

Abstract
Background
Mild cognitive impairment (MCI) is a precursor to Alzheimer’s disease (AD), but not all MCI patients develop AD. Biomarkers for early detection of individuals at high risk for MCI-to-AD conversion are urgently required.
Methods
We used blood-based microRNA expression profiles and genomic data of 197 Japanese MCI patients to construct a prognosis prediction model based on a Cox proportional hazard model. We examined the biological significance of our findings with single nucleotide polymorphism-microRNA pairs (miR-eQTLs) by focusing on the target genes of the miRNAs. We investigated functional modules from the target genes with the occurrence of hub genes though a large-scale protein-protein interaction network analysis. We further examined the expression of the genes in 610 blood samples (271 ADs, 248 MCIs, and 91 cognitively normal elderly subjects [CNs]).
Results
The final prediction model, composed of 24 miR-eQTLs and three clinical factors (age, sex, and APOE4 alleles), successfully classified MCI patients into low and high risk of MCI-to-AD conversion (log-rank test P = 3.44 × 10−4 and achieved a concordance index of 0.702 on an independent test set. Four important hub genes associated with AD pathogenesis (SHC1, FOXO1, GSK3B, and PTEN) were identified in a network-based meta-analysis of miR-eQTL target genes. RNA-seq data from 610 blood samples showed statistically significant differences in PTEN expression between MCI and AD and in SHC1 expression between CN and AD (PTEN, P = 0.023; SHC1, P = 0.049).
Conclusions
Our proposed model was demonstrated to be effective in MCI-to-AD conversion prediction. A network-based meta-analysis of miR-eQTL target genes identified important hub genes associated with AD pathogenesis. Accurate prediction of MCI-to-AD conversion would enable earlier intervention for MCI patients at high risk, potentially reducing conversion to AD.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13195-020-00716-0.
Keywords: Alzheimer’s disease, Biomarkers for early diagnosis, eQTL effect
Background
Mild cognitive impairment (MCI) is an intermediate stage between normal aging and dementia, and its presence is associated with a higher risk of progression to clinically probable Alzheimer’s disease (AD) [1–3]. The annual conversion rate from MCI to AD has been reported as 10 to 15% [4]. After 6 years of follow-up, approximately 80% of MCI patients will have converted to AD (MCI converters [MCI-C]) [4, 5], although some MCI patients remain stable or convert back to normal (MCI non-converters [MCI-NC]) [6]. To date, there are no curative treatments for patients who already have AD, and available treatments are only able to postpone the progression of the disease [7]. Therefore, biomarkers for early detection of MCI-C and prognosis prediction models are both desperately required. These will allow early treatment of patients with MCI before they convert to AD, which could reduce the number of patients with AD.
Multifactorial diseases, such as AD, are induced by a combination of genetic and environmental factors. The heritability of late-onset AD is estimated to be around 60–80% [8]. Therefore, many genetic factors will undoubtedly contribute to the etiopathogenesis and progression of AD. In fact, a large number of genetic factors have already been associated with increased risk for AD. Amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) as causes of autosomal dominant AD [9] and the ε4 allele of apolipoprotein E (APOE ε4) are the strongest known genetic risk factors [10, 11]. Some additional genetic loci (single nucleotide polymorphisms [SNPs]) associated with AD diagnosis have been recently identified by genome-wide association studies [12–14]. However, a large proportion of the heritability remains unexplained, and further investigation of novel genetic factors will be necessary for early detection of MCI-C.
As with genetic factors, microRNAs (miRNAs) have been examined as potential biomarkers for early AD prediction [15, 16]. MicroRNAs are small non-coding RNA molecules of approximately 22 nucleotides that play a key role in the post-transcriptional regulation of gene expression and cell development, including neural cells [17]. Changes in the expression of some miRNAs have been detected in neurons of patients with AD and other neurodegenerative diseases [18–22]. Also, neurite and synapse destruction associated with the pathology of neurodegenerative diseases has been detected in vitro by quantitative analysis of brain-enriched cell-free miRNA in human blood [23]. These miRNAs may have practical clinical use as blood-based biomarkers, which are attractive because they are minimally invasive, cost-effective, and easy to reproduce.
A powerful and useful method to investigate the relationship between SNPs and miRNA expression is to analyze the genome-wide expression quantitative trait loci (miR-eQTL) [24]. Some miR-eQTLs have been associated with disease pathogenesis, such as the pathogenesis of autoimmune disease [25], cancers [26], and neurological disorders [27], indicating that miR-eQTLs can be effective biomarkers for predicting disease prognosis.
Here, we examined comprehensive miRNA expression profiles and genetic variants in 197 MCI patients (83 MCI-C and 114 MCI-NC) and identified miR-eQTLs that could allow earlier diagnosis and therapeutic intervention. Our final prognosis prediction model was constructed based on a Cox proportional hazard model that included 24 miR-eQTLs and three clinical factors (age, sex, and APOE ε4 status). In an independent test set, the model successfully classified the MCI patients into groups at high and low risk of MCI-to-AD conversion. In addition, a network-based meta-analysis using miRNA target genes revealed important hub genes associated with the pathogenesis of AD. Statistically significant differences in expression between disease groups were observed for some of these hub genes. Accurate prediction of MCI-to-AD conversion risk could enable earlier interventions for appropriate patients, potentially leading to a reduction in conversions from MCI to AD.
