

Abstract
Inflammation‐associated malignancies of the gastrointestinal tract (GI), including those of the stomach and colon, collectively rank as the highest cause of cancer‐related deaths worldwide. It has been well documented that the deregulated activation of the archetypal pro‐inflammatory and oncogenic transcription factors nuclear factor‐kappa B (NF‐κB) and signal transducer and activator of transcription (STAT)3 is a common feature of GI cancers that invariably correlates with poor prognosis. Signal transducer and activator of transcription 3 and NF‐κB are key downstream signal transducers of the interleukin (IL)‐6 cytokine and toll‐like receptor (TLR) families, respectively, and until recently, the potential involvement of these two families in the pathogenesis of cancer has been investigated in isolation. However, there is now emerging evidence of the complex interplay between the IL‐6 cytokine and TLR families in GI tract cancers, with a surprising twist in the identification of a non‐immune role for specific TLR family members. In this review, we discuss the molecular mechanisms associated with cross‐talk between the IL‐6 cytokine family/STAT3 signaling network and the TLR family/NF‐κB signaling network, and we address the potential benefit of their therapeutic targeting in gastric and colorectal cancers.
Gastrointestinal (GI) tract cancers affect the stomach, bowel (colon, rectum), esophagus, liver and pancreas, and they collectively rank as the highest cause of cancer mortalities in the world.1 The human GI tract is heavily colonized by microbes, with studies showing that commensal bacteria play an important role in maintaining intestinal homeostasis.2, 3 Despite this, deregulated immune responses are strongly associated with the development of inflammation‐associated carcinogenesis of the GI tract, and classical examples include Helicobacter (H.) pylori‐mediated gastritis and cancer of the stomach (gastric cancer: GC),4 hepatitis and hepatocellular (liver) cancer,5 and inflammatory bowel disease and colitis‐associated colon cancer (CAC).6 Indeed, the recognition that chronic inflammation can be a precancerous stage common to GI tract cancers has led to an explosion in research over the last decade, with a primary aim being to identify key factors of the host immune system that promote and maintain carcinogenic changes within the GI mucosal epithelium. Accordingly, in this review, we will focus on recent developments to identify such factors, with an emphasis on the complex interplay between two central molecular families of the immune system, the interleukin (IL)‐6 cytokine and toll‐like receptor (TLR) families, which display potent pro‐inflammatory and oncogenic properties in the stomach and colon.
Epidemiology of GI Cancer
Of the GI tract malignancies, GC and colorectal (bowel) cancer (CRC) have the highest worldwide incidence rates, with approximately 2 million people diagnosed annually.1 The development of both cancers is a multifactorial process with environmental (e.g. diet, cigarette smoke exposure, microbial infection), epigenetic and genetic factors being implicated.7, 8 With respect to GC, differentiated intestinal‐type adenocarcinoma represents the major histopathological type, accounting for 95% of cases in East Asia, Eastern Europe, and Central and South America. Its etiology is linked primarily to elevated mucosal immune responses following colonization by H. pylori,4 which infects two‐thirds of the world's 7 billion people. However, <3% of H. pylori‐infected Japanese individuals will develop GC,4 which raises a key question as to what host‐derived genetic factors determine susceptibility to intestinal‐type GC. While the identity of such host‐derived susceptibility factors remains ill‐defined, one candidate is the pro‐inflammatory cytokine IL‐1β, for which gene polymorphisms linked to its augmented expression correlate with an increased GC risk in European populations.9 Another candidate that will be discussed in more detail in this review is TLR2; TLR2 gene polymorphisms are associated with heightened H. pylori‐associated GC risk,10 and increased TLR2 gene expression is significantly elevated in GC patients and correlates with reduced GC patient survival.11
Although, approximately 10–30% of CRC cases are inherited and associated with mutations in genes belonging to the DNA mismatch‐repair system (MutS Homolog 2 [MSH2] and MutL Homolog 1 [MLH1]), as well as the adenomatosis polyposis coli (Apc) tumor‐suppressor gene,8 inflammatory bowel disease is widely acknowledged as the primary precancerous lesion associated with two‐thirds of CAC.8 Indeed, there is now compelling clinical evidence and data from mouse disease models indicating quantitative and/or qualitative differences in the intestinal microbes that influence disease severity.12, 13, 14 Moreover, a seminal report by Arthur et al. recently revealed that intestinal inflammation can promote tumorigenesis by modifying the composition of intestinal commensals, resulting in an overrepresentation of genotoxic microbes.15
Involvement of TLRs in GI Cancers
The strong association between the colonization of microbes and inflammation‐associated cancers, such as GC and CRC, has led to intense investigations in recent years into the role of microbial‐sensing, host‐derived factors of the immune system, namely pattern recognition receptors, in the molecular pathogenesis of these GI cancers. In this respect, much attention has focused on the archetypal class of pattern recognition receptors, the TLR family, the members of which act as critical sensors in the immune system to trigger an inflammatory response following their recognition of pathogen‐associated molecular patterns displayed on viral, bacterial and fungal microorganisms.16 To date, 13 TLRs have been identified in mammals, which are expressed either extracellularly or within endosomes of host cells.16 The best characterized TLR are TLR4, which recognizes bacterial lipopolysaccharide, and TLR2, which binds to a broad range of ligands (e.g. lipopeptides, peptidoglycan) due to heterodimerization with TLR1 and TLR6.16 In addition, TLR2 is involved in the recognition of H. pylori to initiate a pro‐inflammatory response.17 Ligand engagement of TLR causes activation of two distinct signaling pathways, myeloid differentiation primary response 88 (MyD88)‐dependent and ‐independent,16, 18 which are necessary for the activation of the nuclear factor‐kappa B (NF‐κB) transcriptional complex and MAPK cascade to induce an inflammatory response.19 Considering NF‐κB has a clearly defined role in the initiation and progression of many cancers,20 it is not surprising that the requirement for MyD88 has been demonstrated in various mouse disease models for intestinal tumorigenesis.21, 22
A speculative role for TLRs in mediating the pathogenesis of GC and CRC has come from a spate of studies that have identified genetic polymorphisms in TLR that are associated with disease risk (Table 1). In GC, the most well‐characterized genetic polymorphisms are TLR2 –196 to –174del and TLR4 Asp299Gly, which display a strong association with H. pylori‐associated non‐cardia GC risk in Asian and Western populations, respectively.10, 23 Interestingly, the TLR4 Asp299Gly mutation is commonly seen in patients with advanced CRC. Furthermore, intestinal epithelial cells that stably expressed TLR4 Asp299Gly displayed enhanced tumorigenicity in a mouse xenograft model compared to the WT allele.24 In further support of these findings, it is of note that TLR2 and TLR4 are commonly overexpressed in human GC and CRC, respectively.11, 25, 26
Table 1.
