

Abstract
Background
Zymomonas mobilis has recently been shown to be capable of producing the valuable platform biochemical, 2,3-butanediol (2,3-BDO). Despite this capability, the production of high titers of 2,3-BDO is restricted by several physiological parameters. One such bottleneck involves the conversion of acetoin to 2,3-BDO, a step catalyzed by 2,3-butanediol dehydrogenase (Bdh). Several Bdh enzymes have been successfully expressed in Z. mobilis, although a highly active enzyme is yet to be identified for expression in this host. Here, we report the application of a phylogenetic approach to identify and characterize a superior Bdh, followed by validation of its structural attributes using a mutagenesis approach.
Results
Of the 11 distinct bdh genes that were expressed in Z. mobilis, crude extracts expressing Serratia marcescens Bdh (SmBdh) were found to have the highest activity (8.89 µmol/min/mg), when compared to other Bdh enzymes (0.34–2.87 µmol/min/mg). The SmBdh crystal structure was determined through crystallization with cofactor (NAD+) and substrate (acetoin) molecules bound in the active site. Active SmBdh was shown to be a tetramer with the active site populated by a Gln247 residue contributed by the diagonally opposite subunit. SmBdh showed a more extensive supporting hydrogen-bond network in comparison to the other well-studied Bdh enzymes, which enables improved substrate positioning and substrate specificity. This protein also contains a short α6 helix, which provides more efficient entry and exit of molecules from the active site, thereby contributing to enhanced substrate turnover. Extending the α6 helix to mimic the lower activity Enterobacter cloacae (EcBdh) enzyme resulted in reduction of SmBdh function to nearly 3% of the total activity. In great contrast, reduction of the corresponding α6 helix of the EcBdh to mimic the SmBdh structure resulted in ~ 70% increase in its activity.
Conclusions
This study has demonstrated that SmBdh is superior to other Bdhs for expression in Z. mobilis for 2,3-BDO production. SmBdh possesses unique structural features that confer biochemical advantage to this protein. While coordinated active site formation is a unique structural characteristic of this tetrameric complex, the smaller α6 helix and extended hydrogen network contribute towards improved activity and substrate promiscuity of the enzyme.
Keywords: Butanediol dehydrogenase; Serratia marcescens; Acetoin; 2,3-Butanediol; Crystallography; Phylogenetics
Background
Petroleum alternatives are critical to maintaining a sustainable economy while satisfying the ever-growing global energy demands. Diols, potentially renewable compounds containing two hydroxyl groups, have wide-ranging applications in chemicals and fuels. 2,3-butanediol (2,3-BDO) is exemplar of industrial diols having been used in liquid fuels, cosmetics and drugs, paints, food additives, and synthetic rubber [1, 2].
With two chiral centers, three isomers of 2,3-BDO are possible. The levo (2R, 3R) and dextro (2S, 3S) isomers are optically active, whereas the meso (2R, 3S) isomer is not [3, 4]. Biological synthesis of 2,3-BDO occurs via three enzymatic conversion steps [5, 6]. The first step involves condensation of two pyruvate molecules by the acetolactate synthase (Als) in a single decarboxylation reaction to produce α-acetolactate. A second decarboxylation step catalyzed by the enzyme acetolactate-decarboxylase produces acetoin. In the presence of NADH, acetoin is reduced to 2,3-BDO by the enzyme butanediol dehydrogenase (Bdh). Under aerobic conditions, the acetoin spontaneously converts to diacetyl, which can be reconverted to acetoin by diacetyl reductase and subsequently reduced to 2,3-BDO. Depending on oxygenation conditions, (3R)-acetoin or (3S)-acetoin is preferably produced by microorganisms. (3R)-acetoin is the product of anaerobic fermentation from acetolactate. However, under aerobic conditions, (3S)-acetoin is produced from diacetyl [7]. Depending on the type of acetoin used, three different stereoisomers of 2,3-BDO, (2R, 3R)-BDO, (2R-3S)-BDO, and (2S, 3S)-BDO are produced [8].
Several bacteria, including Klebsiella sps. Enterobacter sps., Pseudomonas sps., Serratia sp., and Bacillus sps. are known to naturally produce 2,3-BDO. Interestingly, the highest 2,3-BDO concentrations are produced by risk group 2 organisms and are thus not desirable for large-scale production [4, 9]. Organisms, such as Escherichia coli, Lactobacillus lactis, Synechococcus elongatus, and even yeasts such as Saccharomyces cerevisiae have been engineered to produce 2,3-BDO. The 2,3-BDO titers reported are 73.8, 51, 2.38, and 154.3 g/L, respectively, for these organisms [10–13].
Zymomonas mobilis is known primarily for its ethanologenic properties. In comparison to yeasts, which use the Entner–Meyerhof–Parnas pathway for glycolysis, Z. mobilis uses the Entner–Doudoroff (ED) pathway. The ED pathway is found in facultative anaerobes and aerobic microorganisms, leading to higher ethanol yields [14]. Z. mobilis has other advantages, such as high alcohol and pH tolerance, high rate of sugar uptake, low biomass production, and reduced aeration requirement thereby reducing the production costs [15]. Interestingly, Z. mobilis is capable of using both deacetylated-disc refined (DDR) and deacetylated-mechanically refined (DMR) sugar streams for ethanol production [16]. Recently, it was also demonstrated by our laboratory that the ethanol flux could be diverted to 2,3-BDO using pure or mixed sugars as substrates. By introducing the three 2,3-BDO pathway genes followed by promoter replacements and fermentation condition optimizations, the 2,3-BDO titers of > 13 g/L was achieved [15].
One of the bottlenecks for production of 2,3-BDO in Z. mobilis is the competition between NADH oxidase (Ndh) and Bdh to oxidize NADH to NAD+ under oxic conditions. This is in addition to the NADH demand for glycerol and ethanol production by this organism. Depending on the activity of the Bdh enzyme, the conversion of NADH could be shifted towards 2,3-BDO production instead of the respiratory chain. The conversion of acetoin to 2,3-BDO by Bdh is a reversible reaction governed by the pH [17, 18].
Bdh enzymes can be classified into R-acting or S-acting depending on the chirality of the chiral center introduced by the enzyme at the acetoin C2 atom. Whereas the preference for (3R)-acetoin or (3S)-acetoin is imprinted in the geometry of the substrate-binding pocket, R-acting and S-acting Bdh enzymes belong to different protein families and possess different architectures. Recently, the first structure of an R-acting Bdh from Bacillus subtilis was deposited to the Protein Data Bank (PDB, www.rcsb.org, [19]) with the PDB ID 6IE0, while there are numerous structures available for other members of the medium-chain dehydrogenase/reductase (MDR) superfamily [20–23]. Examples of R-acting Bdh enzymes are those from S. cerevisiae, Paenibacillus polymyxa, B. subtilis, Pseudomonas putida, and Bacillus licheniformis [24–27].
S-acting Bdh enzymes belong to the short-chain dehydrogenase/reductase (SDR) superfamily of proteins [28]. The substrates for this superfamily of enzymes vary greatly in size and include glucose, alcohols, and steroids [29]. These enzymes are well studied and several S-acting Bdhs are characterized structurally, such as those of (3R)-acetoin-dependent S-acting Bdh from Klebsiella pneumoniae (PDB ID 1GEG, deposited in 2001) and (3S)-acetoin-dependent S-acting Bdh from Corynebacterium glutamicum (PDB ID 3A28, deposited in 2010) reported by Otagiri and colleagues [30, 31].
In this work, we have focused on identifying the most active Bdh enzyme that can function in Z. mobilis using a phylogenetics approach to enable efficient conversion of acetoin to 2,3-BDO. Furthermore, we have obtained the crystal structure of the Serratia marcescens Bdh (SmBdh) by expressing it in this industrially relevant host and provided comparative analysis against similar Bdh enzymes. We have also carried out structurally guided changes to SmBdh to explain its superiority over lower activity Bdh enzymes from the same family of proteins, followed by biochemical confirmation of the activity of the designed mutants.
Results and discussion
Production of 2,3-BDO is dependent on several factors, some of which include the types of heterologous 2,3-BDO pathway genes used, the levels of oxygen present, the presence of competitive pathways, such as ethanol and glycerol, as well as competing enzymes for NADH conversion. We have concentrated on only one of these parameters, namely the activity of the NADH-consuming enzyme, Bdh, that can compete with Ndh in the presence of oxygen. This biochemical step is considered as one of the bottlenecks in 2,3-BDO production by this organism [15]. Although Z. mobilis has been shown to express several commonly known Bdh enzymes [15], there has been no comprehensive study undertaken to express functional Bdhs from diverse species that could contribute towards improving 2,3-BDO production in this organism. We therefore decided to take a phylogenetic approach to this problem by screening Bdh enzymes across different bacterial kingdoms to identify the best active enzyme that could be engineered in this organism.
Identification of butanediol dehydrogenase sequences for expression in Z. mobilis
A total of 57 protein sequences were included in the list of Bdh for phylogenetic analysis. This list included two sequences each from Azotobacter vinelandii, Acidovorax avenae, Rhodococcus jostii, and Agrobacterium tumefaciens. These sequences belonged to two different classes of Bdh proteins based on their protein sequence lengths. Bifidobacterium asteroides contained three Bdh sequences, two belonging to the short length class and one belonging to the long class. We also included B. subtilis, B. licheniformis, K. pneumoniae, E. cloacae, and S. marcescens, studies for which expression has been carried out in E. coli [13]. Clearly, two distinct clusters were observed on the phylogenetic tree, one cluster (21 sequences) primarily contained the “short length” Bdhs, while the other (36 sequences) contained the “long” Bdhs (Fig. 1). The “short length” Bdh cluster comprised S-acting enzymes (presumably belonging to the SDR superfamily), such as K. pneumoniae, E. cloacae and S. marcescens [18, 32, 33]. The “long” Bdh cluster comprised R-acting enzymes (presumably belonging to the MDR superfamily), such as B. subtilis and C. beijerinckii [33]. Each of these two major clusters were further subdivided into smaller sub-clusters. Based on their clustering pattern, we subdivided the “short length” clusters into two sub-clusters, while subdividing the “long” class of Bdhs into five sub-clusters. Eight protein sequences were selected from the “long” and three sequences were selected from the “short-length” clusters, such that at least one representative protein sequence was selected from each sub-cluster of the phylogenetic tree (Fig. 1).
Fig. 1.