Methods
Clinical samples
All 197 MCI patients and the associated clinical data were distributed from the National Center for Geriatrics and Gerontology (NCGG) Biobank, which collects human biomaterials and data for geriatrics research. The AD and MCI subjects were diagnosed with probable or possible AD based on the criteria of the National Institute on Aging Alzheimer’s Association workgroups [1, 2]. We included patients with probable AD as subjects in this study. The diagnosis of all subjects was made based on medical history, physical examination, diagnostic tests, neurological examination, neuropsychological tests, and brain imaging with magnetic resonance imaging or computerized tomography by one or more experts in dementia who are familiar with its diagnostic criteria (neurologists, psychiatrists, geriatricians, or neurosurgeons). Comprehensive neuropsychological tests included the Mini-Mental State Examination (MMSE), Alzheimer’s Disease Assessment Scale Cognitive Component Japanese version, Logical Memory I and II from the Wechsler Memory Scale–Revised, frontal assessment battery, Raven’s colored progressive matrices, and the Geriatric Depression Scale [28]. For all subjects, the number of the APOE4 alleles (the major genetic risk factor for AD) and the MMSE score were obtained. All subjects were required to be over 60 years of age.
SNP genotyping and data cleaning
All 197 subjects were genotyped using the Affymetrix “Japonica Array” (TOSHIBA, Inc.) [29]. Genotype imputation was performed using IMPUTE2 (version 2.3.2) [30] with the 1000 Genomes Project reference panel. A total of 2,836,104 autosomal SNPs and short indels passed the quality control criteria after imputation (SNP/indel call rate ≥ 0.99 and minor allele frequency ≥ 0.01). To generate a set of independent SNPs, we further performed linkage disequilibrium-based SNP pruning using the statistical analysis program PLINK, version 1.90b [31] with a window size of 50 SNPs, a step of 5 SNPs, and a pairwise r2 threshold of 0.1 (--indep-pairwise 50 5 0.1). Any sites with missing data were discarded. A final subset of 92,878 SNPs was obtained.
miRNA expression
Total RNA was extracted from a blood sample by using the 3D-Gene RNA extraction reagent from a liquid sample kit (Toray Industries, Inc.). The miRNA expression analysis was performed with a 3D-Gene miRNA Labeling kit and a 3D-Gene Human miRNA Oligo Chip (Toray Industries, Inc.), which was designed to detect 2562 miRNA sequences registered in miRBase release 21 (http://www.mirbase.org/). Normalization of miRNA expression was performed in the following steps. Mean and standard deviation (SD) were calculated by using a set of pre-selected negative control signals (background signals) from which the top and bottom 5% of values were removed. Signal values greater than mean + 2SD of the background signals were replaced with log2(signal − mean) and labeled effective signals. The remaining signal values were replaced with the minimum of the effective signals − 0.1. Undetected signal values were replaced by the minimum value of each miRNA. To normalize the signals across different microarrays, a set of pre-selected internal control miRNAs (miR-149-3p, miR-2861, and miR-4463) was used; these miRNAs had been stably detected in more than 500 serum samples. Each miRNA signal value was standardized by dividing it by the average signal of the three internal control miRNA signals [32].
Construction of the prognosis prediction model
All data were strictly separated into a discovery cohort and a validation cohort. To generate datasets of pre-selected SNPs, we used two thirds of the entire discovery cohort to calculate P values in each cross-validation step. The P values corresponding to the SNPs were calculated with the following logistic regression model between MCI-C and MCI-NC:
where XSNP is the SNP genotypes and (β1, β2, …, βn) is the respective coefficients. The logistic regression was implemented using PLINK [31]. From p pre-selected SNPs (p = 100, 200, …, 10,000), we focused on SNP-miRNA pairs with eQTL effects (miR-eQTLs). The SNP-miRNA pairs with adjusted P value (permutation test) < 0.1 were obtained from an in-house miR-eQTL database (data not shown). Using a combination of the miR-eQTLs and clinical factors (age, sex, and number of APOE4 alleles), a prognosis prediction model was constructed based on a Cox proportional hazard model using two thirds of the discovery cohort as defined by:
where h(t| C, I) is the expected hazard at time t, determined by a set of three covariates (C1, C2, C3 ) = (age, sex, number of APOE4 alleles) and m covariates (I1, I2, …, Im ) whose impacts are measured by the respective coefficients (β1, β2, …, βn ). Then, let X = {X1, …, Xp} be the pre-selected SNP genotypes and Y = {Y1, …, Yq} be the miRNA expression values. Let σ = {(x, y)| x ∈ X ∧ y ∈ Y} be any SNP-miRNA pair, and let I = {(x, y) ∈ σ| miR − eQTLs} be the SNP-miRNA pairs with eQTL effects. The adjusted model was then evaluated using the remaining third of the discovery cohort. This process was repeated 3 times (3-fold cross-validation). Based on the average C-index, we determined the optimal value of p, the number of pre-selected SNPs for model construction. The final model was constructed using the entire discovery cohort, and the adjusted model was evaluated on the independent validation cohort.
By using the combination of miR-eQTLs and clinical factors described above, we calculated a prognostic index for each sample in the discovery cohort as defined by:
We classified the samples into two groups (high and low risk) with an optimal cutoff value of the prognostic index [33]. The optimal cutoff value indicated by the minimum P value of the log-rank test when differences between high- and low-risk groups in the discovery cohort were compared. The optimal cutoff value was used to validate our prognosis prediction model. Kaplan-Meier curves were constructed to illustrate differences in survival without MCI-to-AD conversion. The log-rank test was used to compare the different conditions. P values < 0.05 were considered statistically significant. These statistical analyses were conducted using the survival and survminer packages in the statistical software R [34].
Target gene annotation of miRNAs
The functional gene annotation of miRNAs was conducted using miRDB, which includes gene targets that are predicted to be regulated by a comprehensive set of 6709 miRNAs [35]. All gene targets have a prediction score between 0 and 100 assigned by MirTarget V3, with a higher score representing more statistical confidence in the prediction result. Only gene targets with scores of > 90 were used in our analysis.
Network-based meta-analysis
The network-based analysis was performed with NetworkAnalyst [36] and the STRING Interactome database [37], which provides comprehensive information about interactions between proteins, including prediction and experimental interaction data. The confidence cutoff score was set to 900. The PPI network was constructed with a zero-order interaction network analysis (direct interaction only) and graphically generated using Cytoscape v3.7.2 (http://www.cytoscape.org/) [38].
