice. Collectively, these results indicate for the first time that CXCL14 functions as a tumor suppressor in HCC. Our findings highlight the importance of CXCL14 as a potential tumor suppressor in HCC.
We further revealed by western blotting the molecular basis by which CXCL14 exerts tumor‐suppressing activity in HCC by promoting apoptosis, inhibiting proliferation and regulating cell cycle. We observed that caspase‐3, an important intrinsic apoptosis pathway element, was significantly enhanced by CXCL14. Once activated, caspase‐3 processes the PRAP to induce apoptosis. Bak, a proapoptotic multidomain Bcl‐2 protein, was significantly enhanced by CXCL14. Moreover, mitochondrial dysfunction and reactive oxygen species imbalance induce angiogenesis and tumor progression through a CXCL14‐mediated mechanism31, 32 Collectively, these results suggest the antitumor growth effect of CXCL14 may be related to the induction of apoptosis via various apoptosis‐regulating pathways mediated by CXCL14.
The anti‐proliferative effect of CXCL14 in HCC is attributable mostly to the downregulation of PCNA and NF‐κB. PCNA plays a key role in DNA repair, cell proliferation and cell cycle control.33 Nuclear factor‐κB is widely used by eukaryotic cells as a regulator of genes that control cell proliferation and cell survival. Thus, the suppression of cell proliferation by CXCL14 could be a consequence of PCNA and NF‐κB downregulation.
The anti‐tumorigenic function of CXCL14 also contributed to the downregulation of cyclins and CDKs that control the progression of cells through the cell cycle. CDK4, a catalytic subunit of the protein kinase complex that is important for G1 phase progression, binds to cyclin D, then activates retinoblastoma (Rb) family members, resulting in proliferation.34 CDK2, whose activity is restricted to the G1‐S phase of the cell cycle, is essential for the G1/S transition when binding to cyclin E or cyclin A.35 Cyclin B1, expressed predominantly during G2/M phase of the cell cycle, once activated, promotes several early mitosis events.36 Thus, CXCL14's downregulation of cyclins and CDKs indicates the antitumorigenic property of CXCL14.
Next, we assessed the effect of CXCL14 on the migration and invasiveness of HCC cells using the Matrigel model. CXCL14 was found to promote cell migration but inhibit invasion. Reportedly, ectopic CXCL14 enhanced migration and/or invasion by breast cancer cells, pancreatic cancer cells and prostate cancer cells27, 31, 37; however, a recent report regarding breast cancer showed CXCL14 inhibited the invasion of breast cancer cells MDA‐MB‐231HM.25 In our study, CXCL14 significantly decreased the protein levels of MMP‐1 and MMP‐9, which are key mediators of extracellular matrix degradation. The migration of DCs indicates their crucial role in the development of an antitumor immune response at the tumor site.38 Therefore, a possible reason for the inhibitory function of CXCL14 in vivo is that CXCL14 inhibits HCC metastasis but stimulates activated immune cells and enhances immune surveillance.
To determine the clinical relevance of CXCL14 in HCC in vivo, we examined the mRNA level of CXCL14 in 126 primary HCC patients by quantitative RT‐PCR. mRNA expression has a positive association with hepatic cirrhosis, which is the most important risk factor for developing liver cancer. Tumor‐associated fibroblasts play a crucial role in human liver regeneration, cirrhosis and cancer.39 Martin Augsten et al.37, reported that analyses of matched normal and tumor tissue revealed upregulation of CXCL14 in cancer‐associated fibroblasts in a majority of prostate cancers. The studies of Martin identify CXCL14 as a novel autocrine stimulator of fibroblast growth and migration, with multi‐modal tumor‐stimulatory activities. However, the exact association between CXCL14, fibroblasts and hepatic cirrhosis needs further investigation. Hepatitis B and Hepatitis C were the most common causes of hepatic cirrhosis and hepatocellular carcinoma, but no correlation was found between CXCL14 expression and Hepatitis B and Hepatitis C (Table 1).