Phylogenetic tree analysis of butanediol dehydrogenases. The neighbor joining tree was generated using 57 full-length Bdh sequences. The optimal tree with the sum of branch length = 16.08413861 is shown. Boot strap values are shown next to the branches. The tree has been divided into 2 clusters, the medium-chain dehydrogenases/reductases (MDR) and short chain dehydrogenases/reductases (SDR). The MDR and SDR clusters are further subdivided into 5 and 2 sub-clusters, respectively. The sub-clusters are represented by numbers 1 through 7. Red triangles represent the sequences selected for gene synthesis
Screening of butanediol dehydrogenases for protein expression and activity in Z. mobilis
The Z. mobilis strain used in this study was 9C, which was a modified version of strain 8b that was originally developed for improved ethanol production [15, 34]. The 9C strain lacked the chloramphenicol and tetracycline markers that the 8b strain contained. Neither of these strains were engineered for BDH production. Moreover, native Z. mobilis does not contain the BDH pathway to enable 2,3-BDO production. Thus, our study was not designed to carry out expression of the entire 2,3-BDO pathway, instead it was only intended to study expression and activity of the expressed BDH enzyme in this organism. Transformants obtained from the spectinomycin selection plates were confirmed for the presence of the individual bdh genes by PCR analysis. Eight individual transformants were subjected to colony PCR, results of which are shown in Additional file 1. Based on PCR analysis, five independent transformants were selected for protein expression analysis. Because these Bdh proteins were expressed without an epitope tag, we decided to use activity assays as a screen to detect protein expression in the different Z. mobilis transformants. The results of the activity assay are shown in Table 1. We observed that most of the bdh transformants showed an activity of ≤ 2 µmol/min/mg using acetoin and NADH as the substrate and cofactor, respectively. This included 9 of the 11 tested bdh genes (Additional file 2). We used three Z. mobilis strains expressing the E. cloacae, B. subtilis, and Lactococcus lactis bdh genes under the same promoter as controls for which expression and/or 2,3-BDO production has been tested in-house (unpublished data). Two of the Bdh enzymes, namely those from M. luteus and S. marcescens, showed Bdh activity of ~ 3.0 and 9.0 µmol/min/mg, respectively, which was 2- to 4-fold higher than the control strains. Based on the activity analysis, we selected S. marcescens Bdh (SmBdh) as the candidate enzyme for structural characterization. We also carried out SDS-PAGE analysis to determine expression of individual Bdh protein produced by these transformants. Interestingly, only some of the bdh gene transformants showed unique bands at the expected molecular weight based on Coomassie staining (Additional file 3), suggesting different expression levels of the heterologous Bdh proteins. It is likely that some of these heterologous enzymes were not expressed to detectable levels, which may have been a factor in their poor activity levels. Nevertheless, for the purpose of screening, we used their crude extract activities as the standard to determine the best-expressing and active enzyme.
Table 1.
Enzyme activity of 2,3-butanediol dehydrogenase enzymes expressed in Z. mobilis strain 9C
| Sample # | Origin of BDH enzyme | Activity of the crude extracts (µmol/min/mg) | Average activity of the crude extracts (µmol/min/mg) | Sample # | Origin of BDH enzyme | Activity of the crude extracts (µmol/min/mg) | Average activity of the crude extracts (µmol/min/mg) |
|---|---|---|---|---|---|---|---|
| 1 | Negative control | ND | ND | 30 | Sw1 | 1.13 ± 0.18 | 1.15 ± 0.38 |
| 31 | Sw2 | 1.42 ± 0.27 | |||||
| 2 | Ec | 1.79 ± 0.53 | 1.79 ± 0.53 | 32 | Sw3 | 0.68 ± 0.13 | |
| 3 | Bs | 0.34 ± 0.06 | 0.34 ± 0.06 | 33 | Sw4 | 1.71 ± 0.17 | |
| 4 | Ll | 1.73 ± 0.16 | 1.73 ± 0.16 | 34 | Sw5 | 0.81 ± 0.21 | |
| 5 | Dd1 | 1.03 ± 0.18 | 1.92 ± 0.51 | 35 | Ea1 | 1.22 ± 0.11 | 1.75 ± 0.29 |
| 6 | Dd2 | 2.47 ± 0.08 | 36 | Ea2 | 2.08 ± 0.58 | ||
| 7 | Dd3 | 1.85 ± 0.39 | 37 | Ea3 | 1.75 ± 0.10 | ||
| 8 | Dd4 | 1.87 ± 0.29 | 38 | Ea4 | 1.79 ± 0.48 | ||
| 9 | Dd5 | 2.41 ± 0.27 | 39 | Ea5 | 1.94 ± 0.13 | ||
| 10 | Sc1 | 1.71 ± 0.52 | 1.27 ± 0.29 | 40 | Ml1 | 2.41 ± 0.89 | 2.87 ± 0.75 |
| 11 | Sc2 | 1.26 ± 0.48 | 41 | Ml2 | 3.09 ± 0.51 | ||
| 12 | Sc3 | 1.13 ± 0.03 | 42 | Ml3 | 2.49 ± 0.27 | ||
| 13 | Sc4 | 1.42 ± 0.37 | 43 | Ml4 | 2.14 ± 0.08 | ||
| 14 | Sc5 | 0.84 ± 0.05 | 44 | Ml5 | 4.26 ± 0.56 | ||
| 15 | Sm1 | 6.48 ± 0.97 | 8.89 ± 1.43 | 45 | Mo1 | 1.40 ± 0.26 | 1.44 ± 0.29 |
| 16 | Sm2 | 10.95 ± 1.22 | 46 | Mo2 | 1.96 ± 0.18 | ||
| 17 | Sm3 | 9.27 ± 1.21 | 47 | Mo3 | 1.48 ± 0.24 | ||
| 18 | Sm4 | 9.10 ± 1.40 | 48 | Mo4 | 1.07 ± 0.47 | ||
| 19 | Sm5 | 8.67 ± 0.92 | 49 | Mo5 | 1.30 ± 0.38 | ||
| 20 | At1 | 1.18 ± 0.37 | 1.15 ± 0.59 | 50 | Ms1 | 1.34 ± 0.38 | 1.27 ± 0.24 |
| 21 | At2 | 1.18 ± 0.27 | 51 | Ms2 | 1.22 ± 0.29 | ||
| 22 | At3 | 0.52 ± 0.11 | 52 | Ms3 | 1.63 ± 0.29 | ||
| 23 | At4 | 2.22 ± 0.15 | 53 | Ms4 | 0.87 ± 0.33 | ||
| 24 | At5 | 0.68 ± 0.29 | 54 | Ms5 | 1.30 ± 0.20 | ||
| 25 | Av1 | 1.24 ± 0.12 | 1.30 ± 0.35 | 55 | Tg1 | 1.09 ± 0.29 | 1.13 ± 0.39 |
| 26 | Av2 | 1.92 ± 0.11 | 56 | Tg2 | 0.71 ± 0.19 | ||
| 27 | Av3 | 1.40 ± 0.45 | 57 | Tg3 | 1.87 ± 0.47 | ||
| 28 | Av4 | 0.91 ± 0.67 | 58 | Tg4 | 0.90 ± 0.30 | ||
| 29 | Av5 | 1.05 ± 0.38 | 59 | Tg5 | 1.12 ± 0.06 |
Bdh activity assays were performed on whole cell extracts obtained from transformant cultures (µmol/min per mg crude extract). Each row represents activity assays performed in technical triplicates for every transformant. Five independent transformants were tested for each gene. Therefore, average specific activity has been calculated from 3 × 5 = 15 data points
Ec, E. cloacae; Bs, Bacillus subtilis; Ll, Lactococcus lactis; Dd, Dickeya dadantii; Sc, Streptomyces coelicolor; Sm, Serratia marcescens; At, Agrobacterium tumefaciens; Av, Azotobacter vinelandii; Sw, Staphylococcus warneri; Ea, Erwinia amylovora; Ml, Micrococcus luteus; Mo, Myroides odoratimimus; Ms, Mycobacterium smegmatis; Tg, Thermococcus gammatolerans
Crystal structure of SmBdh along with its bound cofactor and substrate molecules
Expression of a high-performing BDH enzyme in Z. mobilis is critical for 2,3-BDO production in this industrially relevant ethanol producer. Although the structural analysis of SmBdh was not the primary intent of this study, considering that this enzyme turned out to be the best expressed and most active enzyme in this organism, it was therefore deemed important to determine the reasons behind the improved functionality of this enzyme. The Z. mobilis expressed WT SmBdh crystallized in the space group P43212 with two protein molecules per asymmetric unit that could be superimposed with r.m.s.d. of 0.271 Å over 1379 atoms. Two molecules found in the asymmetric unit form a tetramer (dimer of dimers) with a symmetry-related dimer (Fig. 2a) with extensive interface surfaces (Table 2, [35]). The tetrameric arrangement is common to the proteins belonging to the SDR superfamily. SmBdh was shown to be a tetramer in solution by Native PAGE [36]. The main difference between the two molecules in the asymmetric unit is that molecule A is found in ‘closed’ conformation (that could be best described by ~ 10 Å distance between Cα-atoms of residues Ala93 and Trp192) and molecule B is found in ‘open’ conformation (the same distance is ~ 14 Å) (Additional file 4, Fig. 2b and c). Upon examination of the electron density maps, we were able to locate NAD + cofactor molecule and acetoin molecules in protein molecule A (‘closed’ conformation Fig. 3a), while in molecule B (‘open’ conformation) the corresponding space is occupied by only adenine diphosphate (Fig. 3b).
Fig. 2.

SmBdh functional complex and key structural features. a SmBdh tetramer. Protein chains A, B, Asymm and Bsymm are shown in cartoon representation and colored grey, yellow, green and cyan, respectively. NAD + , ADP and acetoin molecules are shown in sticks representation. N- and C-termini of the protein chains are marked. b ‘Open’ and ‘closed’ conformations of SmBdh superimposed. Protein chains are shown in cartoon representation and colored grey except for the loop 180–203 containing helices α6 and α7. ‘Open’ conformation of the loop 180–203 is colored purple and ‘closed’ conformation is colored green. Secondary structure elements are labeled. NAD + and acetoin molecules bound in ‘closed’ conformation are shown in sticks representation. Positions of Cα atoms of Ala93 and Trp192 are shown by arrows. c Zoom-in view of NAD + interaction with the ‘closed’ conformation of SmBdh. Side chains of Thr186, Met188, and Thr189 are shown in sticks representation. H-bonds formed upon NAD + binding are shown as yellow dashed lines
Table 2.