TLR family genetic polymorphisms linked to GC and CRC susceptibility
| Cancer | TLR | Polymorphism | Association | Reference |
|---|---|---|---|---|
| GC | TLR2 | –196 to –174 del | TLR2 –196 to –174 del/del is associated with increased risk of GC in Brazilian and Japanese populations | Tahara et al.10; Zeng et al.73 |
| TLR4 | A896G | TLR4 + 896A/G genotype is associated with increased risk of GC in a Brazilian population | De Oliveira et al.74 | |
| TLR5 | rs5744174 | TLR5 rs5744174 C carriers is associated with increased risk of GC | Zeng et al.73 | |
| CRC | TLR2 | C597C | TLR2 + 597C/C displayed a five‐fold decreased risk of CRC | Pimental‐Nunes et al.75 |
| TLR4 | D299G | Associated with three‐fold increased risk of CRC in Europeans | Pimental‐Nunes et al.75 | |
| rs11536898 | TLR4 A/A genotype is associated with a reduced risk of CRC | Slattery et al.76 |
CRC, colorectal (bowel) cancer; GC, gastric cancer; TLR, toll‐like receptor.
More compelling evidence for the role of TLR2 in GC has come from our recent finding that, among the TLR family members, TLR2 is most significantly overexpressed in the gp130 F/F knock‐in GC mouse model for spontaneous gastric inflammation and tumorigenesis.11 Furthermore, we uncovered a novel non‐immune role for TLR2 in promoting gastric growth independent of inflammation, whereby activation of TLR2 within (epithelial) tumor cells rather than infiltrating inflammatory cells promoted gastric tumor cell proliferation and survival via upregulation of anti‐apoptotic genes (e.g. BCL2‐related protein A1 [Bcl2a1], baculoviral IAP repeat containing 3 [Birc3] and B‐cell CLL/lymphoma 3 [Bcl3]).11 With respect to CRC, the use of experimentally induced mouse disease models has primarily implicated TLR4 in promoting disease pathogenesis. For instance, upon azoxymethane (AOM)/dextran sodium sulfate (DSS) treatment, Tlr4 −/− mice failed to develop CAC,27 and a combination of studies involving intestinal epithelial‐driven TLR4 transgene expression in mice and TLR4‐based bone marrow chimeras indicate that TLR4 expression in the epithelial rather than the myeloid compartment promotes AOM/DSS‐induced CAC development.25, 26 Conversely, the development of larger colorectal tumors in AOM/DSS‐treated Tlr2 −/− mice compared to their WT counterparts suggests TLR2 may have a protective role in CAC, although the cell types involved were not explored.28 This latter finding contrasts the reported pro‐tumorigenic (epithelial‐driven) roles of TLR2 in the stomach and TLR4 in the intestine,11, 25, 26 and it will be worthwhile to elucidate whether anti‐tumorigenic TLR2 signaling in the intestine is propagated by the epithelial or non‐epithelial (i.e. inflammatory cells) compartment.
