

Abstract
Induced pluripotent stem cells (iPSCs) have become an essential research platform to study different human diseases once being discovered by Dr. Shinya Yamanaka in 2006. Another breakthrough in biomedical research is the application of CRISPR/Cas9 system for genome editing in mammalian cells. Although numerous studies have been done to develop methods for gene editing in iPSCs, the current approaches suffer from several limitations, including time and labor consuming, low editing efficiency, and potential off-target effects. In the current study, we report an electroporation-mediated plasmid CRISPR/Cas9 delivery approach for genome editing in iPSCs. With this approach, an edited iPSC cell line could be obtained within 2 weeks. In addition, the transit introducing of CRISPR/Cas9 machinery could minimize genomic integration of Cas9 gene, which avoided potential long-term side effects of Cas9 enzyme. We showed that CRISPR/Cas9-mediated genomic editing did not affect pluripotency and differentiation ability of iPSCs. With the quickly evolving of both iPSC and CRISPR/Cas9-mediated genome editing research fields, we believe that our method can significantly facilitate the application of genome editing in iPSCs research.
Keywords: iPSC, CRISPR, Cas 9, genome editing, cardiomyocytes
Introduction
In 2006, Yamanaka successfully created embryonic stem cell-like cells by introducing four critical factors, Oct3/4, Sox2, c-Myc, and KLF4, into somatic cells [1]. Based on the pluripotency of these cells, Dr. Yamanaka named them induced pluripotent stem cells (iPSCs). Indeed, by growing iPSCs in different culture conditions, they can differentiate into various cell types [2–4]. iPSCs can be obtained from many mature somatic cells [5], which avoids ethical concerns raised from using embryonic stem cells. In addition, by isolating somatic cells from human patients and converting them into iPSCs, physicians and researchers can establish patient-specific iPSC models, which can be used for potential high-throughput drug screening for personalized medicine. However, it is still challenging to study specific genetic mutations as causative factors of specific diseases.
To tackle this problem, a reliable, specific, and efficient method is needed for genome editing in patient-derived iPSCs to determine the disease-causing genetic alterations. To date, there are three major genome editing techniques: zinc finger nucleases (ZFNs) [6], transcription activator-like effector nuclease [7], and clustered regularly interspaced short palindromic repeats (CRISPR) [8]. Among them, the CRISPR/Cas9 system has been proven to be the most straightforward and most efficient approach [9]. Thus, it is currently the most commonly used gene editing approach.
However, the conventional protocol to introduce the CRISPR/Cas9 system into iPSCs requires the delivery of Cas9 and guide RNA via viral vectors, RNA, or purified Cas9 protein, which is time and labor consuming [10]. In the current study, we developed a simple and quick protocol to effectively perform genome editing in iPSCs. The whole procedure to obtain the edited cell line takes only 2 weeks (the entire process is summarized in Fig. 1), a result of this significantly simplified protocol. Thus, our method could significantly facilitate the application of CRISPR/Cas9-mediated genome editing in iPSCs.
Fig. 1. Summary of the key steps of CRISPR/Cas9-mediated genome editing in iPSCs.

Overall, it only takes 2 weeks to generate individual clones after CRISPR/Cas9-mediated gene knockout. After electroporation, cells were treated with puromycin for a single day to remove nontransfected cells, and then the surviving cells with GFP fluorescence were recovered in normal medium without puromycin. Because this method relies on the transient endonuclease activity of the gRNA/Cas9 complex to induce double-strand breakage at the genomic target site, this method is focused on the short-term expression of gRNA/Cas9 in puromycin medium followed by dilution of the intracellular plasmids in normal medium along with further propagation.
Materials and methods
Cell culture and maintenance of human pluripotent stem cells
Human iPSCs were purchased from Thermo Fisher (cat.# A18945, Waltham, MA, USA). Urine-derived human iPSCs were purchased from Cellapy Biological Technology Co., Ltd (Beijing, China). Both iPSC lines were cultured on Matrigel-coated plates (Corning REF. 356231, NY, USA) using StemFlex medium (Thermo Fisher cat.# A3349401, Waltham, MA, USA) under conditions of 37 °C, 95% air, and 5% CO2 in a humidified incubator.