Buried area of the protein–protein interfaces in the WT SmBDH tetramer
| Assembly | Surface area, Å2 | Buried area, Å2 | Buried area, % |
|---|---|---|---|
| ABAsymmBsymm | 31,520 | 16,820 | 53.4 |
| ABsymm | 21,020 | 3140 | 14.9 |
| AB | 21,530 | 2640 | 12.3 |
| AAsymm | 21,150 | 2830 | 13.4 |
| BBsymm | 21,940 | 2410 | 11.0 |
| A | 11,992 | ||
| B | 12,174 |
Fig. 3.

Substrate and cofactor interactions within SmBdh molecule. a Omit Fo-Fc electron density map (calculated after NAD + and acetoin molecules removed from the model) shown at 3σ level for the NAD + and acetoin molecules found in the ‘closed’ conformation of the WT SmBdh. b Omit Fo-Fc electron density map (calculated after ADP molecule was removed from the model) shown at 3σ level for the ADP molecule found in the ‘closed’ conformation of the WT SmBdh. c NAD + interactions with the ‘closed’ conformation of the WT SmBdh
The transition from the ‘open’ conformation into the ‘closed’ seems to be coincidental with NAD + binding and includes the shift of the loop 183–203, containing two helices, α6 and α7 (Fig. 2b, c). In the process, the short helix α6 is unfolded, but this is compensated by the hydrogen bonds formed between the NAD + and Thr186Oγ and between the main chain carbonyl of Val184 and Thr189Oγ (Figs. 2c, 3). Additional stabilization is likely provided by the hydrophobic interaction between Met188 side chain and nicotinamide ring (Fig. 2c). Interactions between protein and NAD + are shown in Fig. 3c.
WT SmBdh contains acetoin in the substrate-binding pocket of the ‘closed’ molecule A. Crystallization conditions included NAD + and acetoin, and more acetoin was used for cryoprotection. Omit electron density maps showed three bulges at the C3 atom (Fig. 4a). When only (3R)-acetoin molecule was modeled, a positive peak at the difference map showed up (Fig. 4b). We interpreted this as both (3R)- and (3S)-acetoin molecules bound in the pocket since commercially available acetoin is a racemic mixture and no other small molecules (like ethylene glycol) were added during crystallization or as a cryoprotectant. Out of three “bulges”, only one should be modeled as a methyl group since its surrounding is hydrophobic (side chains of Leu183, Trp192, Phe200, and Thr189Cγ) and no possible hydrogen bond donors or acceptors are available. Thus, this bulge was modeled as C4 atom of both (3R)-acetoin and (3S)-acetoin. Two other bulges, in contrast, have hydrogen bond partners available: Gln247symmNε (3.1 Å), Ser140Oγ (3.1 Å), or a water molecule (2.7 Å) for (3R)-acetoin O3 atom, and Gln247symmOε (3.0 Å) or Ser138 Oγ (3.2 Å) for (3S)-acetoin O3 atom.
Fig. 4.

Placement of acetoin molecules in the active site of SmBdh and its intramolecular interactions. a Placement of R- and S-acetoin molecules in the active site of the WT SmBDH. NAD + (yellow carbons), R- and S-acetoin molecules (purple and cyan carbons), and side chains of surrounding residues (grey carbons) including Gln247 from the symmetry-related molecule (green carbons) are shown in sticks representation. Hydrogen bonds are depicted as yellow dashed lines. Omit Fo-Fc electron density map at 3σ level is shown as a green mesh over acetoin molecules. b When only R-acetoin is modeled in the active site of the WT SmBDH, a strong peak in the Fo-Fc map appears after refinement (shown as a green mesh at 3σ level) indicating incomplete interpretation of the electron density maps. 2Fo-Fc map contoured at 1σ level is also shown
SmBdh belongs to the SDR superfamily of enzymes
SmBdh exhibits the typical single domain short-chain dehydrogenase/reductase (SDR) architecture. A monomer displays a dinucleotide-binding Rossmann fold that includes seven-stranded parallel β-sheet (β3–β2–β1–β4–β5–β6–β7) flanked by three α-helices (α2, α1, and α8) on one side and three α-helices (α3, α4, and α5) on another. There is an additional small lobe on top of a core structure, formed by two short helices, α6 and α7, creating a deep cleft in which the cofactor is bound, and where the active site is located (Figs. 2 and 3). It should be noted that SmBdh has been shown to be an NADH dependent dehydrogenase enzyme [18]. Two α-turns (αt1 and αt2) can also be recognized in this structure.
PDBeFold [37] search returned over 3000 molecules with Z-score between 6 (usually considered to be the lower threshold for likeliness) and 16.6, and sequence identity up to 37%. Top matching structure could be superimposed over SmBdh with r.m.s.d. of 1.03 Å over 239 Cα-atoms (FabG from Bacillus sp., PDB ID 4NBU). While SDR superfamily is vast and includes enzymes active towards various substrates, there are several reported Bdh structures, namely from Burkholderia cenocepacea (BcBdh, PDB ID 4WEO), Burkholderia xenovorans (BxBdh, PDB ID 5JY1), C. glutamicum (PDB ID 3A28), Gluconobacter oxydans (GoBdh, PDB ID 3WTC), and K. pneumoniae (KpBdh, PDB ID 1GEG) that are available in the PDB database. All mentioned enzymes are S-acting or S-installing, introducing 2S chiral center in the acetoin molecule. However, only two enzymes are well characterized with regard to the substrate chiral specificity: Bdh from K. pneumoniae (KpBdh) is (3R)-acetoin dependent [30] and Bdh from C. glutamicum (CgBdh) is (3S)-acetoin dependent [31]. An explanation for the chiral substrate recognition based on the spatial organization of the substrate-binding pocket has been presented before [31].
Key differences in the SmBdh structure lead to improved positioning of acetoin in the active site of the enzyme
Placement of acetoin molecule inside the active site is key to the efficient conversion of this substrate to 2,3-BDO. To determine the structural characteristics that confers this property to the SmBdh enzyme, we compared this protein with similar Bdhs from other organisms. Based on our assessment, the strategic placement of the hydrophobic cluster on the αt1 side of the acetoin binding site (Ile142, Phe148, Leu151) and possible hydrogen bond partner on the α6 side of the acetoin binding site (Trp192) would be preferential for (3S)-acetoin binding in CgBdh (Figs. 2b and 5a). The opposite arrangement: hydrophobic cluster on the α6 side of the acetoin binding site (Ile184, Trp190, Ile193, Phe212) and possible hydrogen bond partner(s) on the αt1 side (Ser139, Gln140) would be preferential for (3R)-acetoin binding in KpBdh (Figs. 2b and 5b). We also analyzed the substrate binding sites of other Bdh enzymes such as those from GoBdh, BcBdh, and BxBdh. Analyzing substrate binding sites in these three Bdh enzymes suggested that GoBdh and BcBdh are likely (3S)-acetoin dependent, whereas BxBdh is likely (3R)-acetoin dependent. For simplicity, we used KpBdh and CgBdh as reference structures to compare the acetoin binding site of SmBdh. SmBdh stands out when compared to these two protein structures. At a first glance, it looks similar to the KpBdh—a hydrophobic cluster made of Leu183, Thr189Cγ, Trp192, and Phe200 located on the α6 side of the groove is well suited for interaction with the C4 atom of the acetoin molecule (Figs. 5b and c). However, Gln140 of the KpBdh is replaced with Val139 in SmBdh. In this protein, another glutamine side chain makes up for that difference (i.e., Gln247symm from a symmetry-related molecule). Other differences then start to accumulate. Whereas in CgBdh and KpBdh the number of partners for possible H-bonds stabilizing O3 atom of acetoin is very limited—one (Trp192Nε) and possibly two (Gln140Nε and Ser139Oγ), respectively, the acetoin binding site in SmBdh has a vast H-bond network better suited to stabilize both acetoin enantiomers. Specifically, 1: Leu151/149 of CgBdh/KpBdh, respectively, is replaced with Ala148 in SmBdh creating a void where a water molecule W71 is now located (Fig. 5c). This water molecule is held in place by Nε of His91 that replaced Ala 92/90 of CgBdh/KpBdh, respectively, and Oδ of Asp145. This water molecule is placed within 2.7 Å from O3 atom of (3R)-acetoin molecule in SmBdh. 2: Ala143/141 of CgBdh/KpBdh, respectively, is replaced with Ser140 in SmBdh and Ser140Oγ is within 3.1 Å from the O3 atom of (3R)-acetoin. Nε of the Gln247symm is situated 3.1 Å away from the O3 atom of (3R)-acetoin (Fig. 5c). 3: For the (3S)-acetoin, its O3 atom is located 2.8 Å away from Oε of the Gln247symm and 3.2 Å away from Ser138Oγ (Fig. 5c). We can suggest that significant improvements in supporting the hydrogen bond network in the active site of SmBdh, in comparison to the other known Bdhs, contributes to more precise positioning of acetoin molecule in the active site and probably better stabilization of the reaction intermediate which could in turn lead to higher turnover of the substrate.
Fig. 5.

Comparison of the acetoin binding sites of S-acting Bdh enzymes. a CgBdh; b KpBdh, and c SmBdh. Carbon backbone are represented in green for CgBdh, cyan for KpBdh and grey for SmBdh. Main chain atoms of the amino acid residues involved in the substrate binding are shown in lines and side chains are shown in sticks. NAD + substrate is shown in sticks with magenta carbons. (3S)-acetoin in CgBdh is represented in cyan backbone and (3R)-acetoin in KpBdh is represented in purple backbone, are modeled following acetoin binging in SmBdh using C1, C2, C3 and O2 atoms of the acetoin molecules in SmBdh as anchors. Available hydrogen bonds are depicted as yellow dashed lines. Hydrogen bonds specific for (3S)-acetoin O3 atom are depicted as cyan dashed lines (panels a and c). W71 indicates a water molecule
Substrate promiscuity of the SmBdh enzyme
Most of the characterized Bdh enzymes show strong preference to either (3S)- or (3R)-acetoin as a substrate [24, 33, 38, 39]. SmBdh was reported to be able to reduce both (3R)- and (3S)-acetoin to 2,3-BDO, although (3R)-acetoin was more readily converted than (3S)-acetoin [18]. On the other hand, SmBdh was able to oxidize meso-2,3-BDO and (2S,3S)-BDO, while oxidation of (2R,3R)-BDO was not detectable. Moreover, its activity towards (2S,3S)-BDO was only 11% of that towards meso-2,3-BDO [18]. This observation was similar to that reported by Médici et al. where (3R)-acetoin was found to be the preferred substrate in the racemic mixture of acetoin. Fifteen percent of the (3S)-acetoin remained unconverted after 24 h in their study [36]. Indeed, in our crystal structure we were able to locate both acetoin enantiomers in the active site. Whereas the positions of all acetoin atoms, with the exception of O3, superimpose quite well; the O3 atoms of (3S)- and (3R)-acetoin molecules are involved in hydrogen bonds with different surrounding residues (Figs. 4, 5c). This finding supports the idea of productive binding of both acetoin enantiomers in the active site. Still, (3S)- and (3R)-acetoin molecules are not treated equally by the SmBdh. We suggest that the differences in the hydrogen bond networks of the (3S)-acetoin and (3R)-acetoin can explain our findings. Specifically, the O3 atom of the (3S)-acetoin is only involved in one hydrogen bond with Gln247symm Oε (or Nε if the side chain is flipped). While Ser138Oγ is located only 3.2 Å away from (3S)-acetoin O3, the geometry is not favorable for the H-bond formation. In contrast, O3 atom of the (3R)-acetoin has three possible partners for H-bond formation: water molecule W71 (2.7 Å), Nε of the Gln247symm (3.1 Å), and Ser140Oγ (3.1 Å). It is possible that the "more complex" H-bond network provides better stabilization of the reaction intermediate, which could in turn lead to higher turnover of the (3R)-acetoin in comparison to (3S)-acetoin.