In mouse models for TLR/MyD88‐driven CRC, a potential confounding issue in assigning the disease‐associated cell type(s) is the divergent nature (i.e. carcinogen‐induced vs genetic) of disease models employed. As an example, in a comparison of Myd88 −/− and Apc Min/+ mice, genetic deletion of Myd88 significantly reduced the colon tumor burden in Apc Min/+ mice, which was associated with increased apoptosis within the tumor epithelium of Apc Min/+.21 The reduced tumor load likely arose from an altered (MyD88‐dependent) gene expression profile in both the epithelial and stromal (i.e. immune cell) compartments, which ultimately impact epithelial cell survival rather than the infiltration of leukocytes, the latter being analogous to the role of TLR2 and MyD88 in the gp130 F/F GC model.11, 29 While these observations and those regarding the role of TLR4 in DSS/AOM‐induced CRC imply that epithelial‐derived TLR4/MyD88 signaling primarily promotes intestinal tumorigenesis, TLR/MyD88 signaling in myeloid cells has recently been shown to support spontaneous CRC induced upon Cre‐mediated Apc allelic loss in the colon.22 To relate these divergent findings to the complex pathogenesis of human CRC, it is most likely that TLR/MyD88 signaling in both non‐immune (epithelial) and immune (myeloid) cell types is required to elicit the initiation and progression of intestinal tumors, a finding which is supported by the oncogenic anti‐apoptotic and pro‐inflammatory properties of the NF‐κB pathway in the intestinal epithelial and myeloid compartments, respectively.30
While the identity of TLR ligands in GC and CRC is also currently unknown, it is worth considering that TLRs not only recognize microbial‐derived products, but also host‐derived products or danger‐associated molecular patterns (DAMP).19 With respect to DAMP, augmented expression of TLR4 non‐microbial ligands, such as high mobility group box 1 (HMGB1) and S100 calcium binding protein A9 (S100A9),31, 32, 33, 34 is often observed in CRC patients, and in GC, the TLR2 agonists versican and serum amyloid A (SAA) are frequently overexpressed.35, 36, 37, 38 Taken together these observations suggest the possible involvement of a combination of pathogen‐associated molecular patterns and DAMPs in TLR‐driven disease pathogenesis, whereby excessive TLR signaling results from the overexpression of TLRs and/or DAMPs, which serve as TLR agonists.
Interleukin (IL)‐6 Family Cytokine Signaling and Cancer
Over the last decade, a wealth of mouse disease models and clinical studies have strongly implicated the IL‐6 family of cytokines, in particular IL‐6 and IL‐11, in malignancies of the digestive system.39, 40 These cytokines bind to their respective α‐receptor subunits, resulting in the activation of the common signal‐transducing gp130 β‐receptor subunit to activate the JAK/signal transducer and activator of transcription (STAT), Ras/MAPK and phosphoinositide‐3‐kinase (PI3K)/Akt signaling pathways.41 With respect to the predominant JAK/STAT pathway, tyrosine residues on the cytoplasmic domain of gp130 are phosphorylated by JAK, which facilitates tyrosine phosphorylation of STAT3.41 STAT3 phosphorylation results in the formation of homodimers or heterodimers with other STAT molecules, predominantly STAT1, to initiate transcription of STAT target genes in the nucleus.41 Furthermore, activation of STAT3 is tightly regulated by suppressor of cytokine signaling (SOCS) 3, which negatively regulates STAT3 activity by targeting the receptor complex for proteosomal degradation.41
Deregulated STAT3 activation is a common feature of numerous epithelial and hematopoietic malignancies,42 and the direct oncogenic potential of STAT3 in vivo has been demonstrated upon transgenic overexpression of an artificial hyperactive STAT3 mutant in the lung.43 Accordingly, much attention has focused on understanding the molecular basis of the pro‐tumorigenic actions of STAT3. The broad diversity of STAT3's oncogenic actions is evidenced by its ability to be a potent transcriptional inducer of genes involved in cell cycle progression, cell survival,44 angiogenesis and inflammation,45 the last of which exemplifies its oncogenic potency in cancers driven by a chronic inflammatory microenvironment.46, 47 In addition, inhibiting STAT3 signaling in hematopoietic‐derived immune cells has also been shown to improve tumor immune surveillance and elicit multi‐component anti‐tumor immunity.48
The gp130/STAT3‐Signaling Axis in GI Cancers
In approximately 50% of human GC, STAT3 is over‐activated,11, 42 and its high activation and/or expression status has been shown to correlate with a lower survival rate for GC patients.49 While the upstream mechanisms leading to increased STAT3 activity in these studies were not reported, a likely candidate is the gp130‐activating cytokine IL‐11, which is frequently overexpressed in human GC.39, 50 Definitive proof that the IL‐11/STAT3‐signaling axis can be pro‐tumorigenic in the stomach was provided by the gp130 F/F mouse model for intestinal‐type GC, which harbors a homozygous phenylalanine knock‐in substitution at tyrosine 757 of the gp130 receptor that abrogates SOCS3 binding, thus resulting in STAT3 hyperactivation. Specifically, these mice spontaneously develop gastritis and gastric tumors within 6–8 weeks, which are then suppressed upon genetic reduction of IL‐11‐dependent STAT3 activation.39, 45 Further in vivo evidence for the importance of STAT3 in gastric tumorigenesis has recently been reported. This study used conditional knock‐out of SOCS3 in GI epithelial cells (T3b‐SOCS3 cKO mice), which spontaneously developed STAT3‐mediated gastritis and gastric tumors within 8 weeks, indicating that SOCS3 in the gastric compartment is an important negative regulator for STAT3‐mediated gastric tumorigenesis.51