Plasmid design
Guide RNA (gRNA) was designed using a website (crisprscan.org) to target exon 3 of UCHL-1, and an expression vector for the single-guide (sg) RNA was constructed with the following oligos: gRNA 2: 5′-CACCGGCAGGTGCTGTCCCGGCTGG-3′ (forward)′, 5′-AAACCCAGCCGGGACAGCACCTGCC-3′ (reverse). For the cloning of sgRNA, 13 μL of forward and reverse gRNA oligos at an initial concentration of 100 μM were mixed. The gRNA oligos were annealed at 60 °C for 1 min, followed by cooling to room temperature to make a double-stranded insertion. The PNK treatment reaction consisted of 26 μL double-stranded insertion, 1 μL PNK (NEB, cat.# M0201S, Ipswich, MA, USA) and 3 μL 10× T4 DNA ligase buffer (Thermo Scientific, cat.# B69, Waltham, MA, USA) and was incubated at 37 °C for 30 min followed by 65 °C for 20 min for heat inactivation. The insertion was confirmed and purified using a 3% agarose gel and a Zymo column (cat.# C1003–50, Irvine, CA, USA). The CRISPR plasmid PX459V2.0-eSpCas9(1.1) was from Addgene (cat.# 108292, Watertown, MA, USA). The restriction digestion reaction contained 1 μg plasmid, 1 μL BbsI (NEB, cat.# R0539S, Ipswich, MA, USA), 5 μL 10× reaction buffer (NEB, cat.# B7203S, Ipswich, MA, USA) and 43 μL ddH2O and was incubated at 37 °C for 2–3 h. Fifty microliters of vector was added to 150 μL ddH2O and 200 μL phenol (Sigma, cat.# P1037, St. Louis, MO, USA), followed by centrifugation at 15,000 rpm for 3 min at room temperature to remove the restriction enzymes. Isopropanol precipitation was then performed, where 200 μL vector, 20 μL 3 M sodium acetate, and 200 μL isopropanol were mixed followed by centrifugation at 15,000 rpm for 5 min at 4 °C. Vectors were cleaned using 70% ethanol and subsequently dissolved in 12.5 μL ddH2O. The dephosphorylation reaction of the vector consisted of 1.5 μL 10× Antarctic phosphatase reaction buffer (NEB, cat.# B0289S, Ipswich, MA, USA), 1 µL Antarctic phosphatase (NEB, cat.# M0289S, Ipswich, MA, USA) and 12.5 μL vectors and was incubated at 37 °C for 1 h followed by 70 °C for 10 min for heat inactivation. Ligation was performed using 1 μL 10× reaction buffer (NEB, cat.# B0202S, Ipswich, MA, USA), 1 μL 10× PEG, 0.5 μL T4 DNA ligase (NEB, cat.# M0202S, Ipswich, MA, USA), 1.5 μL vector, 1 μL ddH2O and 5 μL insert oligos, which was incubated at 16 °C for 1 h. Transformation of competent E. coli cells (strain JM109) with the ligation reaction was performed by the conventional calcium method. Three or more clones were picked for sequencing. The sequencing primer used was the human U6 promoter forward primer: 5′-GACTATCATATGCTTACCGT-3′. A QIAGEN Maxprep kit (REF. 12362, Hilden, Germany) was used to isolate plasmids from 200 mL of bacteria following the manufacturer’s instructions.
Electroporation and selection of iPSCs
Human iPSCs (4 × 105) were cultured in a 6-well plate for 3 days to reach a confluence of 80%. On day 3, 5 μg of the constructed CRISPR vector and 0.5 μg of the GFP plasmid (CRISPR:GFP vector = 10:1) were cotransfected into 1 × 106 human iPSCs using the Neon transfection system (Thermo Scientific, Waltham, MA, USA) with the conditions of 1200 V, 30 ms, and 1 pulse. Sixteen hours after electroporation the selection culture medium was replaced with puromycin (Life Technologies, REF# A11138–03, Carlsbad, CA, USA) at a final concentration of 10 μg/mL. After 24 h of selection, the medium was changed to normal medium, and the cells were cultured for 4 days. Sterile cloning disks (Scienceware, cat.# F37847–0001, Wayne, NJ, USA) were used to pick single iPSC clones into 48-well plates. IPSCs were cultured for 1 week and then transferred to 6-well plates for further analysis.
