

Abstract
Key points
In vascular smooth muscle cells (VSMCs), activation of Ca2+‐permeable store‐operated channels (SOCs) composed of canonical transient receptor potential channel 1 (TRPC1) subunits mediates Ca2+ entry pathways that regulate contraction, proliferation and migration, which are processes associated with vascular disease.
Activation of TRPC1‐based SOCs requires protein kinase C (PKC) activity, which is proposed to phosphorylate TRPC1 proteins to promote channel opening by phosphatidylinositol 4,5‐bisphosphate (PIP2). We investigated the identity of the PKC isoform involved in activating TRPC1‐based SOCs in rat mesenteric artery VSMCs.
TRPC1‐based SOCs were reduced by PKCδ inhibitors and knockdown of PKCδ expression. Store depletion induced interactions between TRPC1 and PKCδ and PKCδ‐dependent phosphorylation of TRPC1. Furthermore, generation of store‐operated interactions between PIP2 and TRPC1 and activation of TRPC1‐based SOCs by PIP2 required PKCδ.
These findings reveal that PKCδ activity has an obligatory role in activating TRPC1‐based SOCs, through regulating PIP2‐mediated channel opening.
Abstract
In vascular smooth muscle cells (VMSCs), stimulation of Ca2+‐permeable canonical transient receptor potential channel 1 (TRPC1)‐based store‐operated channels (SOCs) mediates Ca2+ entry pathways that regulate cell contraction, proliferation and migration, which are processes associated with vascular disease. It is therefore important to understand how TRPC1‐based SOCs are activated. Stimulation of TRPC1‐based SOCs requires protein kinase C (PKC) activity, with store‐operated PKC‐dependent phosphorylation of TRPC1 essential for channel opening by phosphatidylinositol 4,5‐bisphosphate (PIP2). Experimental protocols used to activate TRPC1‐based SOCs suggest that the PKC isoform involved requires diacylglycerol (DAG) but is Ca2+‐insensitive, which are characteristics of the novel group of PKC isoforms (δ, ε, η, θ). Hence, the present study examined whether a novel PKC isoform(s) is involved in activating TRPC1‐based SOCs in contractile rat mesenteric artery VSMCs. Store‐operated whole‐cell cation currents were blocked by Pico145, a highly selective and potent TRPC1/4/5 channel blocker and T1E3, a TRPC1 blocking antibody. PKCδ was expressed in VSMCs, and selective PKCδ inhibitory peptides and knockdown of PKCδ expression with morpholinos oligomers inhibited TRPC1‐based SOCs. TRPC1 and PKCδ interactions and phosphorylation of TRPC1 induced by store depletion were both reduced by pharmacological inhibition and PKCδ knockdown. In addition, store‐operated PIP2 and TRPC1 interactions were blocked by PKCδ inhibition, and PKCδ was required for PIP2‐mediated activation of TRPC1 currents. These results identify the involvement of PKCδ in stimulation of TRPC1‐based SOCs and highlight that store‐operated PKCδ activity is obligatory for channel opening by PIP2, the probable activating ligand.
Keywords: PKC, PIP, store‐operated channels2, TRPC1, vascular smooth muscle
Key points
In vascular smooth muscle cells (VSMCs), activation of Ca2+‐permeable store‐operated channels (SOCs) composed of canonical transient receptor potential channel 1 (TRPC1) subunits mediates Ca2+ entry pathways that regulate contraction, proliferation and migration, which are processes associated with vascular disease.
Activation of TRPC1‐based SOCs requires protein kinase C (PKC) activity, which is proposed to phosphorylate TRPC1 proteins to promote channel opening by phosphatidylinositol 4,5‐bisphosphate (PIP2). We investigated the identity of the PKC isoform involved in activating TRPC1‐based SOCs in rat mesenteric artery VSMCs.
TRPC1‐based SOCs were reduced by PKCδ inhibitors and knockdown of PKCδ expression. Store depletion induced interactions between TRPC1 and PKCδ and PKCδ‐dependent phosphorylation of TRPC1. Furthermore, generation of store‐operated interactions between PIP2 and TRPC1 and activation of TRPC1‐based SOCs by PIP2 required PKCδ.
These findings reveal that PKCδ activity has an obligatory role in activating TRPC1‐based SOCs, through regulating PIP2‐mediated channel opening.
Introduction
Store‐operated channels (SOCs) are Ca2+‐permeable plasmalemmal ion channels activated by depletion of cytosolic endo/sarcoplasmic (ER/SR) Ca2+ stores (Baudel et al. 2020). In vascular smooth muscle cells (VSMCs), SOCs are stimulated by vasoconstrictors that activate the Gαq protein‐coupled receptor signalling pathway leading to phospholipase C (PLC) activity, phosphatidylinositol 4,5‐bisphosphate (PIP2) hydrolysis, inositol 1,4,5‐trisphosphate (IP3) generation and IP3‐mediated depletion of SR Ca2+ stores. As such SOCs induce Ca2+ entry pathways that regulate vasoconstrictor‐mediated contraction, proliferation and migration, and are considered drug targets for treatment of vascular diseases such as hypertension and atherosclerosis.
There is substantial evidence that SOCs represent a diverse family of ion channels with differing molecular composition, activation mechanisms and functions (Albert & Large, 2003; Albert et al. 2007; 2009; Cheng et al. 2013; Prakriya & Lewis, 2015; Ong et al. 2015). In native contractile VSMCs, SOCs have relatively low Ca2+ permeability, linear current–voltage (I–V) rectification properties, a single channel conductance of 2–3 pS and are composed of canonical transient receptor potential (TRPC) channels (Trepakova et al. 2001; Xu & Beech, 2001; Albert & Large, 2002 a; Bergdahl et al. 2005; Liu et al. 2005 a, b ; Xu et al. 2005 a; Albert et al. 2006; Saleh et al. 2006; 2008; 2009 a; Shi et al. 2012a, 2014, 2016, 2017a a). It is proposed that molecular composition involves a heteromeric TRPC1/C5 template with TRPC1 being the critical subunit for conferring activation by store depletion, hence the term TRPC1‐based SOCs (Xu & Beech, 2001; Xu et al. 2005 a; Saleh et al. 2008; Shi et al. 2012 a). In comparison, synthetic VSMCs that are involved in cell proliferation, migration and growth and are associated with more pathological functions exhibit TRPC1‐based SOCs and also SOCs with properties similar to Orai1‐based calcium release‐activated channels (CRACs) such as high Ca2+ permeability, pronounced inward rectification, single channel conductance in the fS range and composition of Orai1 proteins (Berra‐Romani et al. 2008; Beech, 2012; Trebak, 2012, Prakriya & Lewis, 2015). It is important to highlight that, in the present study, we examined activation mechanisms of TRPC1‐based SOCs in freshly isolated and primary cultured single VSMCs and tissue lysates that have a native contractile phenotype (Shi et al. 2016, 2017a a) and probably do not involve Orai1 proteins (Shi et al. 2017 b). These TRPC1‐based SOCs are probably important in regulating contractility and switching of VSMCs from contractile to synthetic phenotypes (Berra‐Romani et al. 2008; Matchkov et al. 2012).
How store depletion stimulates TRPC‐based SOCs is controversial, especially compared to Orai1‐based CRACs for which it is well‐established that store depletion induces the ER/SR store Ca2+ sensor stromal interaction molecule 1 (STIM1) to oligomerize and translocate to the plasma membrane where it interacts with Orai1 to induce channel assembly and gating (Prakriya & Lewis, 2015). However, there is growing evidence that store depletion also activates TRPC‐based SOCs through STIM1‐mediated processes, potentially involving direct interactions between TRPC and Orai1 proteins (Liao et al. 2014), activation of Orai1‐based CRACs as a prerequisite for TRPC1 channel opening (Ambudkar et al. 2017), and direct interactions between TRPC and STIM1 proteins (Worley et al. 2007; Yuan et al. 2009; Lee et al. 2014; Asanov et al. 2015). In native contractile VSMCs, our recent work indicates that store depletion stimulates TRPC1‐based SOCs through a novel STIM1‐mediated Gαq/PLCβ1 pathway, which probably induces channel opening by regulating interactions between protein kinase C and PIP2 (Shi et al. 2016, 2017a a).
There is considerable evidence that protein kinase C (PKC) activity is critical for activation of TRPC1‐based SOCs in native contractile VSMCs (Large et al. 2009; Albert, 2011). PKC inhibitors prevent activation of TRPC1‐based SOCs and reduce store‐operated phosphorylation of TRPC1, and PKC activators stimulate TRPC1‐based SOCs (Saleh et al. 2008, 2009a a; Shi et al. 2012a, 2016). Activation of TRPC1‐based SOCs and phosphorylation of TRPC1 by physiological vasoconstrictors are also reduced by PKC inhibitors (Albert & Large, 2002 b; Saleh et al. 2006; 2009 b; Shi et al. 2012b, 2014, 2016). It is proposed that PKC activity stimulates opening of TRPC1‐based SOCs through phosphorylation of TRPC1 proteins to promote PIP2 binding that acts as the activating ligand (Shi et al. 2014, 2016, 2017 a). As such, activation of TRPC1‐based SOCs by store depletion or PKC activators is inhibited by anti‐PIP2 antibodies and pre‐treatment with PI4‐kinase inhibitors which deplete PIP2 levels (Saleh et al. 2009 a; Shi et al. 2012 b). In addition, activation of TRPC1‐based SOCs by the water soluble PIP2 analogue diC8‐PIP2 is prevented by PKC inhibitors (Saleh et al. 2009 a; Shi et al. 2012 b). These results indicate that interactions between PKC activity and PIP2 have obligatory roles in activation of TRPC1‐based SOCs; PKC cannot activate TRPC1‐based SOCs without PIP2 and vice versa (Baudel et al. 2020). The present study further examines the importance of PKC activity in activating TRPC1‐based SOCs by investigating the PKC isoform(s) involved.