RNA-sequencing data analysis
The quality of the read sequences was assessed by using FastQC (version 0.11.7). Low-quality reads (< Q20) and trimmed reads with adapter sequences (< 50 bp) were discarded by using Cutadapt (version 1.16). The remaining clean sequenced reads were mapped to the human reference genome (GRCh37) by using STAR (2-pass option, version 2.5.2b) [39]. By using the featureCounts program [40] from the subread package (version 1.6.6), read counts for each gene were calculated to generate expression levels. The values of outliers of the read counts (i.e., the top and bottom 5% of read counts for each gene) were changed to the maximum and minimum of the remaining values, respectively. The read counts from each sample were then combined into a count file, on which differential expression analysis was performed by using edgeR [41] (version 3.18.1). Genes with a threshold CPM (counts per million reads mapped) > 1 in more than one fourth of all sequenced samples were used for further analysis. The caclNormFactorsfunction in edgeR [41] was used to obtain trimmed mean of M value normalization factors to account for library sizes. The dispersion was calculated by using the estimateCommonDisp and estimateTagwiseDisp functions in edgeR [41]. The exactTest function in edgeR [41] was applied to obtain genes differentially expressed between disease groups.
Results
Patient characteristics
The study enrolled 197 MCI patients (73 males and 124 females), followed for at least 6 months and up to 7 years (mean ± SD, 971 ± 552 days), during which time 83 (42.1%) converted to AD (i.e., 83 were classified as MCI-C). The remaining 114 patients with MCI (57.9%) were classified as MCI-NC. We divided the 197 patients with MCI into a discovery cohort of 98 individuals (41 MCI-C and 57 MCI-NC) and a validation cohort of 99 individuals (42 MCI-C and 57 MCI-NC). The patient characteristics of each cohort are summarized in Table 1.
Table 1.
Clinical characteristics of the discovery and validation cohorts
| Phenotype | Factor | All | Discovery cohort | Validation cohort |
|---|---|---|---|---|
| MCI-C | Number of subjects | 83 | 41 | 42 |
| Age ± SD | 75.22 ± 6.22 | 75.10 ± 5.64 | 75.33 ± 6.81 | |
| Percentage of male (# patients) | 29 (24) | 41 (17) | 17 (7) | |
| Number of APOE4 alleles (# patients) | 0 (46), 1 (29), 2 (8) | 0 (25), 1 (13), 2 (3) | 0 (21), 1 (16), 2 (5) | |
| Follow-up, mean ± SD (days) | 927.18 ± 535.03 | 1014.07 ± 593.22 | 842.36 ± 462.90 | |
| MCI-NC | Number of subjects | 114 | 57 | 57 |
| Age ± SD | 75.56 ± 6.39 | 75.42 ± 5.75 | 75.70 ± 7.02 | |
| Percentage of male (# patients) | 43 (49) | 44 (25) | 42 (24) | |
| Number of APOE4 alleles (# patients) | 0 (80), 1 (32), 2 (2) | 0 (40), 1 (15), 2 (2) | 0 (40), 1 (17), 2 (0) | |
| Follow-up, mean ± SD (days) | 1002.23 ± 564.49 | 939.70 ± 554.31 | 1064.75 ± 572.52 | |
| Total subjects | 197 | 98 | 99 |
MCI-C mild cognitive impairment converters (to Alzheimer’s disease), MCI-NC mild cognitive impairment non-converters, SD standard deviation
Prognosis prediction model construction
Our prognosis prediction model was based on a Cox proportional hazard method using miR-eQTLs (see details in the “Methods” section). Because evaluating all possible combinations of miR-eQTLs would be too time-consuming and computationally expensive, we generated datasets of pre-selected SNPs. The selection of the SNPs was carried out based on the P value of logistic regression with adjustments for three covariates: age, sex, and the number of APOE4 alleles. Two thirds of the entire discovery cohort was used for the calculation of the P values. From p pre-selected SNPs (p = 100, 200, …, 10,000), we detected effective miR-eQTLs, which were obtained from an in-house miR-eQTL database (data not shown). Using a combination of the miR-eQTLs and the three clinical factors, the prognosis prediction model was constructed based on two thirds of the entire discovery cohort. The adjusted model was evaluated using the remaining third of the discovery cohort. The value of p that yielded highest average concordance index (C-index) across three rounds of cross-validation of the discovery cohort (Fig. 1) was selected as the optimal p. The final prognosis prediction model was constructed from miR-eQTLs detected from this optimal p (9600) pre-selected SNPs and the clinical factors using the entire discovery cohort. The adjusted model was then evaluated on the validation cohort, which was completely independent of the discovery cohort. Twenty-four miR-eQTLs were used in the final model construction, which achieved a C-index of 0.718 in the discovery cohort (Fig. 2a) and of 0.702 in the validation cohort (Fig. 2b). We also calculated a prognostic index for each subject by applying the 24 miR-eQTLs and three clinical factors to our prognosis prediction model (Table 2). We used the prognostic index to divide the discovery cohort into high- and low-risk groups. The optimal cutoff value was detected by using the minimum P value from the log-rank test and comparing the differences in survival without MCI-to-AD conversion as determined by Kaplan-Meier curves (optimal cutoff = 7.85, minimum P = 3.63 × 10−7, Fig. 2a). This adjusted model then successfully classified MCI patients in the validation cohort into groups with low- and high-risk of MCI-to-AD conversion (log-rank test P = 3.44 × 10−4, Fig. 2b).
Fig. 1.