PCR amplification of the Cas9 sequence
Cas9 sequence PCR primers were designed to amplify a 489 bp product evenly flanking the CRISPR/Cas9 site. The primers were as follows: forward primer, 5′-GCTGCCTGAGAAGTACAAAGA-3′ and reverse primer, 5′-TCGTTGGGCAGGTTCTTATC-3′. Genomic DNA was extracted from a 6-well plate using a standard extraction kit (Sigma, cat.# G1N70, St. Louis, MO, USA). PCRs were performed using a TaKaRa Ex Taq kit (cat.# HRR001A, Kusatsu, Shiga, Japan). A total of 100 pg, 10 pg, or 1 pg constructed CRISPR vector and 100 ng human iPSC genomic DNA were used as templates in the PCR. Each 20 μL PCR reaction consisted of 2 μL Takara 10× Ex Taq Buffer (Mg2+ plus), 2 μL dNTP Mix (2.5 mM each), 0.5 μL Takara Ex Taq DNA polymerase (2.5 units), 1 μL of the forward primer and 1 μL reverse primer at an initial concentration of 10 μM, with respective amounts of DNA templates and ddH2O to reach a reaction volume of 20 μL. The PCR conditions were set at 94 °C for 2 min; 35 cycles of 94 °C for 30 s, 57 °C for 30 s and 72 °C for 30 s; and a final extension at 72 °C for 5 min. PCR products were confirmed using 1% agarose gel electrophoresis.
Western blot analysis
The presence of UCHL-1 protein was determined by Western blot. Total protein was extracted from cells using a protein lysis buffer containing protease inhibitors (Roche Life Science, Penzberg, Germany). The denatured proteins were separated by 12% SDS/polyacrylamide gels and were then transferred onto PVDF membranes (Merck Millipore, Berlin, Germany). Membranes were stained with Ponceau S solution (Sigma, cat.# P7170, St. Louis, MO, USA), and images were taken with a Bio-Rad imaging system. The membranes were incubated with the anti-UCHL-1 primary antibody (CST, cat.# 13179, Danvers, MA, USA) at 4 °C overnight. Membranes were washed with PBST and probed with a secondary antibody. UCHL-1 expression was detected by autoradiography using a chemiluminescent substrate (Thermo, cat.# 34080, MA, USA).
PCR primer design and Sanger sequencing
Genomic DNA PCR primers were designed to amplify a 572 bp or 535 bp product flanking the CRISPR/Cas9 cut site. Primers were as follows: primer pair 1: F1 5′-CGCTTTGTGCTGTGTCATTG-3′, R1 5′-TTCCCTGGATAACGGGATAGA-3′ and primer pair 2: F2 5′-CCTGGGCCCCTGCATTTAGC-3′, R2 5′-CCCCGAGGACTCAAGCTG-3′. Genomic DNA was extracted from a 6-well plate using a mammalian genomic DNA extraction kit (Sigma, cat.# G1N70). PCRs were performed using TaKaRa Ex Taq (cat.# HRR001A). Genomic DNA PCR products were ligated with T-vector (Promega cat.# A1360, Madison, WI, USA) following the manufacturer’s instructions. Transformations were performed to construct T-vectors. To detect all possible genome editing results, including heterozygous mutant alleles, more than 12 individual bacterial colonies from each single cell line were selected for mini-preparation using a kit (IBI Scientific, cat.# IB47102, Dubuque, IA, USA). Purified plasmids were submitted to The Ohio State University Sequencing Core for Sanger sequencing.
Immunofluorescence
Cells were washed using PBS and then fixed by using 4% paraformaldehyde for 15 min at room temperature. Fixed cells were permeabilized by 0.1% Triton X-100 for 20 min at room temperature followed by washing with PBS twice. Cells were blocked with 10% donkey or goat serum (the species where the secondary antibodies were raised) for 1 h and then incubated with primary antibodies diluted in PBS plus 0.05% Tween-20 (PBST) at 4 °C overnight. Subsequently cells were thoroughly washed with PBST three times and then incubated with secondary antibodies conjugated with fluorescence dyes for 1 h at room temperature, followed by three washes with PBST. Finally, the stained cells were mounted with DAPI Fluoromount-G (SouthernBiotech, cat.# 0100–20, Birmingham, AL, USA) and sealed on coverslips. Immunofluorescence slides were visualized by a Zeiss confocal microscope (Oberkochen, Germany) or Leica THUNDER model organism imaging system (Wetzlar, Germany). The antibodies used in this study included the Pluripotent Stem Cell Immunocytochemistry Kit (Invitrogen, REF. A25526, Carlsbad, CA, USA) and monoclonal anti-actin (α-Sarcomeric) antibody produced in mouse (Sigma, cat.# A2172, St. Louis, MO, USA).