Shorter α6 helix results in possible improvement in ingress/egress of the cofactor and the substrate/product
Although the general fold of the SmBdh monomer is very common, we identified several features that are unique to this enzyme. Comparing SmBdh structure with other Bdh structures we found no coexistence of both ‘open’ and ‘closed’ conformations in other Bdhs: the active sites in all molecules in the tetramer are either equally populated in the liganded structures or not populated at all. In SmBdh, two molecules of the tetramer are found in an ‘open’ conformation with substrate binding sites empty and two molecules in ‘closed’ conformation with populated substrate binding sites. This observation points to the possibility of a concerted ‘tug-of-war’ motion in the SmBdh tetramer, where closure of active sites of two diagonally opposite molecules leads to expansion of the active sites of the other two molecules for faster replenishment with the cofactor and the substrate (Fig. 2a).
Comparison between SmBdh and Bdhs with known structures shows a shortened α6 helix in SmBdh (also known as FG1 in SDR structure description) (Fig. 6). The α6 helix is six residues long in SmBdh, 10 residues long in BxBdh and 15 residues long in KpBdh, CgBdh, and GoBdh. The shortened α6 helix provides wider opening of the catalytic cleft thus improving access to the NADH/NAD + and acetoin/2,3-BDO binding sites and easing ingress/egress of the cofactor and substrate/product.
Fig. 6.

Comparison of different domains of SmBdh with other known Bdh enzymes. a Comparison of α6 helix of the SmBdh and KpBdh. SmBdh is shown as grey cartoon covered with semi-transparent surface. NAD + and acetoin molecules are shown in sticks representation. KpBdh is shown as cyan cartoon. The larger α6 helix of the KpBdh is covered in cyan mesh to illustrate the additional volume obstructing the catalytic cleft. b Sequence alignment of SmBdh (accession #AFH00999), EcBdh (accession #AFM58915), KpBdh (PDB 1GEG), LlBdh (UniProtKB—D2BQ24), GoBdh (PDB 3WTC), BxBdh (PDB 5JY1), CgBdh (PDB 3A28), BcBdh (PDB 4WEO) using ClustalW program. The 11 amino acid deletion region in SmBDH is shown with a black box. Red lines, alpha helix; blue lines, beta strand; green lines, alpha turn; black line, C-terminal extension in SmBdh
Substrate-binding pocket is formed by two protein molecules in SmBdh, unlike the others
One of the most interesting and exclusive features of SmBdh is the organization of the active site. We found that the substrate-binding pocket is formed by two protein molecules, not a single peptide as found in all other reported Bdh enzymes. The C-terminus of molecule A protrudes into the groove between α7 helix and the α-turn αt1 capping substrate-binding pocket of molecule Asymm and vice versa (Fig. 2a). The side chain of Q247symm is involved in the substrate positioning forming a hydrogen bond with the O3 atom of the acetoin. (Fig. 5). In most SDRs the groove between α7 helix and α-turn αt1 is unobstructed and open to the solvent, but there are examples of active sites with the involvement of the second protein chain, for example: 17β hydroxysteroid dehydrogenase 14 from Homo sapiens (1yde, [40, 41]), alcohol dehydrogenase from Arthrobacter sp. TS-15 (6qhe, [42]), and glucose dehydrogenase 4 from Bacillus megaterium (3auu, [43]). Searching through the PDB, we could not find any other Bdh structure where the active site required the participation of a second protein molecule, so SmBdh is the first Bdh with this feature. The only similar functional aspect of the involvement of C-terminus in active site formation is found in the case of BxBdh, where it protrudes into the same protein molecule instead of a different protein molecule, as in SmBdh.
There are several consequences of this substrate-binding pocket organization. First, SmBdh would only be active as a tetramer because the active site is incomplete unless tetramerization is achieved. In other Bdh enzymes active sites are solely formed by a single protein molecule and the enzymatic activity in theory could occur in the monomeric or dimeric state. Second, the presence of the C-terminal portion of another protein molecule near the substrate-binding pocket creates a cap making the pocket much less accessible from the solvent compared to other Bdh enzymes. Third, higher specificity towards acetoin is another possible consequence as the substrate binding site is restricted in volume and cannot accept any larger substrate. This concept is in good agreement with the observation that SmBdh prefers smaller substrates, such as vicinal diketones/diols that do not contain bulky groups [36]. Tighter and more specific binding of the acetoin molecule could be achieved due to an improved hydrogen bonding network (as explained in the section “Key differences in the SmBdh structure lead to improved positioning of acetoin in the active site of the enzyme”); thereby improving reaction intermediate stabilization and ultimately enhancing catalytic efficiency of the SmBdh.
Mutational studies of the SmBdh
Gln247 plays a crucial role in SmBdh catalysis
Based on structural evidence that we have obtained, Gln247 seems to play an important role in the hydrogen bonding-mediated stabilization of the substrate, as well as better positioning of the substrate in the substrate-binding pocket of the protein. This is important in the context of the overall catalytic ability of this protein; as well as proving to be one of the critical parameters that could make SmBdh a better performing Bdh enzyme in comparison to its closely related homologs. In order to determine the importance of the Gln247 side chain for the catalysis, we developed two mutants of SmBdh: (1) Q247A where this side chain is removed, leaving alanine at position 247 and (2) the double mutant Q247A + V139Q, where the missing glutamine side chain would be reinstated at the position 139 that is present in KpBdh (Fig. 7, Additional files 5 and 6). Whereas Q247A will disrupt the active site of the protein, Q247A + V139Q is expected to restore the active site via a compensatory mechanism resulting in the active site being established by a single protein chain without contribution from a symmetry-related molecule (i.e., from the C-terminus of the opposite molecule in the tetramer).
Fig. 7.

Modeled structures of the active site of SmBdh and its variants along with their substrates. a Omit Fo-Fc electron density map at 3σ level is shown as a green mesh over acetoin molecules in the active site of the WT SmBdh. b Omit Fo-Fc electron density map at 3σ level is shown as a green mesh over glycerol molecule in the active site of the Q247A SmBdh mutant. c Omit Fo-Fc electron density map at 3σ level is shown as a green mesh over ethylene glycol molecule at the active site of the Q247A + V139Q SmBdh double mutant
The SmBdh Q247A and the Q247A + V139Q mutants crystallized in the same space group (P43212) as the WT SmBdh repeating its protein chains arrangement. Two protein molecules per asymmetric unit could be superimposed with r.m.s.d. of 0.275 Å over 1400 atoms (Q247A), and 0.263 Å over 1368 atoms (Q247A + V139Q). As with the WT SmBdh, the primary difference between the two molecules in the asymmetric unit in the mutant structures was that molecule A was found in ‘closed’ conformation (that could be best described by ~ 10 Å distance between Cα-atoms of residues Ala93 and Trp192) and molecule B was found in ‘open’ conformation (the corresponding distance is ~ 15 Å) (Additional file 4, Fig. 2). Upon examination of the electron density maps, we were able to locate NAD + cofactor molecule and a substrate in protein molecule A (‘closed’ conformation), whereas in molecule B (‘open’ conformation) the corresponding space is occupied by only adenine diphosphate (as in Q247A mutant) or remains unoccupied (as in Q247A + V139Q mutant) at all. The substrate binding site was found to be occupied by glycerol molecule in Q247A mutant and ethylene glycol in Q247A + V139Q mutant (Fig. 7).
In the Q247A mutant structure, a glycerol molecule was modeled at the substrate-binding pocket (Fig. 7b). Acetoin was present in the crystallization conditions, but our attempts to use additional acetoin for cryoprotection only led to crystal damage. Therefore, a 50/50 v/v mix of glycerol and ethylene glycol was used for cryoprotection. We postulate that the absence of the Gln247symm side chain led to an increase of the available space for the substrate and therefore the larger glycerol molecule could be bound. In the Q247A + V139Q double mutant structure, an ethylene glycol molecule was modeled at the substrate-binding pocket (Fig. 7c). As with the Q247A mutant, acetoin was present during crystallization, but raising its concentration was damaging to the crystals. A 50/50 v/v mix of glycerol and ethylene glycol was therefore used for cryoprotection. We suggest that the available space in the substrate binding site became reduced compared to the Q247A single mutant, thereby providing insufficient space for glycerol to fit. Instead, ethylene glycol which is comparable in size to acetoin could fit.
We further tested the activity of both these mutants using the NADH consumption assay to determine the effect of these mutations on the function of the protein. In comparison to the WT SmBdh, Q247A mutant showed a ~ 90% loss in activity as predicted by the structure, whereas the double mutant Q247A + V139Q showed ~ 300% improvement in activity in comparison to Q247A mutant (Fig. 8). Although the double mutant did not completely restore the loss of Gln247 activity, significant function was regained by introducing the V139Q mutation in this protein. It can thus be inferred that Gln247 is extremely important for the activity of the protein, which cannot be fully restored by a complementary mutation, such as V139Q. However, this is not a thorough analysis of complementation of the Q247A mutation and there could be other mutations that could help restore full functionality of the Q247A mutation. This analysis also highlights the importance of the C-terminal Gln247 from the opposite molecule in conferring full functionality.
Fig. 8.