Construction workflow of our prognosis prediction model. We generated data sets of pre-selected SNPs based on the P values from logistic regression models. Two thirds of the entire discovery cohort was used to calculate the P values in each cross-validation step (1). Using the top-ranked SNPs, we focused on SNP-miRNA pairs with eQTL effects (miR-eQTLs) (2). Using a combination of miR-eQTLs and clinical factors, we constructed a prognosis prediction model based on a Cox proportional hazard model using two thirds of the discovery cohort. The adjusted model was then evaluated using the remaining one third (3). On the basis of the average C-index from the 3-fold cross-validation, we determined the optimal pre-selected SNPs for model construction (4). The final model was constructed using the entire discovery cohort (5), and the adjusted model was evaluated in the independent validation cohort (6)
Fig. 2.

Kaplan-Meier curves of survival without conversion to AD produced by the prediction models. We calculated a prognostic index for each subject by applying the miR-eQTLs and clinical factors to our prognosis prediction model. a Based on the prognostic index, we divided the samples of the discovery cohort into high (red) and low (blue) risk groups. The optimal cutoff values were detected by using the minimum P value from the log-rank test and comparing the differences in survival without MCI-to-AD conversion as determined by Kaplan-Meier curves (optimal cutoff = 7.85, minimum P = 3.63 × 10−7). b The adjusted model was then evaluated on the validation cohort (log-rank test P = 3.44 × 10−4). c, d Prediction models constructed using only clinical factors (without miR-eQTLs) in the discovery cohort (c) and the validation cohort (d)
Table 2.
Potential biomarkers used in our prognosis prediction model
| Factor | P value† | Coefficient | |
|---|---|---|---|
| Clinical factors | Age | NA | 0.077 |
| Sex | NA | 0.530 | |
| APOE4 | NA | 0.602 | |
| miR-eQTLs | MIMAT0019690—rs6721935 | 0.092 | 0.181 |
| MIMAT0015080—rs12616298 | 0.089 | − 0.170 | |
| MIMAT0005582—rs12997752 | 0.038 | − 0.141 | |
| MIMAT0027499—rs76232851 | 0.093 | 0.042 | |
| MIMAT0019229—rs117574479 | 0.032 | 0.152 | |
| MIMAT0019229—rs116868325 | 9.69 × 10−9 | 0.058 | |
| MIMAT0019045—rs3777118 | 1.10 × 10−4 | 0.034 | |
| MIMAT0021034—rs118073044 | 0.037 | 1.078 | |
| MIMAT0000751—rs118073044 | 0.078 | − 3.163 | |
| MIMAT0001630—rs118073044 | 0.020 | 1.654 | |
| MIMAT0000086—rs117393460 | 0.078 | 0.260 | |
| MIMAT0021033—rs72861163 | 0.037 | 0.337 | |
| MIMAT0030997—rs9507595 | 0.086 | − 0.123 | |
| MIMAT0027602—rs17682567 | 0.072 | − 0.050 | |
| MIMAT0015080—rs117534907 | 0.006 | − 0.032 | |
| MIMAT0023710—rs11855092 | 0.014 | 0.156 | |
| MIMAT0016899—rs79726130 | 0.054 | − 0.436 | |
| MIMAT0028122—rs79726130 | 0.049 | 0.465 | |
| MIMAT0015080—rs35831886 | 1.97 × 10−5 | 1.785 | |
| MIMAT0019229—rs35831886 | 0.008 | − 1.688 | |
| MIMAT0019045—rs117336092 | 0.031 | − 0.016 | |
| MIMAT0019229—rs117099240 | 0.061 | 0.028 | |
| MIMAT0032029—rs149944930 | 0.009 | 0.090 | |
| MIMAT0027487—rs2830386 | 0.069 | 0.125 |
†miR-eQTLs with adjusted P value < 0.1 obtained from an in-house miR-eQTL database were included
Effectiveness of detected miR-eQTLs
To estimate the effectiveness of the 24 miR-eQTLs selected for the MCI-to-AD conversion prediction model, we compared a prediction model including the miR-eQTLs to one including only clinical factors. The prediction model excluding miR-eQTLs achieved lower C-indices (0.611 and 0.586 in the discovery and validation cohorts, respectively; Fig. 2c, d) than the model including miR-eQTLs (Fig. 2a, b). In addition, this prediction model did not successfully divide samples into high- and low-risk groups in the validation cohort (optimal cutoff = 4.39, log-rank test P = 0.255, Fig. 2d). These results show that the detected miR-eQTLs improve the prognosis prediction model.
To ensure the robustness and generality of our findings, we further compared prognosis models with and without miR-eQTLs with a bootstrap resampling technique. This procedure was repeated 10,000 times. The models with miR-eQTLs performed better than those with only clinical factors, and the distributions of log-rank P values were significantly different between the prognosis models with and without miR-eQTLs (P < 0.01, Welch’s t test) (Fig. 3).
Fig. 3.