Cardiac differentiation and calcium imaging
Cardiac differentiation was initiated by using a small-molecule–based protocol [11]. PSCs (passage 20–35) were cultured until 80%–90% confluence, and their medium was replaced with cardiac differentiation basal medium which is consisted of RPMI (cat.# 11875119, Waltham, MA, USA), B27 supplement minus insulin (cat.# A1895601, Waltham, MA, USA). For the first two days, the basal medium was supplemented with 9 μM CHIR-99021 (Selleckchem, cat.# S2924, Houston, TX, USA). The medium was then replaced with cardiac differentiation basal medium plus 5 μM IWR-1 (Sigma, cat.# I0161, St. Louis, MO, USA). Differentiated cells were maintained in cardiac differentiation basal medium for 2 days and substituted with cardiac proliferation medium which is consisted of RPMI (cat.# 11875119, Waltham, MA, USA), B27 supplement (cat.# 17504044, Waltham, MA, USA) for another 4 days. Beating iPSC differentiated cardiomyocytes (PSC-CMs) were typically observed on days 8–12 of differentiation. Beating PSC-CMs were then exposed to glucose-free RPMI medium (cat.# 11879020, Waltham, MA, USA) plus B27 supplement (cat.# 17504044, Waltham, MA, USA) for 4 days for metabolic purification. The purified PSC-CMs were then replated in Matrigel-coated plates in standard cardiac proliferation medium. Live cell calcium imaging was performed using the Cal-520-AM calcium indicator (AAT Bioquest, Sunnyvale, CA, USA). Briefly, the Cal-520 stock solution was added to the culture medium of beating cardiomyocytes to a final concentration of 5 μM. After 60 min of incubation, culture medium without Cal-520 was used to replace the one containing Cal-520 and incubated for 30 min. Live cell calcium imaging was then performed using the THUNDER Imager Model Organism (Leica, Wetzlar, Germany) with a GFP filter.
Statistical analysis
No statistical analysis is required for this study.
Results
Establishment of UCHL-1 knockout iPSC lines
To test our method, we used the CRISPR/Cas9 system to knock out ubiquitin C-terminal hydrolase L1 (UCHL-1) in human iPSCs, a gene that has been extensively studied in our laboratory [12]. First, we generated a plasmid that expresses Cas9 protein and sgRNA (Fig. 2). Then, the constructed CRISPR vectors were electroporated into commercial iPSC cells together with the GFP expression plasmid (GFP:CRISPR ratio was 1:10). The GFP plasmid was used to determine the transfection efficiency and to monitor the loss of the transgene during selection. Sixteen hours after electroporation, approximately 30% of iPSCs expressed GFP (Fig. 3a, left panels), indicating that these cells are positive for the CRISPR plasmid. Transfected iPSCs were then selected in a medium containing 10 μg/mL puromycin for 24 h to remove nontransfected cells (Fig. 3a, middle panels). More than 90% of iPSCs in the culture died after 24 h of treatment with puromycin. The surviving iPSCs were further cultured in normal medium for 4 days. As these iPSCs proliferated, each cell formed a colony that lost GFP fluorescence as a result of dilution and degradation of the intracellular GFP plasmid and, at the same time, the CRISPR plasmid (Fig. 3a, right panels. Note that the colony lost GFP fluorescence.). To remove the nontransfected cells efficiently, we performed a dose-dependent cytotoxicity analysis to obtain an optimal concentration of puromycin. We chose 10 μg/mL puromycin, by which >99% of normal iPSCs died in 24 h (data not shown). An important tip for our method is to treat transfected iPSCs for only 24 h to reduce the chance of internalization of the CRISPR/Cas9 plasmid in the genome. Due to the high efficiency of the CRISPR/Cas9 system, 40 h (16 h of transfection and 24 h of selection) is sufficient for genome editing, while CRISPR plasmids are rarely incorporated into genomic DNA in this time window (Supplementary Fig. S1). Thus, the risk of off-target effects caused by the long-term expression of Cas9 is greatly reduced.
Fig. 2. Design of plasmid containing eSpCas9(1.1) and guide RNA expression elements.