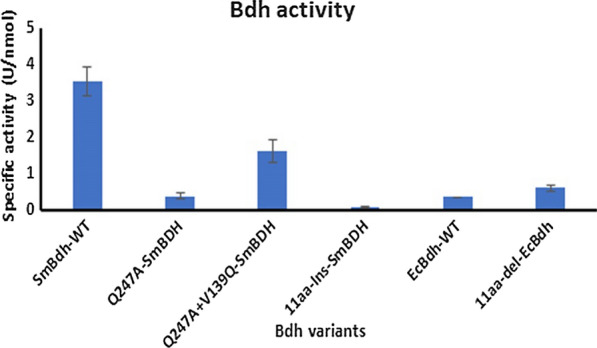
2,3-Butanediol dehydrogenase activity of purified Bdh enzyme variants. Bdh activity was measured by following NADH consumption at 340 nm. Purified proteins were used for the enzyme assay. Reactions were carried out in triplicates and are represented as mean SD
Extending the short α6 helix of SmBdh results in loss in activity
Comparing the different Bdh enzymes, we found that most of the S-acting Bdh enzymes, such as EcBdh, CgBdh, and KpBdh have longer α6 helix structure (Fig. 6), whereas deletion of 11 amino acid residues in the SmBdh leads to a shorter α6 helix along with a shorter linker between the α6 and α7 helices. Based on the activity analysis data (Table 1, Fig. 8), it was clear that SmBdh is much more active than the control enzymes, which include EcBdh. Furthermore, structural analysis also suggested that the shortened α6 helix contributes to the improved ingress or egress of the substrate and/or the product molecules. This result prompted us to investigate the role of this important structural difference in SmBdh with regard to enzyme performance. In order to support this hypothesis, we used two Bdh enzymes—SmBdh with short α6 helix and EcBdh with long α6 helix. Sequence alignments with highlighted differences are shown in Fig. 6b. We designed two mutants: (1) 11aa-Ins-SmBdh, where sequence fragment “RDK” was replaced with “SEAAGKPLGYGTET” to mimic longer α6 helix of EcBdh and (2) 11aa-del-EcBdh, where the amino acid sequence “SEAAGKPLGYGTET” was replaced with “RDK” to mimic the corresponding shorter α6 helix of SmBdh. The residues flanking these regions in EcBdh and SmBdh showed acceptable homology, so we decided to keep the swap region as small as possible. Proteins were expressed in the Z. mobilis 9C strain, purified to homogeneity and subjected to activity analysis using the NADH consumption assay. As expected, activity of the 11aa-Ins-SmBdh was greatly reduced (down to 3% of the WT activity) with the insertion, whereas the activity of 11aa-del-EcBdh increased by ~ 70% in comparison to WT EcBdh (Fig. 8), suggesting that the size of the α6 helix contributes to the unique activity of SmBdh in comparison to the other S-acting Bdhs. Nevertheless, replacing the longer α6 helix in EcBdh with the shorter helix from SmBdh did not render EcBdh similar to SmBdh in terms of its activity; we found that the activity of the 11aa-del-EcBdh was only 17% of WT SmBdh (Fig. 8). This clearly suggests that while the short α6 helix is important for the high activity of SmBdh, there are other structural features that this enzyme possesses that also contribute towards its superior performance.
Conclusions
Based on expression of 11 phylogenetically different Bdh enzymes, we have identified that the S. marcescens Bdh enzyme shows the highest activity when expressed in Z. mobilis. This enzyme has been classified as an S-acting Bdh based on production of meso-2,3-BDO and (2S,3S)-BDO from a racemic mixture of acetoin. We have structurally characterized this enzyme and ascertained the distinct structural features that may be critical for its activity. Specifically: (1) this enzyme is organized as a tetramer with diagonally opposite protein molecules acting in tandem, such that one diagonally opposite pair is bound by the substrate in the closed conformation, with the other two protein molecules in the tetramer found ready to take up new substrate molecules in the open confirmation. This is the first instance of a piston-type function of a Bdh enzyme. (2) The active site of the SmBdh enzyme is formed with the participation of the Gln247 of the opposite molecule from the tetramer, removal of this residue causes severe functional defects to the protein. A complementary mutation that introduces a glutamine partially restores the function of this protein. (3) SmBdh possesses improved hydrogen bond network resulting in better positioning of the substrate in its active site. (4) SmBdh is able to bind to both (3S)- and (3R)-acetoin productively and we were able to locate both substrate molecules in the active site of the crystal structure, thus providing the structural confirmation to the enzyme substrate promiscuity. (5) The presence of a shorter α6 helix provides a wider cleft for efficient entry and exit of the substrate and products, respectively, to SmBdh. We have experimentally verified this hypothesis by introducing residues from a low activity Bdh enzyme (i.e., EcBdh), which resulted in dramatic reduction in activity of this protein. (6) Finally, we have also demonstrated that deletion of residues that shortened the α6 helix of EcBdh can result in dramatic increase in its activity (~ 70%), which could prove to be a game-changer when considering low-active Bdh enzymes for 2,3-BDO production. Overall, this work has identified a superior heterologous Bdh enzyme suitable for expression in Z. mobilis. Moreover, availability of a strain expressing this Bdh enzyme is expected to greatly alleviate one of the major bottlenecks involved in 2,3-BDO production by this organism. The next logical step would be to incorporate the SmBDH into a Z. mobilis strain carrying the other two BDO pathway genes, Als and Aldc. Our future work will involve testing the SmBdh in combination with other Als and Aldc enzymes to identify the best combination of enzymes that would lead to improvement in 2,3-BDO production.
Materials and methods
Strain and growth conditions
Zymomonas mobilis strain 9C (ZM4 derivative strain with xylose-utilizing abilities) was revived from glycerol stocks and grown in rich medium (RM, 10 g/L yeast extract, 2 g/L KH2PO4) containing 5% glucose (RMG) at 30 °C under shaking conditions (120 rpm). Spectinomycin at a concentration of 200 µg/mL was used to maintain transformant colonies.
Phylogenetic analysis of Bdh
Initially, 52 Bdh amino acid sequences were obtained from the NCBI database. These Bdh sequences represented one enzyme per genus under the assumption that Bdh enzymes within the same genus would have similar structures and functions. Two distinct sets of Bdh sequences were formed based on their sequence lengths. One set contained ~ 350 aa, while the other contained ~ 250 aa. In genera containing both sequence types, both the sequences were selected. In the end, 57 Bdh sequences were used for phylogenetic analysis. Full-length Bdh sequences were aligned using ClustalW, and phylogenetic analysis was carried out using the MEGA X software [44]. The unrooted evolutionary tree was generated using the neighbor joining method [45]. A bootstrap value of 1000 replications was used for the analysis [46]. All gaps and ambiguous positions were removed using the pairwise deletion option. The evolutionary distances were calculated using the Poisson correction method [47].
Plasmid construction, transformation, and screening of transformants
Eleven bdh gene sequences were codon-optimized for expression in Z. mobilis (Additional file 2), synthesized and cloned into the vector pEZ15Asp [15] under the control of the pyruvate decarboxylase promoter (PDC) by GenScript (Piscataway, NJ, USA). These included Erwinia amylovora (Ea), Myroides odoratimimus (Ma), Staphylococcus warneri (Sw), Thermococcus gammatolerans (Tg), A. vinelandii (Av), Mycobacterium Smegmatis (Ms), Micrococcus luteus (Ml), A. tumefaciens (At), S. marcescens (Sm), Streptomyces coelicolor (Sc), and Dickeya dadantii (Dd). Prior to transformation, these plasmids were transformed into a Dam- Dcm- E. coli strain to avoid methylation to the DNA. These plasmids were transformed into the Z. mobilis 9C strain using the protocol described in Yang et al. [15]. Briefly, 1 µg of plasmid DNA was mixed with 50 µL of freshly thawed competent cells in a 0.1 cm gap electroporation cuvette on ice. Cells were electroporated using a Bio-Rad Gene Pulser (Bio-Rad Laboratories, Inc. Hercules, CA, USA) using the following settings: 200 Ω, 25 µF, 1.6 kV. The tubes were immediately transferred to ice for cooling. One milliliter of mating medium (MM; 50 g/L glucose, 10 g/L yeast extract, 5 g/L tryptone, 2.5 g/L (NH4)2SO4, and 0.2 g/L K2HPO4 containing 1 mM MgSO4 was added to the tube, the liquid transferred to a 1 mL cryovial and then incubated for 5–6 h at 30 °C to allow cell recovery. Cells were then plated on MM agar containing spectinomycin (200 µg/mL) and incubated in an anaerobic jar (containing a gas pak) at 30 °C for 2 days. Individual colonies were restreaked on MM medium containing spectinomycin followed by colony PCR to check for the presence of the bdh gene. Colony PCR was carried out using the forward primer, SV-90, and the reverse primer, SV-91. All primers used in this study are available in Additional file 7. These primers flanked the promoter and terminator sequences, respectively, thereby amplifying the complete gene cassette.
Construction of Bdh variants for crystal structure assessment
Two different primers SV-161 and SV-162 were used to amplify the pEZ15Asp plasmid containing the Smbdh gene, such that a 6X-histidine tag and a Tobacco etch virus protease cleavage site (ENLYFQG) sequences were introduced, in that order, immediately following the start codon of the Smbdh gene. The PCR product was subjected to DpnI digestion followed by ligation using the In-Fusion Cloning protocol (Takara Bio USA, Inc., Mountain View, CA, USA) to obtain the final plasmid, which will be henceforth referred to as “N-term SmBdh”.
The Q247A SmBdh mutant was generated by amplifying the N-terminal His-TEV-Smbdh containing vector with primers SV-274 and SV-275 to introduce the Q247A mutation within the bdh coding fragment, while amplifying the entire plasmid. This PCR product was then subjected to DpnI treatment followed by recircularization using the In-Fusion Cloning protocol to obtain the final plasmid.
The double mutant, Q247A + V139Q SmBdh, was generated by amplifying the Q247A Smbdh plasmid with primers SV-279 and SV-280. Following DpnI treatment, the PCR product was self-ligated using the In-Fusion Cloning protocol to obtain the final plasmid.
The 11aa-Ins-SmBdh mutant was constructed by amplifying the N-term SmBdh plasmid with primers SV-277 and SV-276 to introduce the Enterobacter cloacae Bdh (EcBdh) 14 amino acid coding sequence “SEAAGKPLGYGTET” replacing 3 amino acids from SmBdh (“RDK”) into the Smbdh gene along with amplification of the entire vector. This PCR product was subjected to DpnI treatment followed by self-ligation using the In-Fusion Cloning protocol. Prior to cloning the 14 amino acid sequence, the respective E cloacae sequence was codon-optimized for expression in Z. mobilis.
