

Abstract
Woodchuck hepatitis virus (WHV)-infected woodchucks have been used for preclinical development of drugs against hepatitis B virus (HBV). However, there is no simple in vivo model to evaluate small amounts of compounds against HBV. To develop such a model, HepAD38 cells, in which HBV replication is regulated by tetracycline (tet), were grown as subcutaneous tumours in nude mice. Mice developing viraemia were then left untreated or given tet in the drinking water. In some of the mice given tet, it was removed and the mice were injected intraperitoneally with phosphate buffer saline (PBS), lamivudine (3TC), clevudine (CLV) or tenofovir dipivoxil fumarate (TDF). Virus DNA titres were measured by real-time PCR during and after drug treatment. In water-fed and PBS-injected mice, virus titres reached ~109 copies/ml serum within 35 days of HepAD38 injection, whereas in tet-treated mice, virus titres remained at 104–105 copies/ml. HBV DNA levels were suppressed by 3TC, TDF and CLV, with the latter two drugs showing more sustained virus suppression compared with 3TC. Combination therapy with CLV plus TDF was much more effective than either drug alone in suppressing virus titre for at least 3 weeks after the end of treatment. There was no demonstrable toxicity to HepAD38 cells in drug-treated mice. Hence, a robust tet-controlled system for HBV replication in vivo was demonstrated, validated with monotherapies against HBV and shown to be useful in assessing combination therapy. This system will be useful for preclinical assessment of small amounts of single or multiple compounds against HBV in vivo.
Keywords: HepAD38 cells, hepatitis B virus, mouse model, nucleoside analogues
Introduction
Chronically hepatitis B virus (HBV)-infected persons are at high risk to develop hepatitis, cirrhosis and hepatocellular carcinoma (HCC; Szmuness, 1978; Tiollais et al., 1985). These diseases cause significant morbidity and mortality in many of the estimated 350 million carriers of HBV worldwide (Tiollais et al., 1985). Interferon-α (IFN-α) was the first drug approved for the treatment of hepatitis B, even though it is expensive, only benefits 20–25% of treated carriers, and has serious side effects (Perillo, 1993; Lok, 1994). Pegylated IFN-α2a is longer lasting, requires fewer injections, and has fewer side effects. The introduction of the nucleoside analogue lamivudine ((−)-β−2′,3′-dideoxy-3′-thiacytidine or 3TC; Doong et al., 1991; Chang et al., 1992) resulted in the partial or total clearance of virus from the blood and in the improvement of liver histology in most patients (Hunt et al., 1999; Suzuki et al., 1999). However, prolonged treatment was associated with the appearance of drug-resistant mutants in up to 20% of patients per year (Dienstag et al., 1999; Zoulim et al., 2006). Other drugs, such as adefovir dipivoxil (Gilson et al., 1999; Jacob et al., 2004), entecavir, and telbivudine (Foster et al., 2005; Han, 2005), are also effective against 3TC-resistant mutants, but rebound viraemia occurs after therapy is withdrawn. The telvivudine-related compound clevudine (1-(2-deoxy-2-fluoro-β-l-arabinofuranosyl)-thymidine, CLV or l-FMAU) showed potent in vitro activity against HBV in HepG2.2.15 cells (Chu et al., 1995) and in chronically infected woodchucks (Jacquard et al., 2004). In combination with emtricitabine (5-fluoro-1-(2R,5S)-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine or FTC), CLV showed synergistic antiviral activity in cell culture (Korba, 1996) and in humans (Lim et al., 2006).
Additional compounds under development include emtricitabine (Furman et al., 1992; Paff et al., 1994; Cui et al., 1996), amdoxovir (DAPD; Furman et al., 2001) and tenofovir dipivoxil fumarate (TDF; Butt, 2006), among others (Hoofnagle et al. 2007). 3TC (Mason et al., 1998), FTC (Cullen et al., 1997), CLV (Aguesse-Germon et al., 1998) and other nucleoside analogues (Zoulim et al., 2002) are also active against HBV-related viruses in ducks and woodchucks (Schinazi et al., 1999). Combination treatment of woodchuck hepatitis virus (WHV) infection with CLV plus FTC demonstrated potent and sustained suppression of viraemia compared with either drug alone (Jacquard et al., 2004; Lim et al., 2006), thereby establishing proof-of-principle for combination therapy in the treatment of chronic HBV.