a Genomic structure of the uchl1 gene and selection of the gRNA targeting site. To assess our strategy, we performed gRNA/Cas9-mediated knockout of the uchl1 gene on the locus chr4:41,256,413–41,268,455 (GRCh38/hg38), size: 12,043 bases, plus-strand orientation (NCBI Ref: NC_000004.12). b Design of the sgRNA/Cas9 expression construct. To construct the UCHL-1 gRNA/Cas9 expression vector, we used the PX459v2.0 vector (Addgene cat.# 108292). A 5′-adjacent region of exon 3 was targeted and is shown as blue letters in the sequence. The guide RNA sequence is underlined, and the PAM sequence (3 bp NGG) is indicated in the black box. The annealed gRNA oligos were subcloned between two BbsI sites downstream of the U6 promoter.
Fig. 3. Early procedures for electroporation, puromycin selection, and analysis of UCHL-1 expression by immunoblotting.

a Representative bright-field (upper panels) and GFP (lower panels) microscope images at the indicated time points after electroporation. b Single colonies of edited cells were analyzed by immunoblotting for the expression of UCHL-1. Ponceau red staining was used to indicate equal loading.
After 4 days of culture, single colonies were picked up with sterile cloning disks and transferred to 48-well plates. After culturing for one more week, the propagated colonies were transferred to 6-well plates for further expansion or analysis. Thus, the whole procedure took <2 weeks (the key steps are summarized in Fig. 1). The efficiency of CRISPR/Cas9-mediated UCHL-1 knockout was determined by Western blotting analysis. We found that the target gene was completely knocked out in 10%–30% of colonies (Fig. 3b). Furthermore, ~20% of colonies showed significantly reduced expression of UCHL-1, indicating the generation of a heterozygous knockout mutant by CRISPR/Cas9-mediated gene editing.
Sanger sequencing results for individual iPSC colonies
To further confirm the genome editing sites of each iPSC cell line showing no UCHL-1 signal by WB (Fig. 3b), we amplified the genomic sequence containing the gRNA target site from each cell line and subcloned it into a T-vector for sequencing. To cover all possible heterozygous mutations, more than fifty individual bacterial colonies per selected iPSC cell clone were picked up, and plasmids were prepared for DNA sequencing. Sequencing analysis showed that various genomic editing events occurred in the selected cell lines, including indel mutations causing frameshift, loss of splicing site causing splice skipping of exon 3, or deletion of the whole exon 3, which would lead to the production of a truncated UCHL-1 protein (Fig. 4).
Fig. 4. Sanger sequencing results show various indel mutations in the selected UCHL-1 knockout clones.

Genomic PCR products from the selected iPSC clones were subcloned in a T-vector, and more than fifty individual colonies were analyzed for sequencing. Genotypes are listed as either homozygous, indicating that both alleles have the same mutation, or heterozygous, indicating that there is a mutation in at least one allele and the other allele contains a different mutation. Specific nucleotide sequences of insertions (underlined uppercases, green), deletions (red dash) and substitutions (lower cases, blue or black) around the editing site are indicated. The guide RNA sequence is underlined, and the PAM sequence is indicated in black boxes in the wt sequence. Additional marked sequences* show detected sequences that were not characterized for allelic features of homo or heterozygosity of the clones.
Interestingly, two clones (clones 1–7 and 1–11) were mutated identically on one allele by a 3-base insertion, causing the insertion of a proline (Pro, P) residue in the UCHL-1 primary sequence. Although this mutation does not cause either frameshift or early termination of translation, we could not detect UCHL-1 protein by repeated immunoblotting analysis (data not shown). Since these clones contain another mutant allele causing frameshift (clone 1–7) or loss of the splicing site of exon 3 (clone 1–11), one possible explanation could be that the alleles with a 3-base insertion may contain additional mutation(s) in a sequence that our current genomic PCR primers cannot detect.
CRISPR/Cas9-mediated genomic editing does not affect the pluripotency and differentiation ability of iPSCs
Next, we tested whether our procedures could affect the pluripotency and differentiation of selected iPSCs. As shown in Fig. 5a, UCHL-1 knockout iPSCs expressed normal levels of OCT4 and SSEA4, two iPSC biomarkers. As expected, we were also able to differentiate iPSC-UCHL-1 knockout cells into cardiomyocytes, which beat spontaneously with normal gap junctions for synchronized calcium signaling, based on further functional analysis (Fig. 5b and Supplemental Movies S1, S2).
Fig. 5. Genomic editing does not affect the pluripotency of iPSCs.