In order to introduce a 6X-histidine tag and a TEV protease cleavage site into the N-terminal end of the E. cloacae Bdh (N-term EcBdh), the untagged version of the Ecbdh gene was first amplified from a previously transformed Z. mobilis strain BC-11B [15] using the primers SV-285 and SV-286. The vector fragment was obtained by amplifying pEZ15Asp-Smbdh vector with primers SV-283 and SV-284 followed by DpnI treatment of the PCR product. Both these fragments were ligated using the In-Fusion Cloning protocol to obtain the Ecbdh gene in the same expression vector, pEZ15Asp. The N-terminal histidine tag along with the TEV site was then introduced by amplifying the pEZ-15Asp-Ecbdh vector with primers SV-293 and SV-294, followed by DpnI treatment of the PCR product and self-ligation by the In-Fusion Cloning protocol.
The 11 amino acid deleted version of the Ecbdh gene (11aa-del-EcBdh) was cloned by amplifying the N-term Ecbdh plasmid with primers SV-302 and SV-303 to amplify the entire plasmid, except the 14 amino acid sequence (“SEAAGKPLGYGTET”) of EcBdh, while replacing this segment with a 3 amino acid sequence (“RDK”) of SmBdh. Following amplification of the entire plasmid, this PCR product was subjected to DpnI treatment followed by circularization using the In-Fusion Cloning protocol.
Bdh protein expression and purification
Following confirmation of the strains for the presence of the bdh genes, individual colonies were inoculated into 5 mL RMG medium containing 200 µg/mL spectinomycin and incubated at 30 °C for 2 days under shaking conditions (120 rpm). An aliquot of this pre-grown culture was then transferred to 20 mL RMG medium containing 200 µg/mL spectinomycin for small scale protein extraction and activity analysis. For large-scale protein purification studies, 750 mL cultures were started in duplicates. In both cases, the starting OD600 of the cultures were adjusted to 0.025. Cultures were incubated at 120 rpm at 30 °C for 3 days. The final OD600 of the cultures were between 3 and 6 for the different cultures. Cultures were then transferred to ice prior to centrifugation at 6000 × g for 5 min to obtain cell pellets. Cells were immediately frozen in liquid nitrogen prior to further processing.
For small-scale protein extraction, cells were thawed on ice and resuspended on 1 mL of 100 mM phosphate buffer (pH 7.0) containing protease inhibitor cocktail (Sigma-Aldrich Corp. St. Louis, MO, USA). Cells were disrupted by sonication using four cycles of 30 s pulses, with intermittent cooling on ice for 30 s between cycles. Cells were then subjected to bead beating for 10 min at 4 °C to ensure complete lysis of the cells. Protein extract was clarified by centrifugation at 13,000 × g for 10 min. Total protein content was estimated using the Bradford reagent protocol (Bio-Rad Laboratories, Inc. Hercules, CA, USA).
For large-scale protein extraction, frozen cell pellets were thawed and resuspended at room temperature with equal volume of buffer (50 mM Tris pH 7.5, 100 mM NaCl and 10 mM imidazole) and lysed using lysozyme and vortexing with glass beads. Specifically, 1 mg/mL lysozyme (Hampton Research, Aliso Viejo, CA, USA), 1 U/mL Pierce Universal Nuclease (Thermo Scientific, Rockford, IL, USA) and EDTA-free protease inhibitor (Thermo Scientific, Rockford, IL,USA according to manufacturer instructions) were added and the lysis mixture was incubated for 30 min at room temperature. Lysis was completed via vortexing with glass beads (0.1 mm diameter, 1:1 volume with the cell pellet) for 3–5 min and cell debris was removed by centrifugation for 15 min at 22,000 × g. The resulting supernatant was loaded into a 5-mL HisTrap FF crude column (GE Life Sciences, Piscataway, NJ, USA) using an Akta FPLC system (GE Life Sciences, Piscataway, NJ, USA) in 50 mM Tris pH 7.5, 100 mM NaCl and 10 mM imidazole. After loading and washing the unbound proteins from the column, the target proteins were eluted using 50 mM Tris pH 7.5, 100 mM NaCl and 250 mM imidazole. Minor impurities were removed by size exclusion chromatography using HiLoad Superdex 75 (26/60) (GE Life Sciences, Piscataway, NJ, USA) in 20 mM Tris pH 7.5 and 100 mM NaCl. Peaks corresponding to the Bdh samples were pooled and concentrated to 5–15 mg/mL. Protein concentration was measured using absorbance at 280 nm in a NanoDrop1000 and the protein-specific extinction coefficients and molecular weights.
Protein crystallization
Initial screening was done in a sitting drop vapor diffusion setup using a 96-well plate with Crystal Screen HT, PEG/Ion HT and Grid Screen Salt HT from Hampton Research (Aliso Viejo, CA, USA). Fifty microlitre of well solution was added to the reservoir with three drops made of 0.2 µL of well solution and 0.1/0.2/0.3 µL of protein solution using a Phoenix crystallization robot (Art Robbins Instruments, Sunnyvale, CA, USA). The final crystals for all three structures were grown at 20 °C using an optimization screen with 0.1 M sodium malonate pH 6–7 and 6–15% w/v polyethylene glycol 3350 as the well solution. The protein solution for WT SmBdh contained 9 mg/mL of protein, 20 mM Tris pH 7.5, 100 mM NaCl, 20 mM NAD+ and 200 mM acetoin. SmBdh Q247A protein solution consisted of 4.2 mg/mL of protein, 20 mM Tris pH 7.5, 100 mM NaCl, 20 mM NAD+ and 20 mM acetoin. SmBdh Q247V + V139Q had 14.5 mg/mL of protein, 20 mM Tris pH 7.5, 100 mM NaCl, 20 mM NAD+ and 20 mM acetoin.
Data collection and processing
Before data collection the SmBdh crystals were flash frozen in a nitrogen gas stream at 100 K followed by data collection using an in-house Bruker X8 MicroStar X-Ray generator with Helios mirrors and a Bruker Platinum 135 CCD detector (Bruker AXS LLC, Madison, WI, USA. SmBdh Q247A and SmBdh Q247V + V139Q crystals were further protected from ice formation by shortly moving them into a well solution drop with 15% (v/v) ethylene glycol and 15% (v/v) glycerol for improved cryo protection. Bruker Suite of programs version 2014-9 (Bruker AXS LLC, Madison, WI, USA) was used for data indexing and processing.
Structure solution and refinement
The CCP4 package of programs [48] was used for converting intensities into structure factors, for project tracking, and access to the individual programs. Five percent of reflections were reserved for Rfree calculations using programs F2MTZ, Truncate, CAD and Unique. The structure of the WT SmBdh was determined via molecular replacement using MOLREP [49]. The initial model was built based on PDB entry 4ni5 with FFAS03 search and ProtMod modeling servers [50–52] using the SCWRL method. The resulting model was refined in REFMAC5 [53] version 5.8.0258 and rebuilt and inspected using Coot version 0.8.0.2 [54]. Structures of the SmBdh Q247A mutant and Q247A + V139Q double mutant were determined via molecular replacement as described above using the WT SmBdh as a model. Ramachandran plot was calculated with MOLPROBITY [55] and root mean square deviations (r.m.s.d.) of bond lengths and angles were calculated using ideal values of Engh and Huber stereochemical parameters [56]. Wilson B-factor was obtained from CTRUNCATE [48] version 1.0.11. All structures have been deposited to the Protein Data Bank with PDB codes 6XEW (WT SmBdh), 6VSP (SmBdh Q247A), and 6XEX (SmBdh Q247A + V139Q). Data collection and refinement statistics are shown in Table 3.
Table 3.
X-ray data collection and refinement statistics
| WT SmBDH | SmBDH Q247A | SmBDH Q247A + V139Q | |
|---|---|---|---|
| Data collection | |||
| Space group | P43212 | P43212 | P43212 |
| Unit cell (Å, °) |
a = b = 108.17, c = 82.88 α = β = γ = 90.00 |
a = b = 108.68, c = 82.95 α = β = γ = 90.0 |
a = b = 108.80, c = 83.11 α = β = γ = 90.0 |
| Wavelength (Å) | 1.54178 | 1.54178 | 1.54178 |
| Temperature (K) | 100 | 100 | 100 |
| Resolution (Å) | 25.0–2.0 (2.1–2.0) | 25.0–1.9 (2.0–1.9) | 25.0–1.8 (1.9–1.8) |
| Unique reflections | 33,823 (4496) | 39,724 (5531) | 46,796 (6911) |
| Rinta | 0.138 (0.689) | 0.109 (0.748) | 0.093 (0.793) |
| Average redundancy | 11.4 (8.8) | 17.3 (10.6) | 17.0 (12.5) |
| < I > / < σ(I) > | 11.2 (2.0) | 18.7 (2.6) | 21.7 (1.9) |
| Completeness (%) | 100 (100) | 100 (100) | 100 (100) |
| Refinement | |||
| R/Rfree | 0.162 (0.214)/0.279 (0.378) | 0.168 (0.340)/0.222 (0.347) | 0.160 (0.210)/0.295 (0.342) |
| Protein atoms | 3736 | 3792 | 3766 |
| Water molecules | 432 | 551 | 558 |
| Other atoms | 141 | 151 | 70 |
| r.m.s.d. from ideal bond lengthb (Å) | 0.010 | 0.009 | 0.010 |
| r.m.s.d. from ideal bond anglesb (°) | 1.497 | 1.486 | 1.612 |
| Wilson B-factor | 21.6 | 19.5 | 21.2 |
| Average B-factor for protein atoms (Å2) | 25.2 | 25.0 | 24.0 |
| Average B-factor for water molecules (Å2) | 33.0 | 32.6 | 33.1 |
| Ramachandran plot statisticsc (%) | |||
| Allowed | 99.8 | 99.8 | 99.8 |
| Favored | 96.9 | 97.2 | 97.3 |
| Number of outliers | 1 | 1 | 1 |
Statistics for the highest resolution bin are shown in parenthesis
aRint = ∑hkl ∑i|Ii(hkl)—‹I(hkl)›|/ ∑hkl ∑i Ii(hkl), where Ii(hkl) is the intensity of an individual reflection and ‹I(hkl)› is the mean intensity of a group of equivalents; the sums are calculated over all reflections with more than one equivalent measured
b[56]
c[55]
Bdh activity analysis
Bdh activity was assayed by following NADH oxidation at 340 nm for 5 min using a molar extinction coefficient of 6.22 mM−1 cm−1. The enzyme reaction was carried out in a total volume of 200 µL and contained 100 mM potassium phosphate buffer (pH 7.0), 0.25 mM NADH and 25 mM acetoin. The reaction was started by addition of the enzyme. All reactions were carried out in microtiter plates and monitored using a FLUOstar Omega plate reader (BMG Labtech GmbH, Ortenburg, Germany). Enzyme activities were represented as unit per nmol of the purified protein. One unit of enzyme was defined as the amount of enzyme that consumed one micromole of NADH per minute. For total proteins, enzyme activities were represented as micromole of NADH consumed per min per mg of total protein.