The PKC family comprises of at least 11 homologous serine/threonine kinases divided into three groups according to their basic structure and activation requirements: conventional PKC isoforms (α, βI, βII and γ) require both Ca2+ and diacylglyerol (DAG), novel PKC isoforms (δ, ε, η and θ) require DAG but are Ca2+‐insensitive, and atypical PKC isoforms (ζ, ι and λ) are activated by lipid mediators such as phosphatidylserine and do not require Ca2+ or DAG (Salamanca & Khalil, 2005; Ringvold & Khalil, 2017).
Many of these PKC isoforms are expressed in VSMCs and are proposed to regulate several physiological and pathological processes, including those reported to involve a role for TRPC1‐based SOCs (Salamanca & Khalil, 2005; Ding et al. 2011; Fan et al. 2014; Ringvold & Khalil, 2017). In native contractile VSMCs, stimulation of TRPC1‐based SOCs and PKC‐dependent phosphorylation of TRPC1 by store‐depletion requires PLCβ1 activity (Shi et al. 2016), DAG activates TRPC1‐based SOCs through a PKC‐dependent mechanism (Saleh et al. 2006; 2008; Large et al. 2009; Albert, 2011), and TRPC1‐based SOCs are activated by store depleting agents that probably increase (e.g. the SR Ca2+‐ATPase inhibitor cyclopiazonic acid), decrease (e.g. the cell‐impermeable and ‐permeable high affinity Ca2+ chelators BAPTA and BAPTA‐AM) or produce little change in [Ca2+]i (e.g. the cell‐permeable low affinity Ca2+ chelator TPEN). These results suggest that the PKC isoform involved requires DAG, although it is probably Ca2+‐insensitive; these are characteristics of the novel group of PKC isoforms. These ideas on the identity of the PKC isoform involved form the basis of the present work, and our findings indicate that the PKCδ novel isoform is the probable candidate involved in activation of TRPC1‐based SOCs in native contractile rat mesenteric artery VSMCs.
Methods
Ethical approval
All animal procedures were carried out in accordance with guidelines laid down by St George's, University of London Animal Welfare Committee, and conform with the principles and regulations described by the Service Project Licence: 70/8512, and also to the principles and regulations of The Journal of Physiology as described by Grundy (2015). Male Wistar rats (8–12 weeks old) were used for the purpose of the present study. Rats were supplied from Charles River (Margate, UK) and housed and maintained in standard sized plastic cages at the Biological Research Facility at St George's, University of London, under a 12:12 h light/dark photocycle, at 18–20 °C and ∼50% relative humidity, with water and laboratory rodent diet (Specialist Dietary Services, UK) available ad libitum. Animals were culled by cervical dislocation in accordance with the UK Animals Scientific Procedures Act of 1986 and as revised by European Directive 2010/63/EU.
Cell isolation and tissue lysates
Mesenteric arteries were dissected and cleaned of adherent fat in physiological salt solution containing (mmol L−1): 126 NaCl, 6 KCl, 10 glucose, 11 Hepes, 1.2 mgCl2, and 1.5 CaCl2 with pH adjusted to 7.2 with 10 mol L−1 NaOH. Single VSMCs were enzymatically dispersed and tissue lysates prepared as described previously (Shi et al. 2016, 2017a a).
PKCδ knockdown
Knockdown of PKCδ was performed by transfection of vessel segments with morpholino anti‐sense oligonucleotides as described previously (Stott et al. 2018). PKCδ morpholino oligomers and a control oligomer containing five mismatched nucleotides (10 μm; Genetools, Philomath, OR, USA) were mixed with lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) in Opti‐MEM and left at room temperature for 2 h. Mesenteric artery segments were cultured with this mix in Dulbecco's modified Eagle's medium F‐12 with 1% penicillin/streptomycin at 37°C for 48 h. Vessel segments were then enzymatically dispersed into single VSMCs or used as tissue lysates as required.
Electrophysiology
Whole‐cell and cell‐attached patch clamp techniques were used to record TRPC1‐based SOCs with an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA) at room temperature (20–23°C) using bath and patch pipette solutions, data analysis and experimental protocols as described previously (Shi et al. 2017 a). In experiments investigating the effects of phorbol 12,13‐dibutyrate, a phorbol ester and PKC activator, and diC8‐PIP2 on whole‐cell cation currents, 750 ms duration voltage ramps from +100 mV to −150 mV were applied every 30 s from a holding potential of 0 mV. The patch pipette solution contained (mm): 126 CsCl, 1.2 MgCl2, 10 nHepes, 11 glucose, 10 BAPTA, 4.8 CaCl2, ∼100 nm free internal Ca2+ concentration as calculated using MaxChelator (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator), 1 Na2ATP, 0.2. NaGTP, pH 7.2 with Tris. The external solution contained (mm): 126 NaCl, 1,5 CaCl2, 10 Hepes, 11 glucose, 0.1. DIDS, 0.1 niflumic acid and 0.005 nicardipine, pH 7.2 with NaOH.
Immunoprecipitation and western blotting
Freshly isolated or cultured vessel segments were prepared for immunoprecipitation, one‐dimensional protein gel electrophoresis and immunoblotting as described previously (Shi et al. 2017 a). The primary antibodies used were: mouse anti‐PKCε (dilution 1:500; sc‐1681; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti‐PKCθ (dilution 1:500; sc‐1680; Santa Cruz Biotechnology), mouse anti‐PKCη (dilution 1:500; sc‐136 036; Santa Cruz Biotechnology) and rabbit anti‐PKCδ (dilution 1:1000; ab182126; Abcam, Cambridge, MA, USA). Rabbit anti‐TRPC1 antibody (1 μg mL−1) was generated by GenScript (Piscataway, NJ, USA) using peptide sequences from a previously characterized putative extracellular region (Xu & Beech, 2001). Visualization were performed using anti‐rabbit and anti‐mouse secondary antibodies conjugated to IRDye 800RD or IRDye 680CW (dilution 1:10 000; Li‐Cor Biosciences, Cambridge, UK) as appropriate and the Odyssey Infrared Imaging System (Li‐Cor Biosciences). Protein band intensities were measured using Image Studio software (Li‐Cor Biosciences) and normalized to smooth muscle actin (dilution 1:2000; #ab5694; Abcam) when quantified.
Immunofluorescence
Single VSMCs were fixed with 4% (w/v) paraformaldehyde for 15 min, cells were treated with 0.1 mol L−1 glycine for 5 min and permeabilized with phosphate‐buffered saline (PBS) containing 0.1% (v/v) Triton X‐100 (PBS‐T) for 15 min at room temperature. Cells were then incubated with PBS‐T containing 1% (w/v) BSA for 1 h at room temperature to block non‐specific binding of antibodies. Immunostaining was performed with mouse anti‐PKCε (dilution 1:500; sc‐1681; Santa Cruz Biotechnology), mouse anti‐PKCθ (dilution 1:200; sc‐1680; Santa Cruz Biotechnology), rabbit anti‐PKCη (dilution 1: 200; ab4134; Abcam), rabbit anti‐PKCδ (dilution 1:1000; ab182126; Abcam) overnight at 4°C. Cells were then washed and incubated with a 488 fluorophore‐conjugated donkey anti‐goat secondary antibody (dilution 1:1000; A‐11 055; Alexa Fluor; Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature. Unbound secondary antibodies were removed by washing with PBS, and nuclei were labelled with 4′,6‐diamidino‐2‐phenylindole mounting medium (Sigma, Poole, UK). Control experiments were performed by omitting either primary or secondary antibodies. Cells were imaged using an A1R confocal microscope (Nikon, Tokyo, Japan). PIP2 immunofluorescence experiments were performed in the presence of saponin with rabbit anti‐PIP2 (sc‐53 412, 1:50; Santa Cruz Biotechnology) as described previously (Edimo et al. 2016).