In conclusion, we have identified a novel functional tumor suppressor gene, CXCL14, which is inactivated by promoter methylation in HCC. CXCL14 induced tumor cell apoptosis through both the mitochondrial and nuclear apoptosis pathways. CXCL14 suppressed tumor cell proliferation through regulation of the cell cycle by downregulation of cyclins and CDKs. CXCL14 may serve as a potential epigenetic biomarker, and re‐expression of this gene may provide a novel molecular target in cancer therapy in the future.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Bisulfite sequencing PCR analysis in HCC cell lines and tissues.
Table S1. DNA sequences of primers used in this study.
Table S2. Correlation between CXCL14 expression and methylation status in HCCs.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81172315/H1617), the research Special Fund For public welfare industry of health and the translational research of early diagnosis and comprehensive treatment in pancreatic cancer (201202007) and the National S&T Major Project (No. 2012ZX10002017).
(Cancer Sci 2013; 104: 1523–1531)
References
- 1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Tung‐Ping Poon R, Fan ST, Wong J. Risk factors, prevention, and management of postoperative recurrence after resection of hepatocellular carcinoma. Ann Surg 2000; 232: 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yuen MF, Hou JL, Chutaputti A. Hepatocellular carcinoma in the Asia pacific region. J Gastroenterol Hepatol 2009; 24: 346–53. [DOI] [PubMed] [Google Scholar]
- 4. El‐Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132: 2557–76. [DOI] [PubMed] [Google Scholar]
- 5. Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology 2005; 42: 1208–36. [DOI] [PubMed] [Google Scholar]
- 6. Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 2003; 37: 429–42. [DOI] [PubMed] [Google Scholar]
- 7. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003; 362: 1907–17. [DOI] [PubMed] [Google Scholar]
- 8. Shrimal A, Prasanth M, Kulkarni AV. Interventional radiological treatment of hepatocellular carcinoma: an update. Indian J Surg 2012; 74: 91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ling TC, Kang JI, Bush DA, Slater JD, Yang GY. Proton therapy for hepatocellular carcinoma. Chin J Cancer Res 2012; 24: 361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008; 48: 1312–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pang RW, Poon RT. From molecular biology to targeted therapies for hepatocellular carcinoma: the future is now. Oncology 2007; 72(Suppl 1): 30–44. [DOI] [PubMed] [Google Scholar]
- 12. Hernandez‐Alcoceba R, Sangro B, Prieto J. Gene therapy of liver cancer. World J Gastroenterol 2006; 12: 6085–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palmer DH, Hussain SA, Johnson PJ. Gene‐ and immunotherapy for hepatocellular carcinoma. Expert Opin Biol Ther 2005; 5: 507–23. [DOI] [PubMed] [Google Scholar]
- 14. Johrer K, Pleyer L, Olivier A, Maizner E, Zelle‐Rieser C, Greil R. Tumour‐immune cell interactions modulated by chemokines. Expert Opin Biol Ther 2008; 8: 269–90. [DOI] [PubMed] [Google Scholar]
- 15. Ben‐Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev 2006; 25: 357–71. [DOI] [PubMed] [Google Scholar]
- 16. Strieter RM, Burdick MD, Mestas J, Gomperts B, Keane MP, Belperio JA. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer 2006; 42: 768–78. [DOI] [PubMed] [Google Scholar]
- 17. Singh S, Sadanandam A, Singh RK. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev 2007; 26: 453–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frederick MJ, Henderson Y, Xu X et al In vivo expression of the novel CXC chemokine BRAK in normal and cancerous human tissue. Am J Pathol 2000; 156: 1937–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hromas R, Broxmeyer HE, Kim C et al Cloning of BRAK, a novel divergent CXC chemokine preferentially expressed in normal versus malignant cells. Biochem Biophys Res Commun 1999; 255: 703–6. [DOI] [PubMed] [Google Scholar]