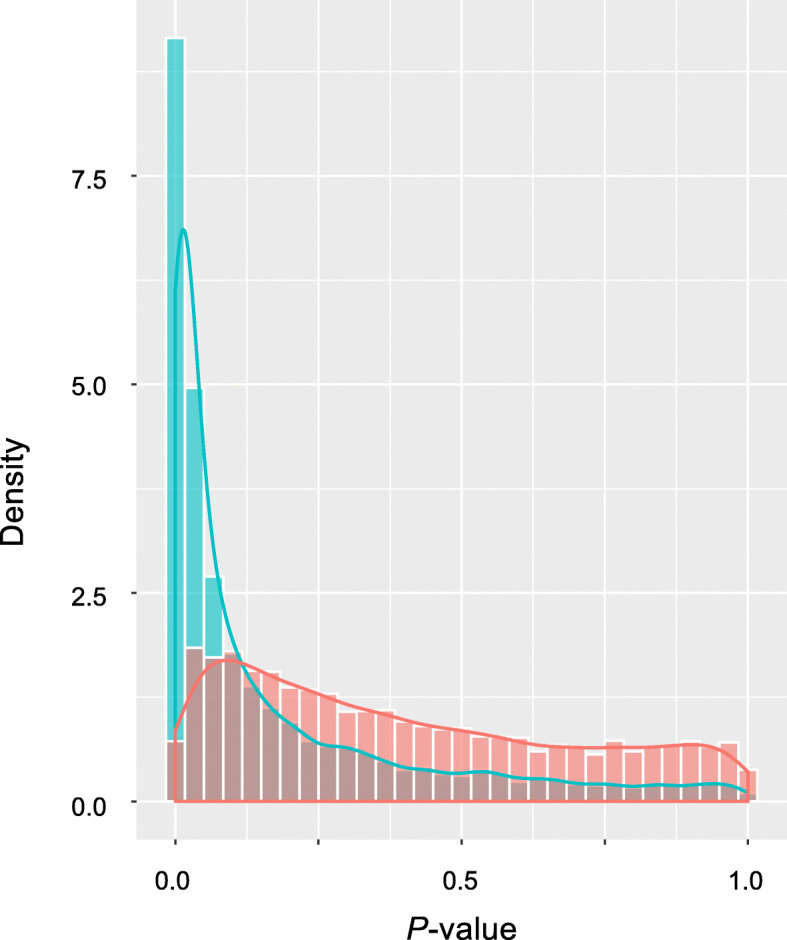
Distribution of log-ranked P values using a bootstrap resampling technique. We compared a prognosis model with miR-eQTLs (blue) to one without miR-eQTLs (red) using a bootstrap resampling technique. This procedure was repeated 10,000 times. The distribution of log-rank P values was significantly different between the prediction models with and without miR-eQTLs (P < 0.01, Welch’s t test)
Functional gene annotations
We next examined the biological significance of our findings with miR-eQTLs by focusing on the target genes of the miRNAs. miRNAs regulate the expression of thousands of mRNAs from protein-coding genes at both the post-transcriptional and translational levels [42–44]. We predicted functional target genes of miRNAs by using the microRNA Target Prediction and Functional Study Database, miRDB [35]. Our 24 miR-eQTLs, which are composed of 20 SNPs and 18 miRNAs (Table 2 and Supplementary Table S1), were predicted to target 778 genes. We attempted to elucidate functional modules from the target genes with the occurrence of hub genes though a large-scale protein-protein interaction (PPI) network analysis. The PPI network analysis was performed by using NetworkAnalyst [36] (http://www.networkanalyst.ca) with the STRING Interactome database [37]. A PPI network generated with 2304 nodes and 3901 edges was obtained. To prune the network down to a more manageable size, we conducted a zero-order interaction network analysis and detected a refined network containing 60 nodes and 66 edges (Fig. 4). This PPI network visualization was performed with Cytoscape software [38]. Four hub genes, GSK3B, PTEN, FOXO1, and SHC1, were detected as functional modules that directly interacted with each other and with ≥ 5 other genes (Fig. 4). These four genes were regulated by the miRNAs MIMAT0032029 (hsa-miR-1249-5p), MIMAT0000086 (hsa-miR-29a-3p), MIMAT0000751 (hsa-miR-330-3p), and MIMAT0021034 (hsa-miR-5006-3p), respectively.
Fig. 4.

Results of the NetworkAnalyst PPI network analysis. Nodes represent genes. Node size corresponds to the number of connected edges. The gene name is displayed for nodes with ≥ 5 edges
Hub gene expression detected by PPI network analysis
Because the four hub genes (GSK3B, PTEN, FOXO1, and SHC1) were detected by PPI network analysis based on blood-based miRNA data, we checked whether these genes are actually expressed in blood cells as well as the brain by using the Human Protein Atlas database [45], which provides quantitative transcriptomics at the tissue and organ level and is publicly accessible at http://www.proteinatlas.org. Two genes (FOXO1 and GSK3B) showed low levels of expression in blood, but all genes showed high levels of expression in the brain (Fig. 5a). To validate these results, we examined the expression of these genes in 610 blood samples [46] (271 ADs, 248 MCIs, and 91 cognitively normal elderly subjects [CNs]), and we investigated the differential gene expression across the disease groups. FOXO1 and GSK3B showed no statistically significant difference in expression between disease groups (Fig. 5b and Supplementary Table S2). However, PTEN showed significantly higher gene expression in AD than in MCI (P = 0.023), and SHC1 showed significantly lower expression in AD than in CN (P = 0.049) (Fig. 5b and Supplementary Table S2). We further investigated whether the genes were differentially expressed between MCI-C and MCI-NC patients (n = 123; 48 MCI-C and 75 MCI-NC), but no genes showed statistically significant differences between the two groups (Fig. 5c and Supplementary Table S3). Thus, while it might be difficult to predict the risk of AD conversion of MCI patients from these genes’ expressions, there is no doubt that these genes are associated with AD pathogenesis.
Fig. 5.

Expression of hub genes detected in the PPI network analysis. a The expression of all hub genes in blood cells (red) and brain tissues (yellow) were checked in the Human Protein Atlas database. An X-axis represents the resulting transcript expression values, denoted normalized expression (NX), which were calculated for each gene in every sample. b, c Gene expression was further examined using our 610 blood samples (271 from patients with AD, 248 from patients with MCI, and 91 from CNs). The difference in gene expression was examined between diseases (b) and between MCI-C and MCI-NC patients (n = 123; 48 MCI-C and 75 MCI-NC) (c). b, c P values are displayed above the boxplots. *Statistically significant differences, “exactTest” function in edgeR; TMM, trimmed mean of M values
Discussion
Early detection of individuals at high risk for MCI-to-AD conversion is important for delivering appropriate early intervention and for better managing the disease. Potential new biomarkers for this early diagnosis and prognosis have been investigated [47–50]. The role of genetic variation and blood-based miRNAs has been reviewed, with emphasis on their impact on the etiopathogenesis of sporadic AD [51, 52]. However, an integrated omics analysis of these genetic variants and miRNA expression has not, to our knowledge, been conducted in AD or other subtypes of dementia. Therefore, in this study, we investigated potential biomarkers by considering both genetic variants and miRNA expression, and we constructed a prognosis prediction model using the identified biomarkers.