Similar to GC, STAT3 is hyperactivated in 90% of colorectal tumor biopsies and correlates with poor prognosis.52 However, unlike the apparent reliance on IL‐11/STAT3 signaling for GC,39 deregulated IL‐6 and IL‐11‐mediated STAT3 hyperactivation both contribute to the pathogenesis of AOM/DSS‐induced CAC.47 With respect to IL‐6, trans‐signaling is the main mode by which IL‐6 promotes CAC,40, 53 whereby proteolytic cleavage of membrane‐bound IL‐6 receptor alpha subunit results in the release of soluble IL‐6 receptor alpha subunit. Upon interacting with IL‐6, IL‐6 receptor alpha subunit enables this complex to associate with gp130 to transduce a signal.54 The importance of IL‐6 trans‐signaling in CAC was demonstrated in DSS‐treated mice, in which administration with the IL‐6 trans‐signaling, antagonist‐soluble gp130Fc resulted in animals with smaller colonic polyps compared to vehicle‐treated animals.53
Tying the Knot Between Cytokine and TLR Signaling in GI Cancers
Despite the overwhelming evidence for a causal link between TLR and IL‐6 cytokine families in GI cancers, the interplay between these two families in the pathogenesis of such inflammation‐associated cancers has not been extensively investigated. An early insight into such potential cross‐talk between these families was provided by the often coexistent over‐activation of STAT3 and NF‐κB, the archetypal transcription factors of the IL‐6 cytokine and TLR families in GI tumors, respectively.55 Upon activation in tumor (i.e. epithelial) cells, both transcription factors induce the expression of overlapping genes involved in cell survival/anti‐apoptosis and cell cycle progression, while their activation in inflammatory cells within the stroma propagates the chronic inflammatory state that supports cellular transformation.11, 30, 56 Evidence for such synergistic interactions between these two transcription factors is provided by the observation that non‐phosphorylated versions of STAT3 and NF‐κB, in response to IL‐6, can form a transcriptional complex that regulates a unique set of inflammatory genes not induced individually by tyrosine phosphorylated STAT3.57
An early example that both TLR and IL‐6 cytokine family members can cooperate to promote oncogenic cellular responses was provided by evidence that the production of IL‐6 is a key event in the MyD88‐dependent pathogenesis of diethylnitrosamine‐induced liver carcinogenesis.58 Given the well‐documented pro‐inflammatory role of IL‐6 as a downstream effector of TLR‐induced inflammation, this finding is not surprising,55 but it nonetheless sets the stage for further investigations into the co‐dependence of these two families in cancer. In this respect, it has recently emerged that IL‐6 family cytokines can influence TLR‐induced cellular processes in the GI tract. Specifically, we recently reported that in two independent GC mouse models characterized by elevated STAT3 activation, gp130 F/F and Gan mice, STAT3 directly induces the transcription of the Tlr2 gene in the gastric epithelium, which upon overexpression promotes proliferation and inhibits apoptosis of gastric epithelial cells (Fig. 1).11 Moreover, increased gene expression of TLR2 and over‐activation of the STAT3 transcription factor correlated with decreased 5‐year survival rates in GC patients.11 Considering the disparity in the role between specific TLRs (e.g. TLR2, TLR4) in GC and CRC, for instance, it will now be of great interest to examine whether such cross‐talk exists in GI cancers between STAT3 and these and other TLR family members.
Figure 1.
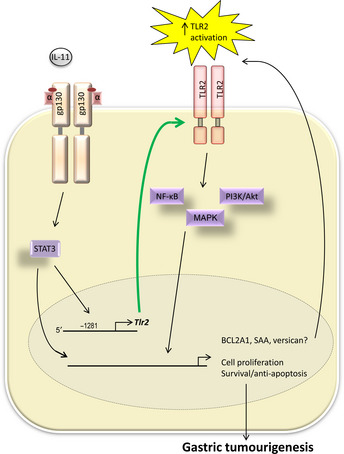
The STAT3‐driven upregulation of TLR2 promotes gastric tumorigenesis by regulating genes involved in cell proliferation and survival. The induction of several TLR2 DAMPs by TLR2 and/or IL‐11/STAT3 signaling pathways may lead to excessive TLR2 activation, resulting in the promotion of gastric epithelial cell growth in a positive feedback loop mechanism. BCL2A1, BCL2‐related protein A1; DAMP, danger‐associated molecular pattern; gp130, glycoprotein 130; IL, interleukin; NF‐κB, nuclear factor‐kappa B; PI3K, phosphoinositide 3‐kinase; SAA, serum amyloid A; STAT, signal transducer and activator of transcription; TLR, toll‐like receptor.
Another consideration in cross‐talk between TLR and IL‐6 cytokine families is the observation that numerous non‐microbial TLR agonists (i.e. DAMPs) are regulated by STAT3. For example, in the context of TLR2 and gastric tumorigenesis, the TLR2 agonists serum amyloid A (SAA) and versican are overexpressed in human GC.38, 59 While versican is upregulated by IL‐11‐induced STAT3 activation in human GC cells,37 SAA is a widely acknowledged STAT3‐inducible gene of the acute phase response.60 In a similar vein, the TLR4 ligands HMGB1 and S100A9, which are upregulated in CRC,31, 33 are also transcriptionally induced by STAT3.61, 62 In contrast to the reported IL‐11/STAT3‐driven role of TLR2 signals on gastric epithelial cell growth in GC,11 it is more likely in CRC that IL‐6/STAT3‐induced upregulation of S100A9 and HMGB1 in infiltrating leukocytes induces TLR4‐mediated inflammatory and proliferative responses to promote tumorigenesis (Fig. 2).
Figure 2.