For phylogenetic screening of the Bdh enzymes, whole cell extracts obtained from the small-scale extraction procedure were used for enzyme assays. A range of concentrations from 1–20 µg total protein were tested for each protein extract. For the Bdh assays involving purified proteins (SmBdh and EcBdh), a range of protein concentrations from 0.05–1 µg were used.
Supplementary information
Additional file 1. PCR analysis of Z. mobilis tranformants containing the different Bdh genes. Eleven Bdh genes were transformed into Z. mobilis strain 9C and plated on RMG medium containing spectinomycin. Eight independent colonies were selected for colony PCR analysis using gene specific primers. Five microliters of the PCR products were run on 1% agarose gel and visualized using FluoChem Gel analyzer. White bar represents sets of 8 lanes for each Bdh gene. Av, Azotobacter vinelandii; Ml, Micrococcus luteus; Ea, Erwinia amylovora; Sw, Staphylococcus warneri; Sc, Streptomyces coelicolor; Sm, Serratia marcescens; Dd, Dickeya dadantii; Tg, Thermococcus gammatolerans; At, Agrobacterium tumefaciens; Ms, Mycobacterium Smegmatis; Mo, Myroides odoratimimus; 9c, Z. mobilis control strain.
Additional file 2. BDH genes selected for expression in Z. mobilis.
Additional file 3. Detection of heterologous Bdh proteins from Z. mobilis transformants using total protein staining. Five independent transformant colonies were selected for protein extraction. Total proteins were separated on 4-12% SDS PAGE gel followed by Coomassie staining. Arrows represent the expected molecular weight of the individual Bdh proteins. Two Bdh protein sets are shown in each PAGE gel. Each protein set is represented by 5 lanes as indicated by the black bars. Bdh protein names are shown above the black bars. Av, Azotobacter vinelandii; Ml, Micrococcus luteus; Ea, Erwinia amylovora; Sw, Staphylococcus warneri; Sc, Streptomyces coelicolor; Sm, Serratia marcescens; Dd, Dickeya dadantii; Tg, Thermococcus gammatolerans; At, Agrobacterium tumefaciens; Ms, Mycobacterium Smegmatis; Mo, Myroides odoratimimus; C, Z. mobilis control strain.
Additional file 4. Ala93Cα-Trp192Cα distance in ‘open’ and ‘closed’ molecules, Å.
Additional file 5. Multiple alignment of N-terminal histidine tagged SmBdh sequences representing the sites of amino acid changes. Three different versions of SmBdh are shown. Boxes indicate the amino acids that were replaced in the other mutants. SmBdh (N-term), wild-type; Q247A-SmBdh (N-term) and Q247A+V139Q-SmBdh (N-term), two different variants of SmBdh.
Additional file 6. 2,3-butanediol dehydrogenase gene sequences used in the study.
Additional file 7. Primers used in this study.
Acknowledgements
This work was authored by the National Renewable Energy Laboratory, managed and operated by Alliance for Sustainable Energy, LLC for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding provided by U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office under contract DE-AC36-08GO28308. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes. YBC was supported by Indo-US Science and Technology forum (IUSSTF)—Bioenergy-Awards for Cutting Edge Research (B-ACER) grant, Govt. of India. The authors would like to thank Dr. Shihui Yang for providing the control plasmids pEZ-15Asp-EcBdh, pEZ-15Asp-LlBdh and pEZ15Asp-KpBdh used in this study. The authors also thank Mary Ann Franden and Yat-Chen Chou of the National Renewable Energy Laboratory for helpful technical discussions.
Abbreviations
- 2,3-BDO
2,3-butanediol
- Bdh
2,3-butanediol dehydrogenase
- NAD
Nicotinamide adenine dinucleotide (oxidized)
- NADH
Nicotinamide adenine dinucleotide (reduced)
- Als
Acetolactate synthase
- Ndh
NADH oxidase
- PDB
Protein data bank
- SDR
Short-chain dehydrogenase/reductase
- PCR
Polymerase chain reaction
- α6
Alpha 6 helix
- KpBdh
Klebsiella pneumoniae Bdh
- GoBdh
Gluconobacter oxydans Bdh
- BcBdh
Burkholderia cenocepacea Bdh
- BxBdh
Burkholderia xenovorans Bdh
- CgBdh
Corynebacterium glutamicum Bdh
- SmBdh
Serratia marcescens Bdh
- EcBdh
Enterobacter cloacae Bdh
- Ea
Erwinia amylovora
- Ma
Myroides odoratimimus
- Sw
Staphylococcus warneri
- Tg
Thermococcus gammatolerans
- Av
Azotobacter vinelandii
- Ms
Mycobacterium smegmatis
- Ml
Micrococcus luteus
- At
Agrobacterium tumefaciens
- Sc
Streptomyces coelicolor
- Dd
Dickeya dadantii
- Gln247
Glutamine 247
- r.m.s.d.
Root mean square deviations
- Q247A
Glutamine 247 to alanine
- V139Q
Valine 139 to glutamine
- RMG
Rich medium containing glucose
Authors’ contributions
VS wrote the paper, designed Bdh constructs, and performed enzyme assays; VL wrote the paper, designed Bdh mutant constructs, crystallized and determined the crystal structures of Bdh proteins; SF, KL, AH and YBC carried out plasmid cloning, transformations, small and large-scale culture growths, protein extractions, and DNA and protein gel electrophoresis; MA was in involved in crystallization and structure determination of the Bdh proteins; SD and MH managed the project and were involved in editing the manuscript. All authors read and approved the final manuscript.
Funding
Funding provided by U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office under contract DE-AC36-08GO28308.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Ethics and approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Provisional patent filed on Bdh modifications.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Venkataramanan Subramanian and Vladimir V. Lunin contributed equally to this work
Contributor Information
Venkataramanan Subramanian, Email: venkat.subramanian@nrel.gov.
Vladimir V. Lunin, Email: vladimir.lunin@nrel.gov
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13068-020-01820-x.
References
- 1.Bialkowska AM. Strategies for efficient and economical 2,3-butanediol production: new trends in this field. World J Microbiol Biotechnol. 2016;32(12):200. doi: 10.1007/s11274-016-2161-x. [DOI] [PubMed] [Google Scholar]
- 2.Hakizimana O, Matabaro E, Lee BH. The current strategies and parameters for the enhanced microbial production of 2,3-butanediol. Biotechnol Rep. 2020;25:e00397. doi: 10.1016/j.btre.2019.e00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prescott SC, Dunn CG. The production and properties of 2,3-butanediol. In: Foster JW, editor. Industrial microbiology. New York: McGraw-Hill; 1949. pp. 487–523. [Google Scholar]
- 4.Sabra W, Groeger C, Zeng AP. Microbial cell factories for Diol production. Adv Biochem Eng Biotechnol. 2016;155:165–197. doi: 10.1007/10_2015_330. [DOI] [PubMed] [Google Scholar]
- 5.Juni E, Heym GA. A cyclic pathway for the bacterial dissimilation of 2, 3-butanediol, acetylmethylcarbinol, and diacetyl. I. General aspects of the 2, 3-butanediol cycle. J Bacteriol. 1956;71(4):425–432. doi: 10.1128/JB.71.4.425-432.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Syu MJ. Biological production of 2,3-butanediol. Appl Microbiol Biotechnol. 2001;55(1):10–18. doi: 10.1007/s002530000486. [DOI] [PubMed] [Google Scholar]
- 7.Xiao Z, Xu P. Acetoin metabolism in bacteria. Crit Rev Microbiol. 2007;33(2):127–140. doi: 10.1080/10408410701364604. [DOI] [PubMed] [Google Scholar]
- 8.Ji XJ, Huang H, Ouyang PK. Microbial 2,3-butanediol production: a state-of-the-art review. Biotechnol Adv. 2011;29(3):351–364. doi: 10.1016/j.biotechadv.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Song CW, Park JM, Chung SC, Lee SY, Song H. Microbial production of 2,3-butanediol for industrial applications. J Ind Microbiol Biotechnol. 2019;46(11):1583–1601. doi: 10.1007/s10295-019-02231-0. [DOI] [PubMed] [Google Scholar]
- 10.Kandasamy V, Liu J, Dantoft SH, Solem C, Jensen PR. Synthesis of (3R)-acetoin and 2,3-butanediol isomers by metabolically engineered Lactococcus lactis. Sci Rep. 2016;6:36769. doi: 10.1038/srep36769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JW, Kim J, Seo SO, Kim KH, Jin YS, Seo JH. Enhanced production of 2,3-butanediol by engineered Saccharomyces cerevisiae through fine-tuning of pyruvate decarboxylase and NADH oxidase activities. Biotechnol Biofuels. 2016;9:265. doi: 10.1186/s13068-016-0677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver JW, Machado IM, Yoneda H, Atsumi S. Cyanobacterial conversion of carbon dioxide to 2,3-butanediol. Proc Natl Acad Sci USA. 2013;110(4):1249–1254. doi: 10.1073/pnas.1213024110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, Chu H, Gao C, Tao F, Zhou Z, Li K, et al. Systematic metabolic engineering of Escherichia coli for high-yield production of fuel bio-chemical 2,3-butanediol. Metab Eng. 2014;23:22–33. doi: 10.1016/j.ymben.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 14.Jeffries TW. Ethanol fermentation on the move. Nat Biotechnol. 2005;23(1):40–41. doi: 10.1038/nbt0105-40. [DOI] [PubMed] [Google Scholar]
- 15.Yang S, Mohagheghi A, Franden MA, Chou YC, Chen X, Dowe N, et al. Metabolic engineering of Zymomonas mobilis for 2,3-butanediol production from lignocellulosic biomass sugars. Biotechnol Biofuels. 2016;9(1):189. doi: 10.1186/s13068-016-0606-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Kuhn E, Jennings E, Nelson N, Tao L, Zhang M, et al. DMR (deacetylation and mechanical refining) processing of corn stover achieves high monomeric sugar concentrations (230 g/L) during enzymatic hydrolysis and high ethanol concentration (>10% v/v) during fermentation without hydrolyzate purification or concentration. Energy Environ Sci. 2016;9:1237–1245. doi: 10.1039/C5EE03718B. [DOI] [Google Scholar]
- 17.Zhang X, Bao T, Rao Z, Yang T, Xu Z, Yang S, et al. Two-stage pH control strategy based on the pH preference of acetoin reductase regulates acetoin and 2,3-butanediol distribution in Bacillus subtilis. PLoS ONE. 2014;9(3):e91187. doi: 10.1371/journal.pone.0091187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Xu Q, Zhan S, Li Y, Lin H, Sun S, et al. A new NAD(H)-dependent meso-2,3-butanediol dehydrogenase from an industrially potential strain Serratia marcescens H30. Appl Microbiol Biotechnol. 2014;98(3):1175–1184. doi: 10.1007/s00253-013-4959-x. [DOI] [PubMed] [Google Scholar]