Proximity ligation assay
Interactions between different proteins were investigated with a Duolink® in situ detection kit (Sigma) as described previously (Shi et al. 2017 a). Single VSMCs were fixed and permeabilized as per immunofluorescence and blocking was performed with Duolink blocking buffer for 1 h at 37°C. Cells were then incubated with appropriate antibodies overnight at 4°C as per immunofluorescence experiments plus mouse anti‐P‐Ser (dilution 1:100; sc‐16B4; Santa Cruz Biotechnology), mouse anti‐P‐Thr (dilution 1:100; sc‐5267; Santa Cruz Biotechnology), mouse anti‐TRPC1 (dilution 1:100; sc‐133 076; Santa Cruz Biotechnology) and rabbit anti‐TRPC5 (T5E3), which was generated by GenScript using peptide sequences from a previously characterized putative extracellular region (1 μg mL−1) ((Xu & Beech, 2001; Xu et al. 2005a, 2005b a,b). The rest of the protocol was conducted in accordance with the manufacturer's instructions. Fluorescent puncta were visualized with an A1R confocal microscope and images were analysed with ImageJ Fiji (https://fiji.sc). The mean number of puncta per cell was calculated by counting the number of particles across a z‐stack of the cell.
Reagents
Pico145 was a generous gift provided by Robin S Bon and David J Beech (University of Leeds, Leeds, UK). The cell permeable PKCδ inhibitor, δV1‐TAT peptide (RRRQRRKKRGY‐SFNSYELGSL) was synthesized by Mimotopes (Wirral, UK). PKCδ non‐permeable δPKC8‐17 peptide inhibitor (SFNSYELGSL) was purchased from AnaSpec (AnaSpec, EGT Corporate Headquarters, Fremont, CA, USA). PKCε peptide inhibitor was purchased from Santa Cruz Biotechnology. All other drugs were purchased from Sigma or Tocris (Abingdon, UK). Agents were dissolved in distilled H2O or 0.1% DMSO. DMSO alone had no effect on whole‐cell or single channel currents.
Statistical analysis
All statistical analysis was performed using Prism, version 8 (GraphPad Software Inc., San Diego, CA, USA). Data were calculated as the mean ± SD, with n indicating the number of data points. Data points were generated from at least three different isolated VSMCs or tissue lysate preparations. To compare between two or more I–V relationships, two‐way ANOVA with Tukey's multiple comparisons test was used, and differences in means at −80 mV are reported. To compare between two data sets, paired or unpaired t tests were used. P < 0.05 was considered statistically significant.
Results
PKC‐dependent store‐operated currents are composed of TRPC1 subunits in rat mesenteric artery VSMCs
In our first series of experiments, we confirmed that PKC‐dependent TRPC1‐based SOCs are functionally expressed in native contractile VSMCs from freshly isolated rat mesenteric arteries using a highly selective and potent TRPC1/C4/C5 channel blocker Pico145 (Rubaiy et al. 2017), an externally‐acting TRPC1 blocking antibody T1E3 (Xu & Beech, 2001, Xu et al. 2005 b) and the pan‐PKC isoform selective inhibitor GF109203X. Figure 1A shows that passive depletion of internal Ca2+ stores, following cell dialysis with a patch pipette solution containing high concentrations of BAPTA and no added Ca2+, induced whole‐cell cation currents that had a relatively linear I–V relationship, an E rev between 0 mV and +20 mV, with a mean control amplitude of −1.15 ± 0.62 pA pF–1 at −80 mV which increased to a mean peak amplitude of −2.74 ± 0.88 pA pF–1 (n = 6, p = 0.0015) and reduced to −0.89 ± 0.35 pA pF–1 by bath application of Pico145 (n = 6, p = 0.0002, two‐way ANOVA, Tukey's multiple comparisons test). Figure 1B and C show that bath application of T1E3 and G109203X also inhibited store‐operated whole‐cell currents, with mean peak amplitudes at −80 mV reduced respectively from −2.96 ± 0.92 pA pF–1 to −0.84 ± 0.43 pA pF–1 (n = 6, p = 0.0001) and from −2.14 ± 0.92 pA pF–1 to −0.91 ± 0.45 pA pF–1 (n = 7, p = 0.0051, two‐way ANOVA, Tukey's multiple comparisons test).
Figure 1. TRPC1 compose PKC‐dependent SOCs in native contractile VSMCs.
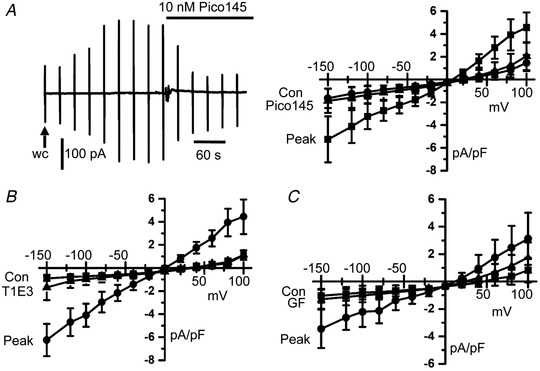
A, representative trace and mean I–V relationships showing that the development of store‐operated whole‐cell currents from control to peak levels in freshly isolated rat mesenteric artery VSMCs following obtaining whole‐cell configuration (wc) was inhibited by bath application of Pico145. Vertical deflections represent currents evoked by voltage ramps from +100 mV to −150 mV (750 ms duration) every 30 s from a holding potential of 0 mV. B and C, Mean data showing that bath applications of T1E3 (1 μg mL−1) and GF109203X (3 μm) inhibited store‐operated whole‐cell currents.
There is considerable evidence that heteromeric TRPC1/C5 molecular structures compose TRPC1‐based SOCs in VSMCs from different rabbit, mouse and human vascular beds (see Introduction). In proximity ligation assay (PLA) studies, Figure 2 shows that fluorescent puncta with a mean number per cell of 51.5 ± 12.6 (n = 14) produced using anti‐TRPC1 and anti‐TRPC5 antibodies were present at (or close to) the plasma membrane in freshly isolated rat mesenteric artery VSMCs.
Figure 2. Expression of TRPC1‐TRPC5 interactions in native contractile VSMCs.
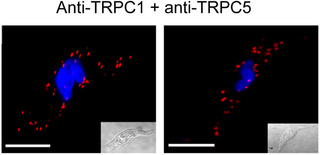
Representative PLA images from two different freshly isolated rat mesenteric artery VSMCs showing the association between TRPC1 and TRPC5. These images, and other PLA images, represent puncta observed across a z‐stack of each cell. Scale bar = 10 μm.
These findings clearly confirm that PKC‐dependent TRPC1‐based SOCs are functionally expressed in native contractile rat mesenteric artery VSMCs, similar to previous studies in VSMCs from different rabbit, mouse and human vascular preparations (see Introduction).
PKCδ is the dominant novel PKC isoform in VSMCs
The PKC isoform involved in activating TRPC1‐based SOCs in VSMCs requires DAG but is Ca2+ insensitive and is probably a member of the novel PKC isoform family (see Introduction). We therefore investigated the expression of novel PKC isoforms in tissue lysates from freshly isolated rat mesenteric arteries. We used brain lysates as positive controls where novel PKC isoforms have been previously identified (Fleegal et al. 2005; Popp et al. 2006; Callender & Newton, 2017; Wang et al. 2019; Yang et al. 2019).
Figure 3 a shows PKCδ expression with relatively low levels or little expression of PKCε, PKCη and PKCθ were found in tissue lysates from rat mesenteric arteries. Using the same anti‐PKC novel isoform antibodies, expression of PKCδ, PKCε, PKCη and PKCθ was present in brain lysates. In addition, Figure 3 b shows that immunocytochemical studies revealed PKCδ staining at (or close to) the plasma membrane of VSMCs with little staining recorded for PKCε, PKCη and PKCθ isoforms.
Figure 3. Expression of novel PKC isoforms in native contractile VSMCs.
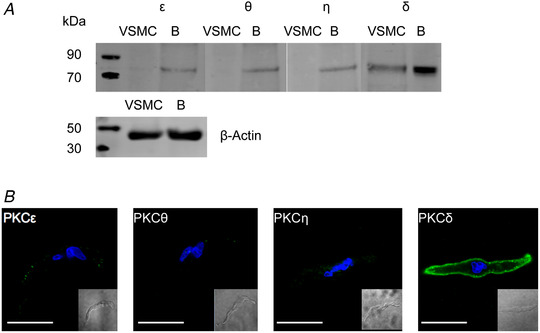
A, representative western blots showing expression of the novel PKC isoforms PKCε, PKCθ, PKCη and PKCδ in rat mesenteric artery VSMCs and brain (B) protein lysates. B, representative immunocytochemical images of rat mesenteric VSMCs immunolabelled with the same anti‐novel PKC isoform antibodies used in (A). Scale bar = 10 μm.
PKCδ activity is essential for activation of TRPC1‐based SOCs
In our next series of experiments, we examined whether pharmacological inhibition of PKCδ and knockdown of PKCδ expression using morpholino technology resulted in an anticipated decrease in activation of TRPC1‐based SOCs. We focused on the PKCδ isoform based on the expression studies detailed above and previous data on the expression and function of PKCδ in vascular smooth muscle (Salamanca & Khalil, 2005; Ringvold & Khalil, 2017).