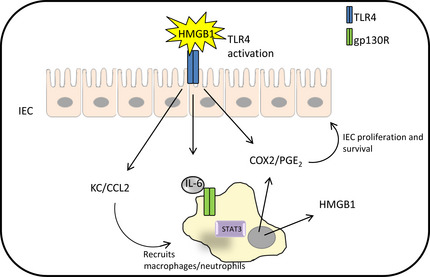
Over‐activation of TLR4 in IEC promotes the recruitment of macrophages and leukocytes to the lamina propria and subsequently results in IL‐6‐mediated STAT3 activation to facilitate the production of COX2/PGE 2 and the TLR4 DAMP HMGB1. The production of COX2/PGE 2 by both IEC and innate immune cells encourages IEC proliferation and survival in CAC. Additionally, STAT3‐driven upregulation of HMGB1 may augment TLR4‐mediated immune and non‐immune signaling in CAC. CAC, colitis‐associated colon cancer; CCL2, chemokine (C‐C motif) ligand 2; COX2, Cyclooxygenase 2; DAMP, danger‐associated molecular patterns; gp130R, glycoprotein 130 receptor; HMGB1, high mobility group box 1; IEC, intestinal epithelial cells; IL, interleukin; PGE, prostaglandin E; STAT, signal transducer and activator of transcription; TLR, toll‐like receptor.
Conclusion
The worldwide 5‐year overall survival rates for advanced GC and CRC patients are generally poor (approximately 25% and 20%, respectively), which is largely the result of both the late detection of these aggressive diseases and limited effectiveness of current treatment options.8, 63 In GC, the perioperative adjuvant chemotherapy significantly improved the 5‐year survival rate of GI cancer patients compared to surgery‐only patients (23% vs 36%), but these survival rates remain unacceptably low.64 These statistics highlight the need for a more comprehensive understanding of the molecular pathogenesis of these malignancies, which is essential to identifying oncogenic gene networks and signaling pathways that serve as both novel biomarkers for early detection and molecular targets for selected therapeutic intervention. A key consideration for the development of new treatments for GI cancers is the heterogeneous nature of these malignancies, which are characterized by molecular and genetic variations in tumors from patients of different ethnicities and geographic locations.6, 65 Such heterogeneity among GI cancers highlights the need for targeted, or personalized, therapies to improve a patient's overall survival.
The emergence of a causal role for the IL‐6 cytokine and TLR families in GI cancers provides an exciting new avenue for their development into bona fide diagnostic and predictive biomarkers, the latter for patient stratification and druggable therapeutic targets. In GC, this is evidenced by the following: (i) the mechanistic link between IL‐11/STAT3 signaling and TLR2 expression; (ii) their co‐upregulation in human GC as potential disease‐associated biomarkers;11, 66 and (iii) the therapeutic efficacy of suppressing gastric tumor growth in the gp130 F/F GC mouse model with the monoclonal antibody blocking antibody (OPN‐301) against TLR2,11 a STAT3 anti‐sense oligonucleotide,39 or the IL‐11‐mutein peptide IL‐11 antagonist.67 Although STAT3, like many other transcription factors, serves as a highly attractive cancer therapy target, numerous strategies developed thus far to successfully interfere with STAT3 transcriptional activity in preclinical disease models (with the possible exception of decoy oligonucleotides) have been hampered in their clinical translation due to difficulties with stable and specific drug delivery.68 Given the lack of clinically proven IL‐11 inhibitors, the recent completion of a phase I study demonstrating the safety and tolerability of fully humanized anti‐TLR2 monoclonal antibody (OPN‐305; Opsona Therapeutics, Dublin, Ireland) paves the way for GC to be examined as a potential clinical target indication for anti‐TLR2‐based adjuvant therapy (Fig. 3).69
Figure 3.
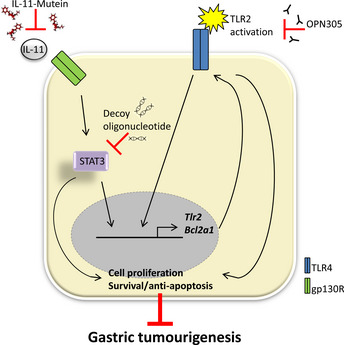
Potential targeted therapies for IL‐11/STAT3‐driven upregulation of TLR2 in GC. BCL2A1, BCL2‐related protein A1; GC, gastric cancer; gp130R, glycoprotein 130 receptor; IL, interleukin; STAT, signal transducer and activator of transcription.
While a definitive link between IL‐11 and TLR2 has been reported for GC,11 such a link between IL‐6 cytokine and TLR family members has yet to be formally proven in CRC (and other GI cancers for that matter). Nonetheless, the increased individual expression of TLR4 and IL‐6 is a common feature of colorectal cancers and is associated with poor prognosis.70, 71 In the near future, it will be of great interest to assess the co‐upregulation of these family members and their downstream signaling pathways in GI cancers. Furthermore, studies have reported the effectiveness of an antibody‐mediated blockade of either TLR4 or IL‐6 to reduce the formation of colorectal tumors in experimentally induced mouse cancer models.25, 53 Unlike TLR4, the clinical efficacy of an IL‐6 blockade has been demonstrated with the humanized anti‐IL‐6 receptor monoclonal antibody tocilizumab in rheumatoid arthritis patients.72 However, such an antagonist has yet to be proven as an effective anticancer agent in clinical trials. With respect to TLR‐related biomarkers and therapeutic targets, other potential candidates include ligands/agonists that signal via TLR. For instance, the TLR2/4 agonists HMGB1 and S100A9 have been touted as potential biomarkers for CRC,31, 33 as they are significantly upregulated in CRC and have been shown to be regulated by STAT3,61, 62 which is hyperactivated in approximately 90% of colorectal tumor biopsies.52 In GC, increased serum levels of SAA and HMGB1 are associated with poor prognostic outcomes,31, 38 and the fact such proteins can be secreted into the bloodstream makes them attractive candidates for GC biomarkers.