- 19.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hedlund J, Jornvall H, Persson B. Subdivision of the MDR superfamily of medium-chain dehydrogenases/reductases through iterative hidden Markov model refinement. BMC Bioinformatics. 2010;11:534. doi: 10.1186/1471-2105-11-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knoll M, Pleiss J. The Medium-Chain Dehydrogenase/reductase Engineering Database: a systematic analysis of a diverse protein family to understand sequence-structure-function relationship. Protein Sci. 2008;17(10):1689–1697. doi: 10.1110/ps.035428.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nordling E, Jornvall H, Persson B. Medium-chain dehydrogenases/reductases (MDR). Family characterizations including genome comparisons and active site modeling. Eur J Biochem. 2002;269(17):4267–4276. doi: 10.1046/j.1432-1033.2002.03114.x. [DOI] [PubMed] [Google Scholar]
- 23.Persson B, Hedlund J, Jornvall H. Medium- and short-chain dehydrogenase/reductase gene and protein families: the MDR superfamily. Cell Mol Life Sci. 2008;65(24):3879–3894. doi: 10.1007/s00018-008-8587-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez E, Fernandez MR, Larroy C, Sola L, Pericas MA, Pares X, et al. Characterization of a (2R,3R)-2,3-butanediol dehydrogenase as the Saccharomyces cerevisiae YAL060W gene product. Disruption and induction of the gene. J Biol Chem. 2000;275(46):35876–35885. doi: 10.1074/jbc.M003035200. [DOI] [PubMed] [Google Scholar]
- 25.Huang M, Oppermann FB, Steinbuchel A. Molecular characterization of the Pseudomonas putida 2,3-butanediol catabolic pathway. FEMS Microbiol Lett. 1994;124(2):141–150. doi: 10.1111/j.1574-6968.1994.tb07276.x. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Zhang L, Li K, Wang Y, Gao C, Han B, et al. A newly isolated Bacillus licheniformis strain thermophilically produces 2,3-butanediol, a platform and fuel bio-chemical. Biotechnol Biofuels. 2013;6(1):123. doi: 10.1186/1754-6834-6-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu C, Ge Y, Cao M, Guo X, Liu P, Gao C, et al. Metabolic engineering of Bacillus licheniformis for production of acetoin. Front Bioeng Biotechnol. 2020;8:125. doi: 10.3389/fbioe.2020.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kavanagh KL, Jornvall H, Persson B, Oppermann U. Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci. 2008;65(24):3895–3906. doi: 10.1007/s00018-008-8588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jörnvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, et al. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34(18):6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 30.Otagiri M, Kurisu G, Ui S, Takusagawa Y, Ohkuma M, Kudo T, et al. Crystal structure of meso-2,3-butanediol dehydrogenase in a complex with NAD+ and inhibitor mercaptoethanol at 1.7 A resolution for understanding of chiral substrate recognition mechanisms. J Biochem. 2001;129(2):205–208. doi: 10.1093/oxfordjournals.jbchem.a002845. [DOI] [PubMed] [Google Scholar]
- 31.Otagiri M, Ui S, Takusagawa Y, Ohtsuki T, Kurisu G, Kusunoki M. Structural basis for chiral substrate recognition by two 2,3-butanediol dehydrogenases. FEBS Lett. 2010;584(1):219–223. doi: 10.1016/j.febslet.2009.11.068. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Wang Y, Zhang L, Ma C, Wang A, Tao F, et al. Biocatalytic production of (2S,3S)-2,3-butanediol from diacetyl using whole cells of engineered Escherichia coli. Bioresour Technol. 2012;115:111–116. doi: 10.1016/j.biortech.2011.08.097. [DOI] [PubMed] [Google Scholar]
- 33.Yan Y, Lee CC, Liao JC. Enantioselective synthesis of pure (R, R)-2,3-butanediol in Escherichia coli with stereospecific secondary alcohol dehydrogenases. Org Biomol Chem. 2009;7(19):3914–3917. doi: 10.1039/b913501d. [DOI] [PubMed] [Google Scholar]
- 34.Zhang M, Eddy C, Deanda K, Finkelstein M, Picataggio S. Metabolic engineering of a pentose metabolism pathway in ethanologenic Zymomonas mobilis. Science. 1995;267(5195):240–243. doi: 10.1126/science.267.5195.240. [DOI] [PubMed] [Google Scholar]
- 35.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Médici R, Stammes H, Kwakernaak S, Otten LG, Hanefeld U. Assessing the stereoselectivity of Serratia marcescens CECT 977 2,3-butanediol dehydrogenase. Catal Sci Technol. 2017;7:1831–1837. doi: 10.1039/C7CY00169J. [DOI] [Google Scholar]
- 37.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 38.Yu M, Huang M, Song Q, Shao J, Ying X. Characterization of a (2R,3R)-2,3-butanediol dehydrogenase from Rhodococcus erythropolis WZ010. Molecules. 2015;20(4):7156–7173. doi: 10.3390/molecules20047156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rados D, Turner DL, Catarino T, Hoffart E, Neves AR, Eikmanns BJ, et al. Stereospecificity of Corynebacterium glutamicum 2,3-butanediol dehydrogenase and implications for the stereochemical purity of bioproduced 2,3-butanediol. Appl Microbiol Biotechnol. 2016;100(24):10573–10583. doi: 10.1007/s00253-016-7860-6. [DOI] [PubMed] [Google Scholar]
- 40.Braun F, Bertoletti N, Moller G, Adamski J, Steinmetzer T, Salah M, et al. First structure-activity relationship of 17beta-hydroxysteroid dehydrogenase type 14 nonsteroidal inhibitors and crystal structures in complex with the enzyme. J Med Chem. 2016;59(23):10719–10737. doi: 10.1021/acs.jmedchem.6b01436. [DOI] [PubMed] [Google Scholar]
- 41.Lukacik P, Keller B, Bunkoczi G, Kavanagh KL, Lee WH, Adamski J, et al. Structural and biochemical characterization of human orphan DHRS10 reveals a novel cytosolic enzyme with steroid dehydrogenase activity. Biochem J. 2007;402(3):419–427. doi: 10.1042/BJ20061319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shanati T, Lockie C, Beloti L, Grogan G, Ansorge-Schumacher MB. Two enantiocomplementary ephedrine dehydrogenases from Arthrobacter sp. TS-15 with broad substrate specificity. ACS Catal. 2019;9(7):6202–6211. doi: 10.1021/acscatal.9b00621. [DOI] [Google Scholar]
- 43.Nishioka T, Yasutake Y, Nishiya Y, Tamura T. Structure-guided mutagenesis for the improvement of substrate specificity of Bacillus megaterium glucose 1-dehydrogenase IV. FEBS J. 2012;279(17):3264–3275. doi: 10.1111/j.1742-4658.2012.08713.x. [DOI] [PubMed] [Google Scholar]
- 44.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 46.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39(4):783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 47.Zuckerkandl E, Pauling L. Molecules as documents of evolutionary history. J Theor Biol. 1965;8(2):357–366. doi: 10.1016/0022-5193(65)90083-4. [DOI] [PubMed] [Google Scholar]
- 48.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 50.Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A. FFAS server: novel features and applications. Nucleic Acids Res. 2011;39(Web Server issue):W38–44. doi: 10.1093/nar/gkr441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A. FFAS03: a server for profile–profile sequence alignments. Nucleic Acids Res. 2005;33(Web Server issue):W284–288. doi: 10.1093/nar/gki418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu D, Jaroszewski L, Li Z, Godzik A. FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics. 2014;30(5):660–667. doi: 10.1093/bioinformatics/btt578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engh RA, Huber R. Accurate bond and angle parameters for X-Ray protein-structure refinement. Acta Crystallographica Section A. 1991;47:392–400. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. PCR analysis of Z. mobilis tranformants containing the different Bdh genes. Eleven Bdh genes were transformed into Z. mobilis strain 9C and plated on RMG medium containing spectinomycin. Eight independent colonies were selected for colony PCR analysis using gene specific primers. Five microliters of the PCR products were run on 1% agarose gel and visualized using FluoChem Gel analyzer. White bar represents sets of 8 lanes for each Bdh gene. Av, Azotobacter vinelandii; Ml, Micrococcus luteus; Ea, Erwinia amylovora; Sw, Staphylococcus warneri; Sc, Streptomyces coelicolor; Sm, Serratia marcescens; Dd, Dickeya dadantii; Tg, Thermococcus gammatolerans; At, Agrobacterium tumefaciens; Ms, Mycobacterium Smegmatis; Mo, Myroides odoratimimus; 9c, Z. mobilis control strain.
Additional file 2. BDH genes selected for expression in Z. mobilis.
Additional file 3. Detection of heterologous Bdh proteins from Z. mobilis transformants using total protein staining. Five independent transformant colonies were selected for protein extraction. Total proteins were separated on 4-12% SDS PAGE gel followed by Coomassie staining. Arrows represent the expected molecular weight of the individual Bdh proteins. Two Bdh protein sets are shown in each PAGE gel. Each protein set is represented by 5 lanes as indicated by the black bars. Bdh protein names are shown above the black bars. Av, Azotobacter vinelandii; Ml, Micrococcus luteus; Ea, Erwinia amylovora; Sw, Staphylococcus warneri; Sc, Streptomyces coelicolor; Sm, Serratia marcescens; Dd, Dickeya dadantii; Tg, Thermococcus gammatolerans; At, Agrobacterium tumefaciens; Ms, Mycobacterium Smegmatis; Mo, Myroides odoratimimus; C, Z. mobilis control strain.
Additional file 4. Ala93Cα-Trp192Cα distance in ‘open’ and ‘closed’ molecules, Å.
Additional file 5. Multiple alignment of N-terminal histidine tagged SmBdh sequences representing the sites of amino acid changes. Three different versions of SmBdh are shown. Boxes indicate the amino acids that were replaced in the other mutants. SmBdh (N-term), wild-type; Q247A-SmBdh (N-term) and Q247A+V139Q-SmBdh (N-term), two different variants of SmBdh.
Additional file 6. 2,3-butanediol dehydrogenase gene sequences used in the study.
Additional file 7. Primers used in this study.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.