Figure 4A reveals that store‐operated whole‐cell TRPC1‐based currents were inhibited by bath application of the cell‐permeable PKCδ inhibitor δV1‐TAT, with mean peak amplitude reduced from −6.18 ± 2.34 pA pF–1 to −1.26 ± 0.57 pA pF–1 at −80 mV (n = 6, p = 0.0001, two‐way ANOVA, Tukey's multiple comparisons test). Figure 4B also shows that following inclusion of the cell‐impermeable PKCδ peptide inhibitor δPKC8‐17 in the patch pipette solution store depletion induced a small whole‐cell TRPC1‐based current with mean control amplitude increasing from −0.97 ± 0.27 pA pF–1 to a mean peak amplitude of −2.01 ± 1.1 pA pF–1 at −80 mV (n = 6, p = 0.0902, two‐way ANOVA, Tukey's multiple comparisons test).
Figure 4. Pharmacological PKCδ inhibitors reduce TRPC1‐based SOCs.

A, representative trace and mean I–V relationship showing that the peak amplitude of store‐operated whole‐cell TRPC1 currents in freshly isolated rat mesenteric artery VSMCs was reduced by bath application of δV1‐TAT. B, a small store‐operated whole‐cell TRPC1 current was induced following inclusion of δPKC8‐17 peptide inhibitor in the patch pipette solution. C, inclusion of εPKC peptide inhibitor in the patch pipette appeared to have little effect on the development of store‐operated whole‐cell TRPC1 currents, which were subsequently inhibited by bath application of δV1‐TAT.
Because expression of PKCε was also observed in VSMCs (Fig. 3) and other studies have reported the expression of PKCε in the vasculature (Salamanca & Khalil, 2005; Ringvold & Khalil, 2017), we investigated the potential contribution of this PKC isoform in activating store‐operated TRPC1‐based SOCs. Figure 4C shows that, following inclusion of a PKCε‐specific peptide inhibitor in the patch pipette solution, store depletion activated whole‐cell TRPC1 currents with a mean control amplitude increasing from −0.84 ± 0.21 pA pF–1 to −3.39 ± 1.42 pA pF–1 at −80 mV (n = 5, p = 0.0006), which was subsequently inhibited by bath application of δV1‐TAT to −0.99 ± 0.56 pA pF–1 (n = 5, p = 0.0012, two‐way ANOVA, Tukey's multiple comparisons test).
Figure 5A shows that incubation of freshly isolated rat mesenteric artery segments with anti‐sense morpholino oligomers against PKCδ for 48 h significantly decreased protein expression of PKCδ by ∼50% compared to a mismatched oligomer (n = 3, p = 0.0202, unpaired t test). Figure 5B shows that, in VSMCs isolated from vessel segments expressing mismatched (scrambled) oligomers, store depletion induced whole‐cell TRPC1 currents with relative linear rectification and a mean control amplitude at −80 mV that increased from −1.07 ± 0.68 pA pF–1 to a mean peak amplitude of −2.57 ± 0.89 pA pF–1 (n = 9, p = 0.0003, two‐way ANOVA, Tukey's multiple comparisons test), which were similar to currents recorded from freshly isolated VSMCs (Figs 1 and 4). By contrast, Figure 5B shows that, in VSMCs expressing specific PKCδ oligomers, store depletion activated small whole‐cell TRPC1 currents, with the control mean amplitude at −80 mV increasing from −1.05 ± 0.59 pA pF–1 to a mean peak amplitude of −1.36 ± 0.69 pA pF–1 (n = 10, p = 0.8240, two‐way ANOVA, Tukey's comparisons test). As such, mean peak amplitudes of store‐operated TRPC1 current at −80 mV were greatly reduced in VSMCs expressing specific PKCδ compared to scrambled oligomers (p = 0.0051, two‐way ANOVA, Tukey's comparisons test).
Figure 5. Knockdown of PKCδ expression reduces activation of TRPC1‐based SOCs.
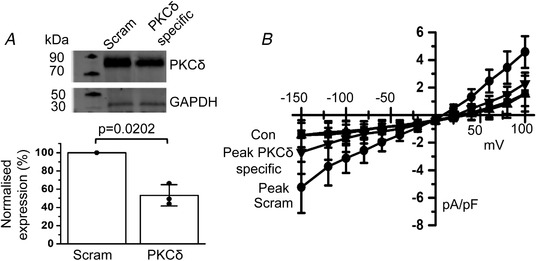
A, representative western blots and mean data showing that PKCδ‐specific morpholino oligomers reduced PKCδ expression in rat mesenteric artery protein lysates compared to scrambled/mismatched oligomers (Scram). Band intensity was normalized to β‐GAPDH to calculate percentage expression (vs. scrambled, n = 3, p = 0.0202, unpaired t test). B, mean I–V relationships showing that the mean peak amplitude of store‐operated whole‐cell TRPC1 currents was reduced in the presence of PKCδ‐specific (n = 10) compared to scrambled (n = 9) morpholino oligomers.
Store depletion induces TRPC1 and PKCδ interactions and PKCδ‐dependent phosphorylation of TRPC1
The above pharmacological and molecular evidence indicates that PKCδ activity is essential for activation of TRPC1‐based SOCs in native contractile VSMCs. Therefore, we investigated whether store depletion induced PKCδ and TRPC1 interactions and PKCδ‐dependent phosphorylation of TRPC1 in freshly isolated rat mesenteric artery tissue lysates and VSMCs.
Immunoprecipitation with anti‐TRPC1 antibodies followed by immunoblotting with an anti‐PKCδ antibody revealed that interactions between these two proteins are absent in unstimulated tissue but occur following incubation with BAPTA‐AM or TPEN (Fig. 6A ). Furthermore, the PLA studies shown in Figure 6B confirmed that interactions between TRPC1 and PKCδ were absent in unstimulated VSMCs, whereasd robust puncta at (or close to) the plasma membrane were induced between TRPC1 and PKCδ following pre‐treatment with BAPTA‐AM and TPEN.
Figure 6. Store depletion induces associations between TRPC1 and PKCδ in native contractile VSMCs.
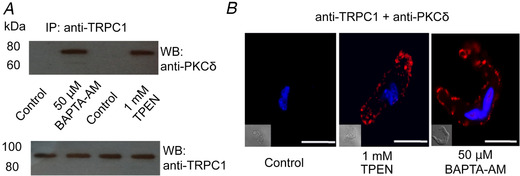
A, representative western blots with anti‐TRPC1 and anti‐PKCδ antibodies following immunoprecipitation of freshly isolated rat mesenteric artery tissue lysates with an anti‐TRPC1 antibody show that pre‐treatment with the BAPTA‐AM and TPEN induced associations between TRPC1 and PKCδ but did not alter TRPC1 expression. B, representative images of proximity ligation assays from freshly isolated rat mesenteric artery VSMCs showing a low level of association between TRPC1 and PKCδ in unstimulated VSMCs, which was greatly increased following pre‐treatment with TPEN and BAPTA‐AM for 10 min. Scale bars = 10 μm.
PLA experiments performed with a mixture of anti‐phosphorylated serine/threonine and anti‐TRPC1 antibodies revealed a low number of fluorescent puncta in unstimulated freshly isolated VSMCs, which is indicative of a low level of basal TRPC1 phosphorylation. Figure 7A shows that the mean number of puncta per cell was increased following pre‐treatment with BAPTA‐AM from 6.1 ± 3.9 (n = 19) to 20.5 ± 9.9 (n = 30, p = 0.0001) and inhibited by coapplication of δV1‐TAT to 8.3 ± 4.2 (n = 25, p = 0.0001, unpaired t test). In addition, Figure 7B shows that transfection of vessel segments with specific PKCδ morpholino oligomers also reduced the BAPTA‐AM‐induced increase in mean puncta number compared to scrambled oligomers from 22.4 ± 10.4 (n = 30) to 7.2 ± 4.9 (n = 25, p = 0.0001, unpaired t test). Importantly, Figure 7A and B shows a similar mean number of puncta using mismatched oligomers compared to non‐transfected freshly isolated VSMCs under unstimulated (8.7 ± 4.3, n = 26 vs. 6.1 ± 3.9, n = 19, p = 0.049) or BAPTA‐AM‐treated conditions (22.4 ± 10.4, n = 30 vs. 20.5 ± 9.9, n = 30, p = 0.4653, unpaired t test), indicating that the transfection process had little effect on this mechanism.
Figure 7. Store depletion induces PKCδ‐dependent phosphorylation of TRPC1.