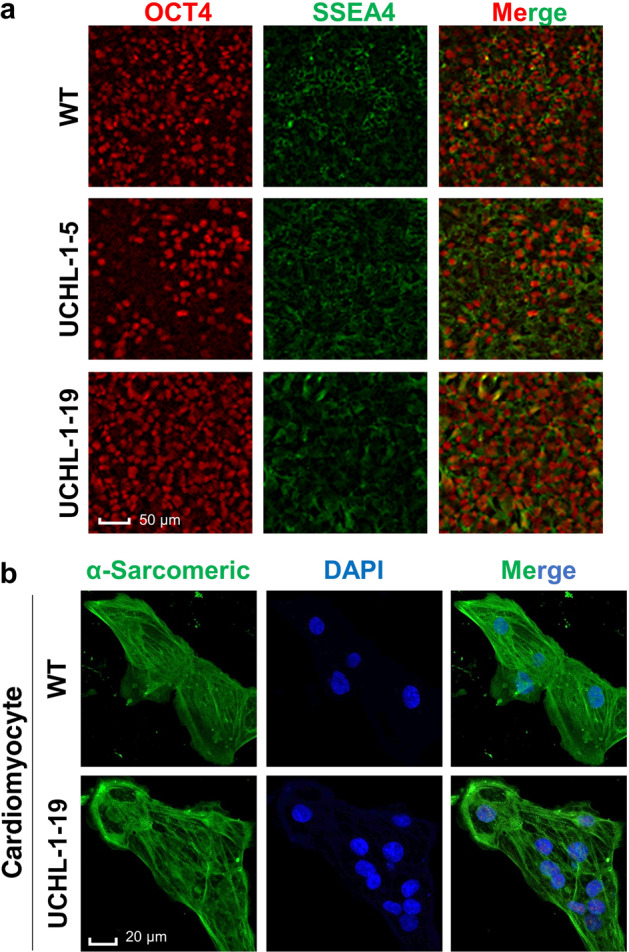
a Immunofluorescent staining of the stem cell biomarkers OCT4 (red) and SSEA4 (green) showed similar signal intensity in iPSC-WT and different colonies of UCHL-1 knockout cells. b Immunofluorescent staining of sarcomeric actinin (green) was performed on iPSC-WT and colony 19 of UCHL-1 knockout cells. DAPI was used to stain nuclei.
Discussion
In the current study, we report a novel method to perform genomic editing with the CRISPR/Cas9 system in iPSCs. This strategy has two features that provide an advantage over conventional cloning procedures. First, this method does not require a time-consuming, long-term selection step using antibiotics, which is required to obtain stable clones integrated with an exogenous gene of interest along with an antibiotic selection marker. We utilized a slightly high concentration and a relatively short period of treatment (24 h) for antibiotic selection to efficiently select sgRNA/Cas9-transfected cells and to achieve quick genomic editing without chromosome integration of the Cas9 gene. This significantly reduces the experimental procedure by at least 2 weeks to several months depending on the cell lines and experimental conditions used. Second, since this method does not require integration of the Cas9 gene in the chromosome of each selected clone, it is free from any potential risk caused by Cas9 off-target effects after the establishment of stable clones. With a single colony pickup, we found that >10% of single colonies are single clone knockout cells. Thus, we were able to generate a single clonal iPSC cell line within as short as 2 weeks.
In the current study, we demonstrated a new method of CRISPR/Cas9-mediated gene knockout in iPSCs. However, we anticipate that this method can be adopted in additional applications, including gene knockdown [13], activation [14], and prime editing [15]. In addition, the action of CRISPR/Cas9 in this method is transient, which will significantly reduce potential off-target effects, in contrast to the conventional methods of using lasting CRISPR/Cas9 action. Furthermore, we are using our new method to include a recently reported Cas9 enzyme that has high-fidelity editing [16], and we anticipate further reduced off-target effects. However, deep sequencing is required to determine the off-target effects of our method [17, 18], which will be the direction of future studies. Last but not least, we are also working on another CRISPR/Cas9 system that uses Cas9 nickase with a D10A mutation or a H840A mutation [19, 20] to reduce off-target effects by avoiding the introduction of double-strand breaks.
Overall, we have developed a novel method to perform CRISPR/Cas9-mediated genome editing in iPSCs, which takes <2 weeks to obtain a single clone of edited cells. Importantly, the Cas9 gene does not integrate into the genome of iPSCs, nor does it alter the pluripotency or differentiation of iPSCs. We believe this method could be useful for promoting CRISPR/Cas9-mediated genome editing in iPSCs to study the molecular causes of different human diseases.