In summary, given the relative paucity of effective therapies against GI cancers, there is a high and largely unmet demand to identify novel therapeutic targets. Until now, the therapeutic targeting of TLR and IL‐6 cytokine family members in a range of genetic and preclinical cancer and inflammatory mouse disease models has been considered in isolation. While these therapies are generally efficacious, the potential for translation of such approaches to the clinic now needs to be fully and rigorously investigated. Future studies may benefit substantially by employing a combinatorial approach against both families.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
The authors kindly thank members from the Centre for Innate Immunity and Infectious Diseases (Monash University, Clayton, Vic., Australia), in particular Rebecca Smith, for fruitful discussion and proofreading of this manuscript. This work was supported by grants from the National Health and Medical Research Council of Australia and Association for International Cancer Research (UK), as well as the Operational Infrastructure Support Program by the Victorian Government of Australia.
(Cancer Sci, doi: 10.1111/cas.12205, 2013)
References
- 1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008 v2.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10. International Agency for Research on Cancer, 2010. [Cited 15 Mar 2013.] Available from URL: http://globocan.iarc.fr. [Google Scholar]
- 2. Cherbuy C, Honvo‐Houeto E, Bruneau A et al Microbiota matures colonic epithelium through a coordinated induction of cell cycle‐related proteins in gnotobiotic rat. Am J Physiol Gastrointest Liver Physiol 2010; 299: G348–57. [DOI] [PubMed] [Google Scholar]
- 3. Hooper LV. Bacterial contributions to mammalian gut development. Trends Microbiol 2004; 12: 129–34. [DOI] [PubMed] [Google Scholar]
- 4. Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12: 354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV‐ and HCV‐associated hepatocellular carcinoma. Nat Rev Cancer 2013; 13: 123–35. [DOI] [PubMed] [Google Scholar]
- 6. Goel GA, Kandiel A, Achkar JP, Lashner B. Molecular pathways underlying IBD‐associated colorectal neoplasia: therapeutic implications. Am J Gastroenterol 2011; 106: 719–30. [DOI] [PubMed] [Google Scholar]
- 7. Milne AN, Carneiro F, O'Morain C, Offerhaus GJ. Nature meets nurture: molecular genetics of gastric cancer. Hum Genet 2009; 126: 615–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cunningham D, Atkin W, Lenz HJ et al Colorectal cancer. Lancet 2010; 375: 1030–47. [DOI] [PubMed] [Google Scholar]
- 9. El‐Omar EM, Carrington M, Chow WH et al The role of interleukin‐1 polymorphisms in the pathogenesis of gastric cancer. Nature 2001; 412: 99. [DOI] [PubMed] [Google Scholar]
- 10. Tahara T, Arisawa T, Wang F et al Toll‐like receptor 2–196 to 174del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci 2007; 98: 1790–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tye H, Kennedy CL, Najdovska M et al STAT3‐driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell 2012; 22: 466–78. [DOI] [PubMed] [Google Scholar]
- 12. Mirande C, Kadlecikova E, Matulova M et al Dietary fibre degradation and fermentation by two xylanolytic bacteria Bacteroides xylanisolvens XB1A and Roseburia intestinalis XB6B4 from the human intestine. J Appl Microbiol 2010; 109: 451–60. [DOI] [PubMed] [Google Scholar]
- 13. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular‐phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104: 13780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maslowski KM, Vieira AT, Ng A et al Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009; 461: 1282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arthur JC, Perez‐Chanona E, Muhlbauer M et al Intestinal inflammation targets cancer‐inducing activity of the microbiota. Science 2012; 338: 120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 2010; 11: 373–84. [DOI] [PubMed] [Google Scholar]
- 17. Mandell L, Moran AP, Cocchiarella A et al Intact gram‐negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll‐like receptor 2 but not toll‐like receptor 4. Infect Immun 2004; 72: 6446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jenkins KA, Mansell A. TIR‐containing adaptors in Toll‐like receptor signalling. Cytokine 2010; 49: 237–44. [DOI] [PubMed] [Google Scholar]
- 19. Kawai T, Akira S. Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011; 34: 637–50. [DOI] [PubMed] [Google Scholar]
- 20. Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature 2006; 441: 431–6. [DOI] [PubMed] [Google Scholar]
- 21. Rakoff‐Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 2007; 317: 124–7. [DOI] [PubMed] [Google Scholar]
- 22. Grivennikov SI, Wang K, Mucida D et al Adenoma‐linked barrier defects and microbial products drive IL‐23/IL‐17‐mediated tumour growth. Nature 2012; 491: 254–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hold GL, Rabkin CS, Chow WH et al A functional polymorphism of toll‐like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology 2007; 132: 905–12. [DOI] [PubMed] [Google Scholar]