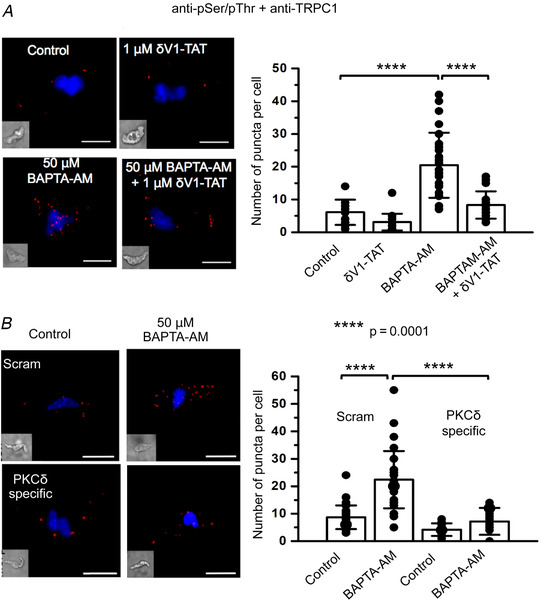
A, representative and mean data from proximity ligation assays showing pre‐treatment with BAPTA‐AM (n = 30) for 10 min greatly increased the number of puncta per cell representing associations between TRPC1 and phosphorylated serine and threonine residues compared to control values (n = 19, p = 0.0001), which was reduced in the presence of δV1‐TAT (n = 25, p = 0.0001, unpaired t test). B, representative and mean data from proximity ligation assays showing that BAPTA‐AM‐induced associations between TRPC1 and phosphorylated serine and threonine residues were reduced in VSMCs from vessels transfected with PKCδ‐specific (n = 24, p = 0.0001) compared to scrambled morpholino oligomers (n = 30, p = 0.0001, unpaired t test). Scale bars = 10 μm. **** p = 0.0001.
Taken together, these results indicate that store depletion induces interactions between TRPC1 and PKCδ in VSMCs, and that these interactions cause PKCδ‐dependent phosphorylation of TRPC1.
PKCδ mediates store‐operated interactions between PIP2 and TRPC1, as well as PIP2‐evoked TRPC1 currents
In our previous work, we identified that PIP2 has an obligatory role in the activation of TRPC1‐based SOCs in VSMCs, potentially as the activating ligand, and that this process requires PKC‐dependent phosphorylation of TRPC1 (see Introduction). Therefore, we investigated whether store‐operated interactions between PIP2 and TRPC1 require PKCδ activity using immunocytochemistry and PLA, and also whether PKCδ is essential for activation of store‐operated whole‐cell TRPC1 currents by PIP2.
In freshly isolated rat mesenteric artery VSMCs, Figure 8A shows that PIP2 immunostaining was highly expressed at (or close to) the plasma membrane in unstimulated cells and BAPTA‐AM and δV1‐TAT had no obvious effect on this distribution. With respect to the staining produced with anti‐PIP2 antibodies being related to endogenous PIP2, Figure 8A shows that pre‐treatment with high concentrations of wortmannin (20 μm), which inhibits PI‐4/PI‐5 kinases to reduce PIP2 recycling and promote PIP2 depletion, produced a considerable reduction in the PIP2 signal (Suh & Hille, 2005; Saleh et al. 2009 a; Shi et al. 2014). In control experiments, TRPC1 immunostaining was unaffected by BAPTA‐AM, δV1‐TAT or wortmannin treatment (Fig. 8A ). We next adapted this immunostaining protocol for PLA experiments. Figure 8B reveals that there was a low level of interactions between PIP2 and TRPC1 in unstimulated VSMCs, which were increased by BAPTA‐AM from a mean puncta number per cell of 7.6 ± 5.2 (n = 23) to 34.6 ± 14.1 (n = 24, p = 0.0001) and inhibited in the presence of δV1‐TAT or following pre‐treatment of wortmannin, respectively, to 6.5 ± 2.9 (n = 20, p = 0.0001) and 6.1 ± 3.7 (n = 19, p = 0.0001, unpaired t test).
Figure 8. Store‐operated interactions between TRPC1 and PIP2 require PKCδ.
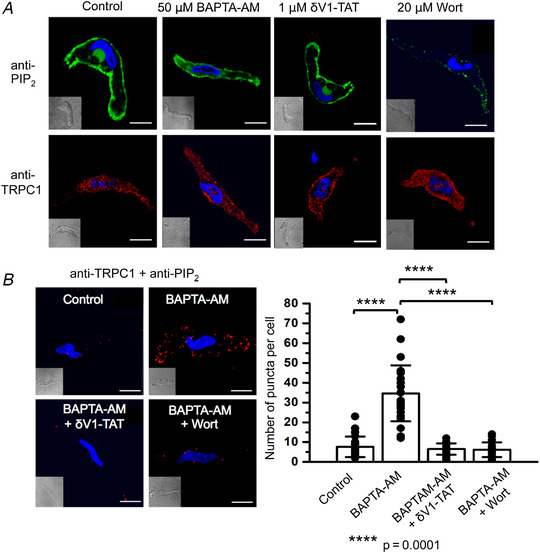
A, representative images from freshly isolated rat mesenteric VSMCs showing expression of PIP2 (green) and TRPC1 (red) following pretreatment with BAPTA‐AM for 10 min, wortmannin (Wort) for 20 min and δV1‐TAT for 10 min. B, representative images and mean data from PLA experiments showing that control interactions between TRPC1 and PIP2 (n = 23) were greatly increased by pre‐treatment with BAPTA‐AM for 10 min (n = 23, p = 0.0001), and this was reduced by coapplication with δV1‐TAT (n = 20, p = 0.0001) and wortmannin (n = 19, p = 0.0001, unpaired t test). Scale bars = 10 μm.
In freshly isolated VSMCs, Figure 9A shows that bath application of the phorbol ester 12,13‐dibutyrate (PDBu) increased whole‐cell non‐selective cation currents from a mean control amplitude of −0.71 ± 0.13 pA pF–1 to a mean peak amplitude of −2.12 ± 0.8 pA pF–1 at −80 mV (n = 8, p = 0.0001, two‐way ANOVA, Tukey's comparisons test). By contrast, inclusion of diC8‐PIP2 in the patch pipette solution had no effect on whole‐cell cation currents (−80 mV, −0.75 ± 0.18 vs. −1.0 ± 0.08, n = 5, p = 0.6151, two‐way ANOVA, Tukey's comparisons test). Figure 9B shows that dialysing cells with diC8‐PIP2 in the patch pipette solution increased PDBu‐activated whole‐cell cation currents, with mean peak amplitude at −80 mV increased from −2.12 ± 0.8 pA pF–1 (n = 8) (Fig. 9A ) to −2.88 ± 0.78 pA pF–1 at −80 mV (n = 7, p = 0.0291, unpaired t test). Figure 9B also shows that PDBu‐evoked whole‐cell cation currents induced in the presence of diC8‐PIP2 were inhibited by bath applications of T1E3 and δV1‐TAT, respectively, to −1.13 ± 0.24 pA pF–1 (n = 5, p = 0.0001) and −0.84 ± 0.38 pA pF–1 at −80 mV (n = 7, p = 0.0001 two‐way ANOVA, Tukey's comparisons test) indicating that these currents were mediated by TRPC1 and required PKCδ activity.
Figure 9. PIP2 increases PDBu‐induced whole‐cell TRPC1 currents.
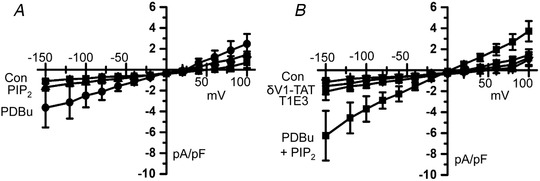
A, mean I–V relationships from freshly isolated rat mesenteric artery VSMCs showing that bath application of PDBu but not inclusion of diC8‐PIP2 in the patch pipette solution induced a whole‐cell current. B, mean I–V relationships showing that, following cell dialysis with diC8‐PIP2, PDBu‐evoked whole‐cell currents had a greatly increased peak amplitude compared to currents induced by PDBu alone (A) and were inhibited by T1E3 and δV1‐TAT.
Noradrenaline‐evoked TRPC1 channels require PKCδ
The present study clearly demonstrates that the PKCδ isoform is important for activation of TRPC1 channels by agents that deplete internal Ca2+ stores. Therefore, in the final series of experiments, we investigated the physiological significance of these findings by examining whether PKCδ is involved in activation of TRPC1 channels by the vasoconstrictor and α1‐adrenoceptor agonist methoxamine (MO). Figure 10 shows that, in unstimulated VSMCs, there was a low level of spontaneous 2 pS TRPC1‐based channel activity in cell‐attached patches held at −80 mV, and that mean peak open probability (NPo) was increased following bath application of MO from 0.14 ± 0.07 to 0.51 ± 0.2 (n = 6, p = 0.0035) and was significantly reduced by subsequent coapplication of δV1‐TAT to 0.11 ± 0.08 (n = 6, p = 0.0029, paired t test).
Figure 10. Methoxamine‐evoked TRPC1 channel activity is mediated by PKCδ.
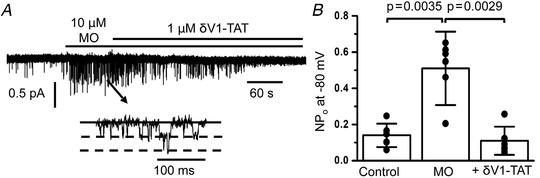
A and B, representative trace and mean open probability (NPo) data showing that bath application of methoxamine evoked TRPC1 channel activity in cell‐attached patches held at −80 mV (n = 6, p = 0.0035), which was reduced by coapplication of δV1‐TAT (n = 6, p = 0.0029, paired t test).