We detected an optimal parameter set for the prediction model by using cross-validation of the discovery cohort. The final model was then constructed with the optimal parameters using the complete discovery cohort. The adjusted model was then evaluated on an independent validation cohort using the C-index to discriminate the accuracy of the prognosis prediction model and using survival without MCI-to-AD conversion as determined with Kaplan-Meier curves. Our final prediction model produced similar C-indices (0.718 and 0.702) in the discovery and validation cohorts and successfully classified MCI patients into two groups, low and high, in terms of risk of MCI-to-AD conversion (log-rank test P = 3.63 × 10−7 and 3.44 × 10−4). These results demonstrate that miR-eQTLs could be efficient biomarkers for the early detection of individuals at high risk of MCI-to-AD conversion, although replication studies using a larger number of samples are necessary. Bootstrap resampling indicated that prediction models with miR-eQTLs had superior performance to those with clinical factors only (without miR-eQTLs). These results provide evidence that our findings can be efficiently applied to early prediction of MCI-to-AD conversion, which is expected to contribute to practical clinical use in MCI-to-AD conversion in the near future.
Our findings represent SNP-miRNA pairs with eQTL effects. Each SNP was associated with variation of miRNA expression levels. These miRNAs act as post-transcriptional regulators of their mRNA targets through mRNA degradation and/or translational repression. Therefore, annotation of the mRNA gene targets can lead to functional characterization of our findings. The functional gene annotation of miRNAs was conducted using miRDB [35], and four functionally important modules (i.e., hub genes, GSK3B, PTEN, FOXO1, and SHC1) were detected from large-scale PPI network analysis. FOXO1 is a member of the evolutionarily conserved FOXO family of transcription factors. Although only FOXO3 has been analyzed in AD models so far, the gene has been reported to induce cell death in response to amyloid beta plaques and to influence the function of the peripheral and central nervous systems [53]. GSK3B has been identified as a tau protein kinase I, which phosphorylates tau at several sites [54]. This abnormally hyperphosphorylated tau is generally observed in the brains of patients with AD [55]. Although PTEN is best known as a tumor suppressor gene, this gene has also been reported to be associated with other diseases, including diabetes and AD [56]. Liang et al. reported that SHC1 might play a key role in the progression of AD [57]. Also, Zheng et al. recently ranked 500 genes according to their potential association with AD risk, and SHC1 was in the top 20 [58]. In this study, we further investigated the expression of these hub genes in CN, MCI, and AD blood samples. Whereas FOXO1 and GSK3B showed low levels of expression in all of the samples, PTEN and SHC1 showed a significant difference in gene expression in the blood between diseases. These results suggest that these identified hub genes are associated with the pathogenesis of AD.
We have proposed an MCI-to-AD conversion prediction model based on a Cox proportional hazard method using SNP-miRNA pairs with eQTL effects. Our proposed model may enable early detection of patients at high risk of MCI-to-AD conversion. However, further refinement of this model, using a larger number of samples, will be required before it can be used in health care. Omics analyses of genetic variations, such as SNPs, insertions and deletions (indels), and gene expression, will play an important role in the further improvement of this prognosis prediction model.
Limitations
There are limitations of the current analyses. As it is too difficult to collect many MCI converters and MCI non-converters, our prediction model is constructed using Japanese MCI patients with limited sample size. In the future, we will perform further investigations with larger sample size and will further validate the effectiveness of this classifier.
Conclusions
Our final prediction model successfully classified MCI patients into low and high risk of MCI-to-AD conversion and achieved a high concordance index on an independent test set. Important hub genes associated with AD pathogenesis were also identified in a network-based meta-analysis of miR-eQTL target genes. Accurate prediction of MCI-to-AD conversion would enable earlier intervention for MCI patients at high risk, potentially reducing conversion to AD.
Supplementary Information
Additional file 1: Supplementary Table S1. SNP genotypes included in miR-eQTLs as potential biomarkers.
Additional file 2: Supplementary Table S2. Gene expression in 610 blood samples.
Additional file 3: Supplementary Table S3. Gene expression in 123 MCI blood samples.
Acknowledgements
We thank the NCGG Biobank for providing the study materials, clinical information, and technical support. We also thank ELSS editors for English corrections.
Authors’ contributions
D.S. developed the method and performed the analyses; S.A., A.S., K. B, T.T., and T.O. provided the technical assistance; S.H., T. Sugimoto, T. Sakurai, and S.N. contributed to the data acquisition and the analyses; D.S. wrote the manuscript; D.S. and K.O. organized this work. All authors contributed to and approved the final manuscript.
Funding
This study was supported by “The Japan Foundation for Aging and Health” and “Takeda Science Foundation” (to D. S.); The Research Funding for Longevity Sciences from the National Center for Geriatrics and Gerontology (29-45 to K.O., and 30-29 to D.S.); a grant for Research on Dementia from the Japanese Ministry of Health, Labor and Welfare (to K.O.); and grants from the Japan Agency for Medical Research and Development (AMED) (Grant Number JP18kk0205009) (to S.N.).
Availability of data and materials
All microarray data (2562 miRNAs) and clinical information from this study are publicly available through the Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (NCBI) and accessible through GEO series accession number GSE150693 at http://www.ncbi.nlm.nih.gov/projects/geo/. The other datasets used or analyzed in the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was approved by the ethics committee of the NCGG. The design and performance of the current study, which involves human subjects, were clearly described in a research protocol. All participation was voluntary, and participants completed informed consent in writing before registering with the NCGG Biobank.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Daichi Shigemizu and Kouichi Ozaki contributed equally to this work.