- 20. Song EY, Shurin MR, Tourkova IL, Gutkin DW, Shurin GV. Epigenetic mechanisms of promigratory chemokine CXCL14 regulation in human prostate cancer cells. Cancer Res 2010; 70: 4394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shellenberger TD, Wang M, Gujrati M et al BRAK/CXCL14 is a potent inhibitor of angiogenesis and a chemotactic factor for immature dendritic cells. Cancer Res 2004; 64: 8262–70. [DOI] [PubMed] [Google Scholar]
- 22. Shurin GV, Ferris RL, Tourkova IL et al Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol 2005; 174: 5490–8. [DOI] [PubMed] [Google Scholar]
- 23. Sleeman MA, Fraser JK, Murison JG et al B cell‐ and monocyte‐activating chemokine (BMAC), a novel non‐ELR alpha‐chemokine. Int Immunol 2000; 12: 677–89. [DOI] [PubMed] [Google Scholar]
- 24. Komori R, Ozawa S, Kato Y, Shinji H, Kimoto S, Hata R. Functional characterization of proximal promoter of gene for human BRAK/CXCL14, a tumor‐suppressing chemokine. Biomed Res 2010; 31: 123–31. [DOI] [PubMed] [Google Scholar]
- 25. Gu XL, Ou ZL, Lin FJ et al Expression of CXCL14 and its anticancer role in breast cancer. Breast Cancer Res Treat 2012; 135: 725–35. [DOI] [PubMed] [Google Scholar]
- 26. Schwarze SR, Luo J, Isaacs WB, Jarrard DF. Modulation of CXCL14 (BRAK) expression in prostate cancer. Prostate 2005; 64: 67–74. [DOI] [PubMed] [Google Scholar]
- 27. Wente MN, Mayer C, Gaida MM et al CXCL14 expression and potential function in pancreatic cancer. Cancer Lett 2008; 259: 209–17. [DOI] [PubMed] [Google Scholar]
- 28. Tessema M, Klinge DM, Yingling CM, Do K, Van Neste L, Belinsky SA. Re‐expression of CXCL14, a common target for epigenetic silencing in lung cancer, induces tumor necrosis. Oncogene 2010; 29: 5159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gu X, Wang H, Wang A et al An intronic polymorphism rs2237062 in the CXCL14 gene influences HBV‐related HCC progression in Chinese population. Mol Biol Rep 2012; 39: 797–803. [DOI] [PubMed] [Google Scholar]
- 30. Razin A. CpG methylation, chromatin structure and gene silencing‐a three‐way connection. EMBO J 1998; 17: 4905–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pelicano H, Lu W, Zhou Y et al Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14‐mediated mechanism. Cancer Res 2009; 69: 2375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maehata Y, Ozawa S, Kobayashi K et al Reactive oxygen species (ROS) reduce the expression of BRAK/CXCL14 in human head and neck squamous cell carcinoma cells. Free Radic Res 2010; 44: 913–24. [DOI] [PubMed] [Google Scholar]
- 33. Gramantieri L, Trere D, Chieco P et al In human hepatocellular carcinoma in cirrhosis proliferating cell nuclear antigen (PCNA) is involved in cell proliferation and cooperates with P21 in DNA repair. J Hepatol 2003; 39: 997–1003. [DOI] [PubMed] [Google Scholar]
- 34. Paternot S, Bockstaele L, Bisteau X, Kooken H, Coulonval K, Roger PP. Rb inactivation in cell cycle and cancer: the puzzle of highly regulated activating phosphorylation of CDK4 versus constitutively active CDK‐activating kinase. Cell Cycle 2010; 9: 689–99. [DOI] [PubMed] [Google Scholar]
- 35. Tsai LH, Harlow E, Meyerson M. Isolation of the human cdk2 gene that encodes the cyclin A‐ and adenovirus E1A‐associated p33 kinase. Nature 1991; 353: 174–7. [DOI] [PubMed] [Google Scholar]
- 36. Kimura K, Hirano M, Kobayashi R, Hirano T. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science 1998; 282: 487–90. [DOI] [PubMed] [Google Scholar]
- 37. Augsten M, Hagglof C, Olsson E et al CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi‐modal stimulator of prostate tumor growth. Proc Natl Acad Sci USA 2009; 106: 3414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol 2001; 2: 123–8. [DOI] [PubMed] [Google Scholar]
- 39. Cesselli D, Beltrami AP, Poz A et al Role of tumor associated fibroblasts in human liver regeneration, cirrhosis, and cancer. Int J Hepatol 2011; 2011: 120925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Bisulfite sequencing PCR analysis in HCC cell lines and tissues.
Table S1. DNA sequences of primers used in this study.
Table S2. Correlation between CXCL14 expression and methylation status in HCCs.