Discussion
The present study reveals that activity of PKCδ, a member of the novel PKC isoform subgroup, is essential for activation of TRPC1‐based channels evoked by store depletion and α1‐adrenoceptor stimulation in native contractile rat mesenteric artery VSMCs. It is proposed that PKCδ‐dependent phosphorylation of TRPC1 is required for TRPC1 channel activation by PIP2, the probable activating ligand. These results provide further evidence that PKC activity and PIP2 are obligatory for activation of TRPC1‐based SOCs in VSMCs, ion channels implicated in regulating contraction, proliferation and migration that are processes linked to hypertension and atherosclerosis. As such, regulating PKCδ activity and PIP2 levels may comprise therapeutic targets for these and other vascular diseases.
TRPC1‐based SOCs require PKC activity
Our results show that the well‐established protocol for inducing store depletion, the inclusion of a high concentration of BAPTA in the patch pipette solution, evoked a whole‐cell non‐selective cation current with a relatively linear I–V relationship and an E rev of between 0 mV and +20 mV in freshly isolated rat mesenteric artery VSMCs, which is similar to currents previously recorded in rabbit and mouse VSMCs (Shi et al. 2012a, 2014, 2016, 2017a, 2017b a,b). Considerable evidence obtained using pharmacological agents, antibodies as blocking agents and TRPC1 knockout mice indicates that these store‐operated currents are mediated by a heteromeric TRPC1/TRPC5 molecular template (Xu & Beech, 2001; Xu et al. 2005 a; Saleh et al. 2008; Shi et al. 2012 a). In further support of this proposal, the present study shows that store‐operated whole‐cell currents were inhibited by Pico145, a highly selective and potent TRPC1/C4/C5 channel blocker (Rubaiy et al. 2017) and T1E3, a TRPC1 antibody raised against an extracellular pore region and known to act as a blocking agent (Xu & Beech, 2001; Xu et al. 2005 b). In addition, using PLA, the present study reveals that TRPC1 and TRPC5 co‐localize within <40 nm of each other at (or close to) the plasma membrane of VSMCs. It should be noted that the electrophysiological and pharmacological properties of these store‐operated currents in freshly isolated VSMCs exhibiting a contractile phenotype are considerably different from those of Orai1‐based CRACs (Prakriya & Lewis, 2015), indicating that Orai1 proteins are probably not involved, as reported previously (Shi et al. 2017 b).
Previous studies show that PKC activity and PKC‐mediated phosphorylation of TRPC1 are essential for activation of TRPC1‐based SOCs in native contractile VSMCs (Albert & Large, 2002 b; Saleh et al. 2006, 2008; Large et al. 2009; Albert, 2011; Shi et al. 2012a, 2014, 2016). The present study provides further support for an excitatory role for PKC, with the pan‐PKC isoform selective inhibitor GF109203X inhibiting store‐operated whole‐cell TRPC1 currents in freshly isolated rat mesenteric artery VSMCs. Therefore, identifying the specific PKC isoform responsible for activating TRPC1‐based SOCs was the main aim of the present study.
PKCδ is the PKC isoform responsible for activation of TRPC1‐based SOCs and store‐operated TRPC1 phosphorylation
The PKC family is subdivided into conventional PKC (α, β and γ), novel PKC (δ, ϵ, η and θ) and atypical PKC (ζ and ι/κ) isoforms (Salamanca & Khalil, 2005; Ringvold & Khalil, 2017). Given that DAG analogues activate TRPC1 channels through a PKC‐dependent mechanism in VSMCs (Saleh et al. 2006; 2008; Large et al. 2009; Albert, 2011), and that the Ca2+ store depletion protocols used to activate TRPC1‐based SOCs probably increase, decrease or do not alter [Ca2+]i, the PKC isoform(s) responsible is probably belongs to the novel subfamily (see Introduction).
Western blotting and immunocytochemical analysis demonstrated that PKCδ is expressed in native contractile rat mesenteric artery VSMCs and was distributed at (or close to) the plasma membrane. There was greater expression of PKCδ compared to other novel PKC isoforms in VSMCs and tissue lysates; however, caution is warranted when suggesting that PKCδ is the dominant novel PKC isoform in VSMCs because expression variations may be linked to potential differences between the affinity and specificity of the anti‐novel PKC isoform antibodies used.
Inhibition of PKCδ activity via well‐characterized selective PKCδ peptide inhibitors and knockdown of PKCδ expression using morpholino oligomers resulted in a significant reduction in the development and peak amplitude of store‐operated whole‐cell TRPC1 currents. Moreover, immunoprecipitation and PLA studies revealed that store depletion induced interactions between TRPC1 and PKCδ. Importantly, PLA experiments showed that store depletion induced puncta formation between anti‐TRPC1 and anti‐phosphorylated serine and threonine antibodies that was greatly reduced by PKCδ peptide inhibitors and PKCδ selective morpholino oligomers, which indicates that store depletion induces PKCδ‐dependent phosphorylation of TRPC1. Taken together, these findings provide strong evidence for the PKCδ isoform being essential in the activation of TRPC1‐based SOCs in native contractile VSMCs.
PKCδ has also been reported to participate in store‐operated Ca2+ entry in airway smooth muscle (Gao et al. 2012), proliferation and migration of VSMCs (Ding et al. 2011; Fan et al. 2014), development of the myogenic response (Kashihara et al. 2008), and membrane insertion of TRPM4 channels and associated vasoconstriction (Crnich et al. 2010; Garcia et al. 2011). Interestingly, TRPC1 has also been associated with many of these vascular functions (Earley & Brayden, 2015) and, accordingly, in the future, it may be useful to investigate the functional association between TRPC1 and PKCδ activities in the vasculature. By contrast to the results outlined above, PKC activity has an inhibitory action on Orai1‐based CRACs with PKC inhibitors including GF109203X and knockdown of PKCβ isoform increasing Orai1 activity (Kawasaki et al. 2010), which further indicates that Orai1 proteins or Orai1‐based CRACs are not involved in the activation of TRPC1‐based SOCs in native contractile VSMCs.
PKCδ activity is required for PIP2‐mediated activation of TRPC1 channels
We have previously described the obligatory requirement of PIP2 in the activation of TRPC1‐based SOCs and proposed that PKC‐dependent phosphorylation of TRPC1 is required for this phospholipid to act as the activating ligand (Large et al. 2009; Saleh et al. 2009a, 2009b a,b; Albert, 2011; Shi et al. 2012 a,2012b, 2014). In the present study, using PLA, store depletion induces interactions between PIP2 and TRPC1 at (or close to) the plasma membrane of VSMCs that are reduced by PKCδ peptide inhibitor. To provide evidence that endogenous PIP2 is involved in these results, we showed that a high concentration of wortmannin (20 μm) that depletes PIP2 levels by inhibiting PI‐4/PI‐5 kinase‐mediated PIP2 synthesis (Suh & Hille, 2005) reduced PIP2 immunostaining and store‐operated associations between TRPC1 and PIP2. It should be noted that a high concentration of wortmannin also inhibits myosin light chain kinase (MLCK) and PI(3) kinase, although previous findings have shown that lower concentrations (<1 μm) inhibiting MLCK and PI‐3 kinase but not depleting PIP2 levels had no effect on activation of TRPC1 channel activity in VSMCs (Saleh et al. 2009 a; Shi et al. 2014). The PKC activator PDBu activated whole‐cell TRPC1 currents that were increased in amplitude when the water soluble PIP2 analogue diC8‐PIP2 was included in the patch pipette solution and were inhibited by a PKCδ peptide inhibitor. Cell dialysis with diC8‐PIP2 failed to activate any whole‐cell currents on its own, as reported previously (Albert et al. 2008), which indicates that the effects of PDBu and PIP2 are not merely additive but probably represent synergism between the two molecules. Taken together, these results re‐enforce the idea that PIP2 is the activating ligand of TRPC1‐based SOCs, and that this requires PKCδ‐dependent phosphorylation of TRPC1.
The proposed excitatory roles of PKC and PIP2 on TRPC1‐based SOCs are opposite to the action of these molecules in the activation of non‐TRPC1‐containing channels (e.g. TRPC3/C6/C7 channels) in native contractile VSMCs (Venkatachalam et al. 2003; Albert & Large, 2004; Large et al. 2009; Shi et al. 2010; Albert, 2011). This subgroup of TRPC channels, known as receptor‐operated channels (ROCs), is activated by receptor‐mediated generation of DAG, which leads to channel opening via PKC independent mechanisms, with subsequent PKC activity induced by DAG producing channel inhibition. Interestingly, stimulation of TRPC1‐based SOCs is proposed to inhibit TRPC6‐based SOCs through inducing Ca2+ influx and PKC activity, which suggests the involvement of a conventional PKC isoform (Shi et al. 2010). A distinct role of PIP2 on TRPC3/C6/C7‐based ROCs is less clear, with both inhibitory and excitatory actions on channel activity being proposed (Albert et al. 2008; Imai et al. 2012). These findings further indicate that TRPC1‐based SOCs and TRPC3/C6/C7‐based ROCs form distinct channel structures with differing activation mechanisms involving differing PKC isoforms, and probably distinct functions in VSMCs.