References
- 1.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabuas-Pereira M, Baldeiras I, Duro D, Santiago B, Ribeiro MH, Leitao MJ, Oliveira C, Santana I. Prognosis of early-onset vs. late-onset mild cognitive impairment: comparison of conversion rates and its predictors. Geriatrics (Basel). 2016;1(2):11. [DOI] [PMC free article] [PubMed]
- 5.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 6.Lovell MA. A potential role for alterations of zinc and zinc transport proteins in the progression of Alzheimer’s disease. J Alzheimers Dis. 2009;16(3):471–483. doi: 10.3233/JAD-2009-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siedlecki-Wullich D, Catala-Solsona J, Fabregas C, Hernandez I, Clarimon J, Lleo A, Boada M, Saura CA, Rodriguez-Alvarez J, Minano-Molina AJ. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):46. doi: 10.1186/s13195-019-0501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 9.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18(5):421–430. doi: 10.1038/gim.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pericak-Vance MA, Bebout JL, Gaskell PC, Jr, Yamaoka LH, Hung WY, Alberts MJ, Walker AP, Bartlett RJ, Haynes CA, Welsh KA, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48(6):1034–1050. [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hagg S, Athanasiu L, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–413. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moreno-Grau S, de Rojas I, Hernandez I, Quintela I, Montrreal L, Alegret M, Hernandez-Olasagarre B, Madrid L, Gonzalez-Perez A, Maronas O, et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: the GR@ACE project. Alzheimers Dement. 2019;15(10):1333-47. [DOI] [PubMed]
- 14.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–430. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shigemizu D, Akiyama S, Asanomi Y, Boroevich KA, Sharma A, Tsunoda T, Matsukuma K, Ichikawa M, Sudo H, Takizawa S, et al. Risk prediction models for dementia constructed by supervised principal component analysis using miRNA expression data. Commun Biol. 2019;2:77. doi: 10.1038/s42003-019-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shigemizu D, Akiyama S, Asanomi Y, Boroevich KA, Sharma A, Tsunoda T, Sakurai T, Ozaki K, Ochiya T, Niida S. A comparison of machine learning classifiers for dementia with Lewy bodies using miRNA expression data. BMC Med Genet. 2019;12(1):150. doi: 10.1186/s12920-019-0607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiol Rev. 2011;91(3):827–887. doi: 10.1152/physrev.00006.2010. [DOI] [PubMed] [Google Scholar]
- 18.Satoh J. MicroRNAs and their therapeutic potential for human diseases: aberrant microRNA expression in Alzheimer’s disease brains. J Pharmacol Sci. 2010;114(3):269–275. doi: 10.1254/jphs.10R11FM. [DOI] [PubMed] [Google Scholar]
- 19.Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, Kelnar K, Kemppainen J, Brown D, Chen C, et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14(1):27–41. doi: 10.3233/JAD-2008-14103. [DOI] [PubMed] [Google Scholar]
- 20.Tacutu R, Budovsky A, Yanai H, Fraifeld VE. Molecular links between cellular senescence, longevity and age-related diseases - a systems biology perspective. Aging (Albany NY) 2011;3(12):1178–1191. doi: 10.18632/aging.100413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lusardi TA, Phillips JI, Wiedrick JT, Harrington CA, Lind B, Lapidus JA, Quinn JF, Saugstad JA. MicroRNAs in human cerebrospinal fluid as biomarkers for Alzheimer’s disease. J Alzheimers Dis. 2017;55(3):1223–1233. doi: 10.3233/JAD-160835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiedrick JT, Phillips JI, Lusardi TA, McFarland TJ, Lind B, Sandau US, Harrington CA, Lapidus JA, Galasko DR, Quinn JF, et al. Validation of MicroRNA biomarkers for Alzheimer’s disease in human cerebrospinal fluid. J Alzheimers Dis. 2019;67(3):875–891. doi: 10.3233/JAD-180539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheinerman KS, Tsivinsky VG, Crawford F, Mullan MJ, Abdullah L, Umansky SR. Plasma microRNA biomarkers for detection of mild cognitive impairment. Aging (Albany NY) 2012;4(9):590–605. doi: 10.18632/aging.100486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325(5945):1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wohlers I, Bertram L, Lill CM. Evidence for a potential role of miR-1908-5p and miR-3614-5p in autoimmune disease risk using integrative bioinformatics. J Autoimmun. 2018;94:83–89. doi: 10.1016/j.jaut.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Xiao W, Zhong Y, Wu L, Yang D, Ye S, Zhang M. Prognostic value of microRNAs in lung cancer: a systematic review and meta-analysis. Mol Clin Oncol. 2019;10(1):67–77. doi: 10.3892/mco.2018.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devanna P, Chen XS, Ho J, Gajewski D, Smith SD, Gialluisi A, Francks C, Fisher SE, Newbury DF, Vernes SC. Next-gen sequencing identifies non-coding variation disrupting miRNA-binding sites in neurological disorders. Mol Psychiatry. 2018;23(5):1375–1384. doi: 10.1038/mp.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawai Y, Miura R, Tsujimoto M, Sakurai T, Yamaoka A, Takeda A, Arahata Y, Washimi Y, Kachi T, Toba K. Neuropsychological differentiation between Alzheimer’s disease and dementia with Lewy bodies in a memory clinic. Psychogeriatrics. 2013;13(3):157–163. doi: 10.1111/psyg.12019. [DOI] [PubMed] [Google Scholar]
- 29.Kawai Y, Mimori T, Kojima K, Nariai N, Danjoh I, Saito R, Yasuda J, Yamamoto M, Nagasaki M. Japonica array: improved genotype imputation by designing a population-specific SNP array with 1070 Japanese individuals. J Hum Genet. 2015;60(10):581–587. doi: 10.1038/jhg.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. Plos Genet. 2009;5(6):e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slifer SH. PLINK: key functions for data analysis. Curr Protoc Hum Genet. 2018;97(1):e59. doi: 10.1002/cphg.59. [DOI] [PubMed] [Google Scholar]