Supplementary information
{kind=link}
Acknowledgements
This study was supported by internal fund from The Ohio State University.
Author contributions
HZ, JKK, NGD, SZW, and BCG designed research; BCG, KHC, PC, and XDP performed research; BCG, HZ, and JKK contributed new analytical tools and reagents; BCG, JKK, and HZ analyzed data; HZ, SZW, JKK and BCG wrote the paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Nian-guo Dong, Email: dongnianguo@hotmail.com.
Jae-Kyun Ko, Email: Jae-Kyun.Ko@osumc.edu.
Hua Zhu, Email: Hua.Zhu@osumc.edu.
Supplementary information
The online version of this article (10.1038/s41401-020-0452-0) contains supplementary material, which is available to authorized users.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci. 2017;20:753–9. doi: 10.1038/nn.4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ullah I, Abu-Dawud R, Busch JF, Rabien A, Erguen B, Fischer I. VEGF-supplemented extracellular matrix is sufficient to induce endothelial differentiation of human iPSC. Biomaterials. 2019;216:119283. doi: 10.1016/j.biomaterials.2019.119283. [DOI] [PubMed] [Google Scholar]
- 4.Ueda T, Kaneko S. In vitro differentiation of T Cell: from CAR-modified T-iPSC. Methods Mol Biol. 2019;2048:85–91. doi: 10.1007/978-1-4939-9728-2_10. [DOI] [PubMed] [Google Scholar]
- 5.Varli HS, Alkan F, Demirbilek M, Turkoglu N. A virus-free vector for the transfection of somatic cells to obtain IPSC. J Nanopart Res. 2019;21:237. [Google Scholar]
- 6.Liu Q, Segal DJ, Ghiara JB, Barbas CF. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc Natl Acad Sci USA. 1997;94:5525–30. doi: 10.1073/pnas.94.11.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng GP, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7:171–92. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brookhouser N, Raman S, Potts C, Brafman DA. May i cut in? Gene editing approaches in human induced pluripotent stem cells. Cells. 2017;6:5. 10.3390/cells6010005. [DOI] [PMC free article] [PubMed]
- 10.Wang G, Yang LH, Grishin D, Rios X, Ye LY, Hu Y. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat Protoc. 2017;12:88–103. doi: 10.1038/nprot.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao MT, Chen HD, Liu Q, Shao NY, Sayed N, Wo HT, et al. Molecular and functional resemblance of differentiated cells derived from isogenic human iPSCs and SCNT-derived ESCs. Proc Natl Acad Sci USA. 2017;114:E11111–E20. doi: 10.1073/pnas.1708991114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo YH, He JF, Yang CL, Orange M, Ren XC, Blair N, et al. UCH-L1 promotes invasion of breast cancer cells through activating Akt signaling pathway. J Cell Biochem. 2018;119:691–700. doi: 10.1002/jcb.26232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glass Z, Lee M, Li YM, Xu QB. Engineering the delivery system for CRISPR-based genome editing. Trends Biotechnol. 2018;36:173–85. doi: 10.1016/j.tibtech.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoger E, Carroll KJ, McAnally J, Tan W, Liaw N, Iyer LM, et al. CRISPR-based gene activation for transcriptional reprograming of mammalian cardiomyocytes. Circ Res. 2019;125:A372. [Google Scholar]
- 15.Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–8. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stajic E, Kielkowska A, Murovec J, Bohanec B. Deep sequencing analysis of CRISPR/Cas9 induced mutations by two delivery methods in target model genes and the CENH3 region of red cabbage (Brassica oleracea var. capitata f. rubra). Plant Cell Tiss Org. 2019;139:227–35. [Google Scholar]
- 18.Slesarev A, Viswanathan L, Tang YT, Borgschulte T, Achtien K, Razafsky D. CRISPR/CAS9 targeted CAPTURE of mammalian genomic regions for characterization by NGS. Sci Rep. 2019;9:587. doi: 10.1038/s41598-019-39667-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao B, Yin SG, Hu Y, Sun MX, Wei JQ, Huang ZH, et al. Epigenetic editing by CRISPR/dCas9 in Plasmodium falciparum. Proc Natl Acad Sci USA. 2019;116:255–60. doi: 10.1073/pnas.1813542116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.