Computation predication of potential PKCδ‐dependent phosphorylation sites within the TRPC1 sequence using GPS 3.0 (Xue et al. 2008) reveals five intracellular serine residues, with Ser619 and Ser752 at the C‐terminal domain being of significance in that both of these sites are close to a known PIP2‐binding domain (Kwon et al. 2007). Hence, it is possible that PKCδ‐dependent phosphorylation of one or both sites increases PIP2 binding to TRPC1, leading to channel opening. Other studies have demonstrated that protein kinase A, protein kinase G and calmodulin kinase II have inhibitory actions on TRPC1‐based SOCs in VSMCs (Liu et al. 2005 a; Albert et al. 2006; Chen et al. 2011), and therefore TRPC1 phosphorylation by these kinases might inhibit TRPC1‐based SOCs by reducing TRPC1 and PIP2 interactions. Similar roles for kinase activities with respect to modulating lipid–protein interactions are well‐established in the regulation of K+ channel subtypes (Logothetis et al. 2015).
One important aspect is how PKCδ activity is stimulated by store depletion. Our proposed hypothesis based on previous work indicates that store depletion induces STIM1–TRPC1 interactions, which stimulate a Gq‐PLCβ1 pathway that generates DAG and PKC activity, leading to PKC‐dependent phosphorylation of TRPC1 (Baudel, 2020). PKC‐dependent phosphorylation of TRPC1 leads to a dissociation of interactions between TRPC1 and the PIP2 binding protein myristoylated alanine‐rich C‐kinase substrate (MARCKS), with the latter molecule then releasing PIP2 into the local environment, allowing it to act as the channel activating ligand. The present study indicates that the PKCδ isoform is probably an essential PKC isoform in this process and this will be important to investigate in detail in future studies.
Physiological relevance of PKCδ‐mediated TRPC1 channels
TRPC1 channel activity induced by the α1‐adrenoceptor agonist and vasoconstrictor methoxamine was reduced by a PKCδ peptide inhibitor, which suggests that a similar store‐operated activation process involving PKCδ may be used by physiological stimulants of TRPC1 channels. This is further supported by findings showing that PLCβ1 and STIM1 knockdown both significantly inhibit noradrenaline‐evoked TRPC1 channel activity (Shi et al. 2016, 2017b b) and noradrenaline induces an increase in PKC‐dependent phosphorylation of TRPC1 and stimulates MARCKS to release PIP2, which is then available to interact with TRPC1 (Shi et al. 2014). It was previously proposed that vasoconstrictor agents may also activate TRPC1‐based SOCs independently of store deletion (Albert & Large, 2002 b; Saleh et al. 2006; Large et al. 2009; Saleh et al. 2009 b; Shi et al. 2010, 2012b b; Albert, 2011), which suggests that TRPC1 channels may also act as ROCs. The current and previous findings certainly suggest that a significant contribution to physiological stimulation of TRPC1 channels probably occurs via a store‐operated pathway.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
MMAB, JS, WL and AA all contributed to the conception or design of the work, as well as the analysis of data or the interpretation of data for the work, and were involved in drafting the work or revising it critically for important intellectual content. MMAB and JS were involved in the acquisition of data. All authors approved final version of the manuscript submitted for publication and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Biotechnology and Biological Sciences Research Council (BB/J007226/1 and BB/M018350/1 to AA).
Supporting information
Statistical Summary Document
Biography
Miguel Martín‐Aragón Baudel obtained his PhD from Edinburgh Napier University under the supervision of Mark Darlison and Amy Poole before moving to St George's, University of London, to work as a postdoctoral research assistant with Anthony Albert. He is currently working at the University of California, Davis, with Manuel Navedo, continuing his research interests in the ion channel‐mediated mechanisms driving vascular contraction and with a role in diseases such as hypertension and diabetes.

Edited by: Ian Forsythe & Pawel Ferdek
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Albert AP & Large WA (2002a). A Ca2+‐permeable non‐selective cation channel activated by depletion of internal Ca2+ stores in single rabbit portal vein myocytes. J Physiol 538, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP & Large WA (2002b). Activation of store‐operated channels by noradrenaline via protein kinase C in rabbit portal vein myocytes. J Physiol 544, 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP & Large WA (2003). Store‐operated Ca2+‐permeable non‐selective cation channels in smooth muscle cells. Cell Calcium 33, 345–356. [DOI] [PubMed] [Google Scholar]
- Albert AP & Large WA (2004). Inhibitory regulation of constitutive transient receptor potential‐like cation channels in rabbit ear artery myocytes. J Physiol 560, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Liu M & Large WA (2006). Dual effect of calmodulin on store‐operated Ca2+ ‐permeable cation channels in rabbit portal vein myocytes. Br J Pharmacol 148, 1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Saleh SN, Peppiatt‐Wildman CM & Large WA (2007). Multiple activation mechanisms of store‐operated TRPC channels in smooth muscle cells. J Physiol 583, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Saleh SN & Large WA (2008). Inhibition of native TRPC6 channel activity by phosphatidylinositol 4,5‐bisphosphate in mesenteric artery myocytes. J Physiol 586, 3087–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Saleh SN & Large WA (2009). Identification of canonical transient receptor potential (TRPC) channel proteins in native vascular smooth muscle cells. Curr Med Chem 16, 1158–1165. [DOI] [PubMed] [Google Scholar]
- Albert AP (2011). Gating mechanisms of canonical transient receptor potential channel proteins: role of phosphoinositols and diacylglycerol. Adv Exp Med Biol 704, 391–411. [DOI] [PubMed] [Google Scholar]
- Ambudkar IS, de Souza LB & Ong HL (2017). TRPC1, orai1, and STIM1 in SOCE: friends in tight spaces. Cell Calcium 63, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanov A, Sampieri A, Moreno C, Pacheco J, Salgado A, Sherry R & Vaca L (2015). Combined single channel and single molecule detection identifies subunit composition of STIM1‐activated transient receptor potential canonical (TRPC) channels. Cell Calcium 57, 1–13. [DOI] [PubMed] [Google Scholar]
- Baudel MASM, Shi J, Large WA & Albert AP (2020). Insights into activation mechanisms of store‐operated TRPC1 channels in vascular smooth muscle. Cells 9, pii: E179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ (2012). Orai1 calcium channels in the vasculature. Pflugers Arch 463, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergdahl A, Gomez MF, Wihlborg AK, Erlinge D, Eyjolfson A, Xu SZ, Beech DJ, Dreja K & Hellstrand P (2005). Plasticity of TRPC expression in arterial smooth muscle: correlation with store‐operated Ca2+ entry. Am J Physiol Cell Physiol 288, C872–C880. [DOI] [PubMed] [Google Scholar]
- Berra‐Romani R, Mazzocco‐Spezzia A, Pulina MV & Golovina VA (2008). Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am J Physiol Cell Physiol 295, C779–C790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callender JA & Newton AC (2017). Conventional protein kinase C in the brain: 40 years later. Neuronal Signal 1, NS20160005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IS, Dai ZK, Welsh DG, Chen IJ & Wu BN (2011). Protein kinases modulate store‐operated channels in pulmonary artery smooth muscle cells. J Biomed Sci 18, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Ong HL, Liu X & Ambudkar IS (2013). Contribution and regulation of TRPC channels in store‐operated Ca2+ entry. Curr Top Membr 71, 149–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crnich R, Amberg GC, Leo MD, Gonzales AL, Tamkun MM, Jaggar JH & Earley S (2010). Vasoconstriction resulting from dynamic membrane trafficking of TRPM4 in vascular smooth muscle cells. Am J Physiol Physiol 299, C682–C694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding RQ, Tsao J, Chai H, Mochly‐Rosen D & Zhou W (2011). Therapeutic potential for protein kinase C inhibitor in vascular restenosis. J Cardiovasc Pharmacol Ther 16, 160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S & Brayden JE (2015). Transient receptor potential channels in the vasculature. Physiol Rev 95, 645–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edimo WE, Ramos AR, Ghosh S, Vanderwinden JM & Erneux C (2016). The SHIP2 interactor Myo1c is required for cell migration in 1321 N1 glioblastoma cells. Biochem Biophys Res Commun 476, 508–514. [DOI] [PubMed] [Google Scholar]
- Fan H‐C, Fernández‐Hernando C & Lai J‐H (2014). Protein kinase C isoforms in atherosclerosis: pro‐ or anti‐inflammatory? Biochem Pharmacol 88, 139–149. [DOI] [PubMed] [Google Scholar]
- Fleegal MA, Hom S, Borg LK, & Davis TP (2005). Activation of PKC modulates blood‐brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol 289, H2012–H2019. [DOI] [PubMed] [Google Scholar]
- Gao Y, Zou J, Geng S, Zheng J & Yang J (2012). Role of protein kinase C in the activation of store‐operated Ca2+ entry in airway smooth muscle cells. J Huazhong Univ Sci Technol Med Sci 32, 303–310. [DOI] [PubMed] [Google Scholar]