- 24. Eyking A, Ey B, Runzi M et al Toll‐like receptor 4 variant D299G induces features of neoplastic progression in Caco‐2 intestinal cells and is associated with advanced human colon cancer. Gastroenterology 2011; 141: 2154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fukata M, Shang L, Santaolalla R et al Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis‐associated tumorigenesis. Inflamm Bowel Dis 2011; 17: 1464–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fukata M, Hernandez Y, Conduah D et al Innate immune signaling by Toll‐like receptor‐4 (TLR4) shapes the inflammatory microenvironment in colitis‐associated tumors. Inflamm Bowel Dis 2009; 15: 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fukata M, Chen A, Vamadevan AS et al Toll‐like receptor‐4 promotes the development of colitis‐associated colorectal tumors. Gastroenterology 2007; 133: 1869–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lowe EL, Crother TR, Rabizadeh S et al Toll‐like receptor 2 signaling protects mice from tumor development in a mouse model of colitis‐induced cancer. PLoS ONE 2010; 5: e13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kennedy CL, Najdovska M, Tye H et al Differential role of MyD88 and Mal/TIRAP in TLR2‐mediated gastric tumourigenesis. Oncogene 2013; doi: 10.1038/onc.2013.205 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 30. Greten FR, Eckmann L, Greten TF et al IKKbeta links inflammation and tumorigenesis in a mouse model of colitis‐associated cancer. Cell 2004; 118: 285–96. [DOI] [PubMed] [Google Scholar]
- 31. Lee H, Song M, Shin N et al Diagnostic significance of serum HMGB1 in colorectal carcinomas. PLoS ONE 2012; 7: e34318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luo Y, Chihara Y, Fujimoto K et al High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur J Cancer 2013; 49: 741–51. [DOI] [PubMed] [Google Scholar]
- 33. Kim HJ, Kang HJ, Lee H et al Identification of S100A8 and S100A9 as serological markers for colorectal cancer. J Proteome Res 2009; 8: 1368–79. [DOI] [PubMed] [Google Scholar]
- 34. Kallberg E, Vogl T, Liberg D et al S100A9 interaction with TLR4 promotes tumor growth. PLoS ONE 2012; 7: e34207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute‐phase serum amyloid A. J Immunol 2008; 181: 22–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim S, Takahashi H, Lin WW et al Carcinoma‐produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009; 457: 102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Z, Zhang J, Miao L et al Interleukin‐11 promotes the progress of gastric carcinoma via abnormally expressed versican. Int J Biol Sci 2012; 8: 383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu C, Pan C, Shen J, Wang H, Yong L. Identification of serum amyloid A in the serum of gastric cancer patients by protein expression profiling. Oncol Lett 2012; 3: 1259–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ernst M, Najdovska M, Grail D et al STAT3 and STAT1 mediate IL‐11‐dependent and inflammation‐associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest 2008; 118: 1727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grivennikov S, Karin E, Terzic J et al IL‐6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis‐associated cancer. Cancer Cell 2009; 15: 103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller‐Newen G, Schaper F. Principles of interleukin (IL)‐6‐type cytokine signalling and its regulation. Biochem J 2003; 374: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9: 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu L, Du H, Li Y, Qu P, Yan C. Signal transducer and activator of transcription 3 (Stat3C) promotes myeloid‐derived suppressor cell expansion and immune suppression during lung tumorigenesis. Am J Pathol 2011. Oct; 179: 2131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bromberg JF, Wrzeszczynska MH, Devgan G et al Stat3 as an oncogene. Cell 1999; 98: 295–303. [DOI] [PubMed] [Google Scholar]
- 45. Judd LM, Bredin K, Kalantzis A, Jenkins BJ, Ernst M, Giraud AS. STAT3 activation regulates growth, inflammation, and vascularization in a mouse model of gastric tumorigenesis. Gastroenterology 2006; 131: 1073–85. [DOI] [PubMed] [Google Scholar]
- 46. Jenkins BJ, Grail D, Nheu T et al Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF‐beta signaling. Nat Med 2005; 11: 845–52. [DOI] [PubMed] [Google Scholar]
- 47. Bollrath J, Phesse TJ, von Burstin VA et al gp130‐mediated Stat3 activation in enterocytes regulates cell survival and cell‐cycle progression during colitis‐associated tumorigenesis. Cancer Cell 2009; 15: 91–102. [DOI] [PubMed] [Google Scholar]
- 48. Kortylewski M, Kujawski M, Wang T et al Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005; 11: 1314–21. [DOI] [PubMed] [Google Scholar]
- 49. Kim DY, Cha ST, Ahn DH et al STAT3 expression in gastric cancer indicates a poor prognosis. J Gastroenterol Hepatol 2009; 24: 646–51. [DOI] [PubMed] [Google Scholar]
- 50. Ellmark P, Ingvarsson J, Carlsson A, Lundin BS, Wingren C, Borrebaeck CA. Identification of protein expression signatures associated with Helicobacter pylori infection and gastric adenocarcinoma using recombinant antibody microarrays. Mol Cell Proteomics 2006; 5: 1638–46. [DOI] [PubMed] [Google Scholar]