- 32.Shimomura A, Shiino S, Kawauchi J, Takizawa S, Sakamoto H, Matsuzaki J, Ono M, Takeshita F, Niida S, Shimizu C, et al. Novel combination of serum microRNA for detecting breast cancer in the early stage. Cancer Sci. 2016;107(3):326–334. doi: 10.1111/cas.12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshihara K, Tsunoda T, Shigemizu D, Fujiwara H, Hatae M, Fujiwara H, Masuzaki H, Katabuchi H, Kawakami Y, Okamoto A, et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin Cancer Res. 2012;18(5):1374–1385. doi: 10.1158/1078-0432.CCR-11-2725. [DOI] [PubMed] [Google Scholar]
- 34.RDC T . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2009. [Google Scholar]
- 35.Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–D152. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santiago JA, Potashkin JA. Network-based metaanalysis identifies HNF4A and PTBP1 as longitudinally dynamic biomarkers for Parkinson’s disease. Proc Natl Acad Sci U S A. 2015;112(7):2257–2262. doi: 10.1073/pnas.1423573112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 41.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heneghan HM, Miller N, Kelly R, Newell J, Kerin MJ. Systemic miRNA-195 differentiates breast cancer from other malignancies and is a potential biomarker for detecting noninvasive and early stage disease. Oncologist. 2010;15(7):673–682. doi: 10.1634/theoncologist.2010-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asaga S, Kuo C, Nguyen T, Terpenning M, Giuliano AE, Hoon DS. Direct serum assay for microRNA-21 concentrations in early and advanced breast cancer. Clin Chem. 2011;57(1):84–91. doi: 10.1373/clinchem.2010.151845. [DOI] [PubMed] [Google Scholar]
- 44.Roth C, Rack B, Muller V, Janni W, Pantel K, Schwarzenbach H. Circulating microRNAs as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res. 2010;12(6):R90. doi: 10.1186/bcr2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 46.Shigemizu D, Mori T, Akiyama S, Higaki S, Watanabe H, Sakurai T, Niida S, Ozaki K. Identification of potential blood biomarkers for early diagnosis of Alzheimer’s disease through RNA sequencing analysis. Alzheimers Res Ther. 2020;12(1):87. doi: 10.1186/s13195-020-00654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khanna S, Domingo-Fernandez D, Iyappan A, Emon MA, Hofmann-Apitius M, Frohlich H. Using multi-scale genetic, neuroimaging and clinical data for predicting Alzheimer’s disease and reconstruction of relevant biological mechanisms. Sci Rep. 2018;8(1):11173. doi: 10.1038/s41598-018-29433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y, Bi Q, Wang X, Hu X, Li H, Li X, Ma T, Lu J, Chan P, Shu N, et al. Prediction of conversion from amnestic mild cognitive impairment to Alzheimer’s disease based on the brain structural connectome. Front Neurol. 2018;9:1178. doi: 10.3389/fneur.2018.01178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moscoso A, Silva-Rodriguez J, Aldrey JM, Cortes J, Fernandez-Ferreiro A, Gomez-Lado N, Ruibal A, Aguiar P, Alzheimer’s Disease Neuroimaging I Prediction of Alzheimer’s disease dementia with MRI beyond the short-term: implications for the design of predictive models. Neuroimage Clin. 2019;23:101837. doi: 10.1016/j.nicl.2019.101837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sorensen A, Blazhenets G, Rucker G, Schiller F, Meyer PT, Frings L, Alzheimer’s Disease Neuroimaging I Prognosis of conversion of mild cognitive impairment to Alzheimer’s dementia by voxel-wise Cox regression based on FDG PET data. Neuroimage Clin. 2019;21:101637. doi: 10.1016/j.nicl.2018.101637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swarbrick S, Wragg N, Ghosh S, Stolzing A. Systematic review of miRNA as biomarkers in Alzheimer’s disease. Mol Neurobiol. 2019;56(9):6156–6167. doi: 10.1007/s12035-019-1500-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bertram L, Tanzi RE. Alzheimer disease risk genes: 29 and counting. Nat Rev Neurol. 2019;15(4):191–192. doi: 10.1038/s41582-019-0158-4. [DOI] [PubMed] [Google Scholar]
- 53.Santo EE, Paik J. FOXO in neural cells and diseases of the nervous system. Curr Top Dev Biol. 2018;127:105–118. doi: 10.1016/bs.ctdb.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325(3):167–172. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- 55.Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, Hanger D, Mulot S, Marquardt B, Stabel S, et al. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol. 1994;4(12):1077–1086. doi: 10.1016/S0960-9822(00)00246-3. [DOI] [PubMed] [Google Scholar]
- 56.Gupta A, Dey CS. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol Biol Cell. 2012;23(19):3882–3898. doi: 10.1091/mbc.e12-05-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang D, Han G, Feng X, Sun J, Duan Y, Lei H. Concerted perturbation observed in a hub network in Alzheimer’s disease. Plos One. 2012;7(7):e40498. doi: 10.1371/journal.pone.0040498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng C, Xu R. The Alzheimer’s comorbidity phenome: mining from a large patient database and phenome-driven genetics prediction. JAMIA Open. 2019;2(1):131–138. doi: 10.1093/jamiaopen/ooy050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Table S1. SNP genotypes included in miR-eQTLs as potential biomarkers.
Additional file 2: Supplementary Table S2. Gene expression in 610 blood samples.
Additional file 3: Supplementary Table S3. Gene expression in 123 MCI blood samples.
Data Availability Statement
All microarray data (2562 miRNAs) and clinical information from this study are publicly available through the Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (NCBI) and accessible through GEO series accession number GSE150693 at http://www.ncbi.nlm.nih.gov/projects/geo/. The other datasets used or analyzed in the current study are available from the corresponding author on reasonable request.