- Garcia ZI, Bruhl A, Gonzales AL & Earley S (2011). Basal protein kinase Cδ activity is required for membrane localization and activity of TRPM4 channels in cerebral artery smooth muscle cells. Channels 5, 210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Itsuki K, Okamura Y, Inoue R & Mori MX (2012). A self‐limiting regulation of vasoconstrictor‐activated TRPC3/C6/C7 channels coupled to PI(4,5)P₂‐diacylglycerol signalling. J Physiol 590, 1101–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashihara T, Nakayama K & Ishikawa T (2008). Distinct roles of protein kinase C isoforms in myogenic constriction of rat posterior cerebral arteries. J Pharmacol Sci 108, 446–454. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Ueyama T, Lange I, Feske S & Saito N (2010). Protein kinase C‐induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store‐operated Ca2+ channel. J Biol Chem 285, 25720–25730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Hofmann T & Montell C (2007). Integration of phosphoinositide‐ and calmodulin‐mediated regulation of TRPC6. Mol Cell 25, 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Large WA, Saleh SN & Albert AP (2009). Role of phosphoinositol 4,5‐bisphosphate and diacylglycerol in regulating native TRPC channel proteins in vascular smooth muscle. Cell Calcium 45, 574–582. [DOI] [PubMed] [Google Scholar]
- Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, So I, Shin DM, Muallem S & Yuan JP (2014). Molecular determinants mediating gating of transient receptor potential canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J Biol Chem 289, 6372–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Abramowitz J & Birnbaumer L (2014). The TRPC family of TRP channels: roles inferred (mostly) from knockout mice and relationship to ORAI proteins. Handb Exp Pharmacol 223, 1055–1075. [DOI] [PubMed] [Google Scholar]
- Liu M, Large WA & Albert AP (2005a) Stimulation of beta‐adrenoceptors inhibits store‐operated channel currents via a cAMP‐dependent protein kinase mechanism in rabbit portal vein myocytes. J Physiol 562 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Albert AP & Large WA (2005b). Facilitatory effect of Ins(1,4,5)P3 on store‐operated Ca2+‐permeable cation channels in rabbit portal vein myocytes. J Physiol 566, 161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis DE, Petrou VI, Zhang M, Mahajan R, Meng XY, Adney SK, Cui M & Baki L (2015). Phosphoinositide control of membrane protein function: a frontier led by studies on ion channels. Annu Rev Physiol 77, 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matchkov VV, Kudryavtseva O & Aalkjaer C (2012). Intracellular Ca²⁺ signalling and phenotype of vascular smooth muscle cells. Basic Clin Pharmacol Toxicol 110, 42–48. [DOI] [PubMed] [Google Scholar]
- Ong HL & Ambudkar IS (2015). Molecular determinants of TRPC1 regulation within ER‐PM junctions. Cell Calcium 58, 376–386. [DOI] [PubMed] [Google Scholar]
- Popp RL, Velasquez O, Bland J & Hughes P (2006). Characterization of protein kinase C isoforms in primary cultured cerebellar granule cells. Brain Res 1083, 70–84. [DOI] [PubMed] [Google Scholar]
- Prakriya M & Lewis RS (2015). Store‐operated calcium channels. Physiol Rev 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringvold HC & Khalil RA (2017). Protein kinase C as regulator of vascular smooth muscle function and potential target in vascular disorders. Adv Pharmacol 78, 203–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubaiy HN, Ludlow MJ, Henrot M, Gaunt HJ, Miteva K, Cheung SY, Tanahashi Y, Hamzah N, Musialowski KE, Blythe NM, Appleby HL, Bailey MA, McKeown L, Taylor R, Foster R, Waldmann H, Nussbaumer P, Christmann M, Bon RS, Muraki K & Beech DJ (2017). Picomolar, selective, and subtype‐specific small‐molecule inhibition of TRPC1/4/5 channels. J Biol Chem 292, 8158–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamanca DA & Khalil RA (2005). Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol 70, 1537–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh SN, Albert AP, Peppiatt CM & Large WA (2006). Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J Physiol 577, 479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh SN, Albert AP, Peppiatt‐Wildman CM & Large WA (2008). Diverse properties of store‐operated TRPC channels activated by protein kinase C in vascular myocytes. J Physiol 586, 2463–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh SN, Albert AP & Large WA (2009a). Obligatory role for phosphatidylinositol 4,5‐bisphosphate in activation of native TRPC1 store‐operated channels in vascular myocytes. J Physiol 587, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh SN, Albert AP & Large WA (2009b). Activation of native TRPC1/C5/C6 channels by endothelin‐1 is mediated by both PIP3 and PIP2 in rabbit coronary artery myocytes. J Physiol 587, 5361–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Ju M, Saleh SN, Albert AP & Large WA (2010). TRPC6 channels stimulated by angiotensin II are inhibited by TRPC1/C5 channel activity through a Ca2+‐ and PKC‐dependent mechanism in native vascular myocytes. J Physiol 588, 3671–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Ju M, Abramowitz J, Large WA, Birnbaumer L & Albert AP (2012a). TRPC1 proteins confer PKC and phosphoinositol activation on native heteromeric TRPC1/C5 channels in vascular smooth muscle: comparative study of wild‐type and TRPC1−/− mice. FASEB J 26, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Ju M, Large WA & Albert AP (2012b). Pharmacological profile of phosphatidylinositol 3‐kinases and related phosphatidylinositols mediating endothelin(A) receptor‐operated native TRPC channels in rabbit coronary artery myocytes. Br J Pharmacol 166, 2161–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Birnbaumer L, Large WA & Albert AP (2014). Myristoylated alanine‐rich C kinase substrate coordinates native TRPC1 channel activation by phosphatidylinositol 4,5‐bisphosphate and protein kinase C in vascular smooth muscle. FASEB J 28, 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Miralles F, Birnbaumer L, Large WA & Albert AP (2016). Store depletion induces Gαq‐mediated PLCβ1 activity to stimulate TRPC1 channels in vascular smooth muscle cells. FASEB J 30, 702–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Miralles F, Birnbaumer L, Large WA & Albert AP (2017a). Store‐operated interactions between plasmalemmal STIM1 and TRPC1 proteins stimulate PLCβ1 to induce TRPC1 channel activation in vascular smooth muscle cells. J Physiol 595, 1039–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Miralles F, Kinet JP, Birnbaumer L, Large WA & Albert AP (2017b). Evidence that Orai1 does not contribute to store‐operated TRPC1 channels in vascular smooth muscle cells. Channels 11, 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott JB, Barrese V, Suresh M, Masoodi S & Greenwood IA (2018). Investigating the role of G protein βγ in Kv7‐dependent relaxations of the rat vasculature. Arterioscler Thromb Vasc Biol 38, 2091–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC & Hille B (2005). Regulation of ion channels by phosphatidylinositol 4,5‐bisphosphate. Curr Opin Neurobiol 15, 370–378. [DOI] [PubMed] [Google Scholar]
- Trebak M (2012). STIM/Orai signalling complexes in vascular smooth muscle. J Physiol 590, 4201–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepakova ES, Gericke M, Hirakawa Y, Weisbrod RM, Cohen RA & Bolotina VM (2001). Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J Biol Chem 276, 7782–7790. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Zheng F & Gill DL (2003). Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J Biol Chem 278, 29031–29040. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kim JM, Schmit MB, Cho TS, Fang C & Cai H (2019). A bed nucleus of stria terminalis microcircuit regulating inflammation‐associated modulation of feeding. Nat Commun 10, 2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley PF, Zeng W, Huang GN, Yuan JP, Kim JY, Lee MG & Muallem S (2007). TRPC channels as STIM1‐regulated store‐operated channels. Cell Calcium 42, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SZ & Beech DJ (2001). TrpC1 is a membrane‐spanning subunit of tore‐operated Ca2+ channels in native vascular smooth muscle cells. Circ Res 88, 84–87. [DOI] [PubMed] [Google Scholar]
- Xu SZ, Boulay G, Flemming R & Beech DJ (2005a). E3‐targeted anti‐TRPC5 antibody inhibits store‐operated calcium entry in freshly isolated pial arterioles. Am J Physiol Heart Circ Physiol 291, H2653–H2659. [DOI] [PubMed] [Google Scholar]
- Xu S‐Z, Zeng F, Lei M, Li J, Gao B, Xiong C, Sivaprasadarao A & Beech DJ (2005b). Generation of functional ion‐channel tools by E3 targeting. Nat Biotechnol 23, 1289–1293. [DOI] [PubMed] [Google Scholar]
- Xue Y, Ren J, Gao X, Jin C, Wen L & Yao X (2008). GPS 2.0, a tool to predict kinase‐specific phosphorylation sites in hierarchy. Mol Cell Proteomics 7, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WC, Wang Q, Chi L‐T, Wang Y‐Z, Cao H‐L & Li W‐Z (2019). Therapeutic hypercapnia reduces blood‐brain barrier damage possibly via protein kinase Cε in rats with lateral fluid percussion injury. J Neuroinflammation 16, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Kim MS, Zeng W, Shin DM, Huang G, Worley PF & Muallem S (2009). TRPC channels as STIM1‐regulated SOCs. Channels 3, 221–225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical Summary Document
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.