- 51. Inagaki‐Ohara K, Mayuzumi H, Kato S et al Enhancement of leptin receptor signaling by SOCS3 deficiency induces development of gastric tumors in mice. Oncogene 2012; doi: 10.1038/onc.2012.540 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 52. Corvinus FM, Orth C, Moriggl R et al Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 2005; 7: 545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matsumoto S, Hara T, Mitsuyama K et al Essential roles of IL‐6 trans‐signaling in colonic epithelial cells, induced by the IL‐6/soluble‐IL‐6 receptor derived from lamina propria macrophages, on the development of colitis‐associated premalignant cancer in a murine model. J Immunol 2010; 184: 1543–51. [DOI] [PubMed] [Google Scholar]
- 54. Rose‐John S, Scheller J, Elson G, Jones SA. Interleukin‐6 biology is coordinated by membrane‐bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol 2006; 80: 227–36. [DOI] [PubMed] [Google Scholar]
- 55. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF‐kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 2010; 21: 11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mansell A, Jenkins BJ. Dangerous liaisons between interleukin‐6 cytokine and toll‐like receptor families: a potent combination in inflammation and cancer. Cytokine Growth Factor Rev 2013; 24: 249–56. [DOI] [PubMed] [Google Scholar]
- 57. Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL‐6 and activates transcription by binding to NFkappaB. Genes Dev 2007; 21: 1396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Naugler WE, Sakurai T, Kim S et al Gender disparity in liver cancer due to sex differences in MyD88‐dependent IL‐6 production. Science 2007; 317: 121–4. [DOI] [PubMed] [Google Scholar]
- 59. Theocharis AD, Vynios DH, Papageorgakopoulou N, Skandalis SS, Theocharis DA. Altered content composition and structure of glycosaminoglycans and proteoglycans in gastric carcinoma. Int J Biochem Cell Biol 2003; 35: 376–90. [DOI] [PubMed] [Google Scholar]
- 60. Hagihara K, Nishikawa T, Sugamata Y et al Essential role of STAT3 in cytokine‐driven NF‐kappaB‐mediated serum amyloid A gene expression. Genes Cells 2005; 10: 1051–63. [DOI] [PubMed] [Google Scholar]
- 61. Lee MJ, Lee JK, Choi JW et al Interleukin‐6 induces S100A9 expression in colonic epithelial cells through STAT3 activation in experimental ulcerative colitis. PLoS ONE 2012; 7: e38801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu H, Yao YM, Yu Y, Dong N, Yin HN, Sheng ZY. Role of Janus kinase/signal transducer and activator of transcription pathway in regulation of expression and inflammation‐promoting activity of high mobility group box protein 1 in rat peritoneal macrophages. Shock 2007; 27: 55–60. [DOI] [PubMed] [Google Scholar]
- 63. Meyer HJ, Wilke H. Treatment strategies in gastric cancer. Dtsch Arztebl Int 2011; 108: 698–705; quiz 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cunningham D, Allum WH, Stenning SP et al Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006; 355: 11–20. [DOI] [PubMed] [Google Scholar]
- 65. Ooi CH, Ivanova T, Wu J et al Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet 2009; 5: e1000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Deng JY, Sun D, Liu XY, Pan Y, Liang H. STAT‐3 correlates with lymph node metastasis and cell survival in gastric cancer. World J Gastroenterol 2010; 16: 5380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Harmegnies D, Wang XM, Vandenbussche P et al Characterization of a potent human interleukin‐11 agonist. Biochem J 2003; 375: 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sen M, Thomas SM, Kim S et al First‐in‐human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov 2012; 2: 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McCormack W, Oshima M, Tan P, Jenkins B. Toll‐like receptor 2: therapeutic target for gastric carcinogenesis. Oncotarget 2012; 3: 1260–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang EL, Qian ZR, Nakasono M et al High expression of Toll‐like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer 2010; 102: 908–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kantola T, Klintrup K, Vayrynen JP et al Stage‐dependent alterations of the serum cytokine pattern in colorectal carcinoma. Br J Cancer 2012; 107: 1729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Smolen JS, Beaulieu A, Rubbert‐Roth A et al Effect of interleukin‐6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double‐blind, placebo‐controlled, randomised trial. Lancet 2008; 371: 987–97. [DOI] [PubMed] [Google Scholar]
- 73. Zeng HM, Pan KF, Zhang Y et al Genetic variants of toll‐like receptor 2 and 5, Helicobacter pylori infection, and risk of gastric cancer and its precursors in a chinese population. Cancer Epidemiol Biomarkers Prev 2011; 20: 2594–602. [DOI] [PubMed] [Google Scholar]
- 74. de Oliveira JG, Silva AE. Polymorphisms of the TLR2 and TLR4 genes are associated with risk of gastric cancer in a Brazilian population. World J Gastroenterol 2012; 18: 1235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pimentel‐Nunes P, Teixeira AL, Pereira C et al Functional polymorphisms of Toll‐like receptors 2 and 4 alter the risk for colorectal carcinoma in Europeans. Dig Liver Dis 2013; 45: 63–9. [DOI] [PubMed] [Google Scholar]
- 76. Slattery ML, Herrick JS, Bondurant KL, Wolff RK. Toll‐like receptor genes and their association with colon and rectal cancer development and prognosis. Int J Cancer 2012; 130: 2974–80. [DOI] [PMC free article] [PubMed] [Google Scholar]