Part of the problem in developing new drugs against HBV is the lack of suitable in vivo models with sustained virus replication and associated liver disease, which could be targeted. Given the narrow range of hosts for HBV, it is impractical to evaluate antiviral drug combinations in HBV-infected chimpanzees, due to their cost and limited availability. As mentioned above, HBV-like viruses exist in ground squirrels, woodchucks and ducks (Summers et al., 1978; Marion et al., 1980; Mason et al., 1980; Schinazi et al., 1999), and these systems have been used to evaluate new drugs against hepadnaviruses (Korba et al., 1996, 2000; Tsiquaye et al., 1996; Nicoll et al., 1998); however, these systems are limited. As these models replicate animal pathogens, HBV-specific therapies cannot be tested. Woodchucks, for example, are expensive, often coinfected with other pathogens that may alter response to therapy and only breed once a year. Alternatively, several laboratories have made HBV-transgenic mice, but such models have not been used extensively for preclinical characterization of lead compounds or for testing combination therapies (Julander et al., 2003; Iyer et al., 2004; Jacob et al., 2004; Anderson et al., 2005; Li et al., 2005). Other laboratories have successfully transplanted human, tupaia (tree shrew) or woodchuck hepatocytes into uPA/SCID or uPA/RAG-2 mice, and showed that these hepatocytes could be successfully infected with HBV or WHV (Dandri et al., 2001, 2005; Meuleman et al., 2005). However, these are difficult systems to work with and have not been widely used. Some years ago, HepAD38 cells were constructed, in which HBV replication was under control of the tetracycline (tet) repressor. In the presence of tet, undetectable levels of virus were observed in cell culture supernatant, whereas in the absence of tet high titres of HBV were observed (Ladner et al., 1997). Subsequent studies have shown the utility of HepAD38 cells for drug discovery and development against HBV (Bijsterbosch et al., 2001; Shi et al., 2003; Ying et al., 2003). In this report, HepAD38 cells transplanted subcutaneously into nude mice resulted in the development of high virus titres in blood. Treatment of these mice with drugs against HBV demonstrated significant antiviral activity in vivo, suggesting this simple small animal model could be used to assess new therapeutic approaches and combination therapies against hepatitis B.
Materials and methods
Cell culture
HepAD38 cells were obtained from ATCC (Rockville, MD, USA) and cultured exactly as described in Ladner et al. (1997). Freshly trypsinized cells were washed twice in phosphate buffer saline (PBS), trypan blue stained and counted. Cultures with >90% viability were used for injections into mice.
Mice
Young adult, outbred nu/nu mice used for this work were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN, USA).
Study design
The protocol presented below is summarized in Figure 1. Each mouse was retro-orbitally bled at day 0 and then injected subcutaneously at a single site with freshly prepared 1×107 HepAD38 cells. By day 21, most tumours were palpable, and mice were bled to assess initial virus DNA titre. Mice with titres of 2–6×105 virus genome equivalents/ml serum were then entered into the drug study. These mice were divided into five groups containing five mice each. In group 1, mice were kept on a normal diet of water and solid food for another 35 days (days 21–56 after injection of HepAD38 cells). In group 2, mice were treated as in group 1 except that tet (2.5 mg/ml final concentration; Sigma Chemical Co, St. Louis, MO, USA) was added to the drinking water for days 21–56. In groups 3–5, tet (2.5 mg/ml) was added to the drinking water for 10 days (days 21–31 following the injection of HepAD38 cells). The mice were then taken off tet and injected intraperitoneally once a day for 6 consecutive days (days 31–36) with PBS (group 3), 3TC (group 4) or TDF (group 5). The dose of 3TC or TDF used was 100 mg/kg per day, which was comparable to those used in the clinic for these drugs (Yurdaydin et al., 2005; Yan et al., 2006). From day 36 to 56, all mice were fed a normal diet. In addition to the prebleed on day 0, mice were bleed on days 21, 31, 36, 41, 51 and 56. At each time point, 10 μl of fresh serum samples were tested for human alanine aminotransferase (ALT) using a commercially available kit (ALT/AST 50, Sigma Chemical Co). HBV DNA titres were determined by real-time PCR (see below). On day 56, all mice were sacrificed by carbon dioxide anesthesia and then cervical dislocation. The tumours were removed, their wet weights determined, and then samples from three different regions were formalin-fixed and paraffin-embedded. The embedded samples were then used for standard hematolylin & eosin (H&E) staining. All mice were housed in a BL-3 facility and treated by trained personnel in accordance with university policy and approved animal protocols.
Figure 1.
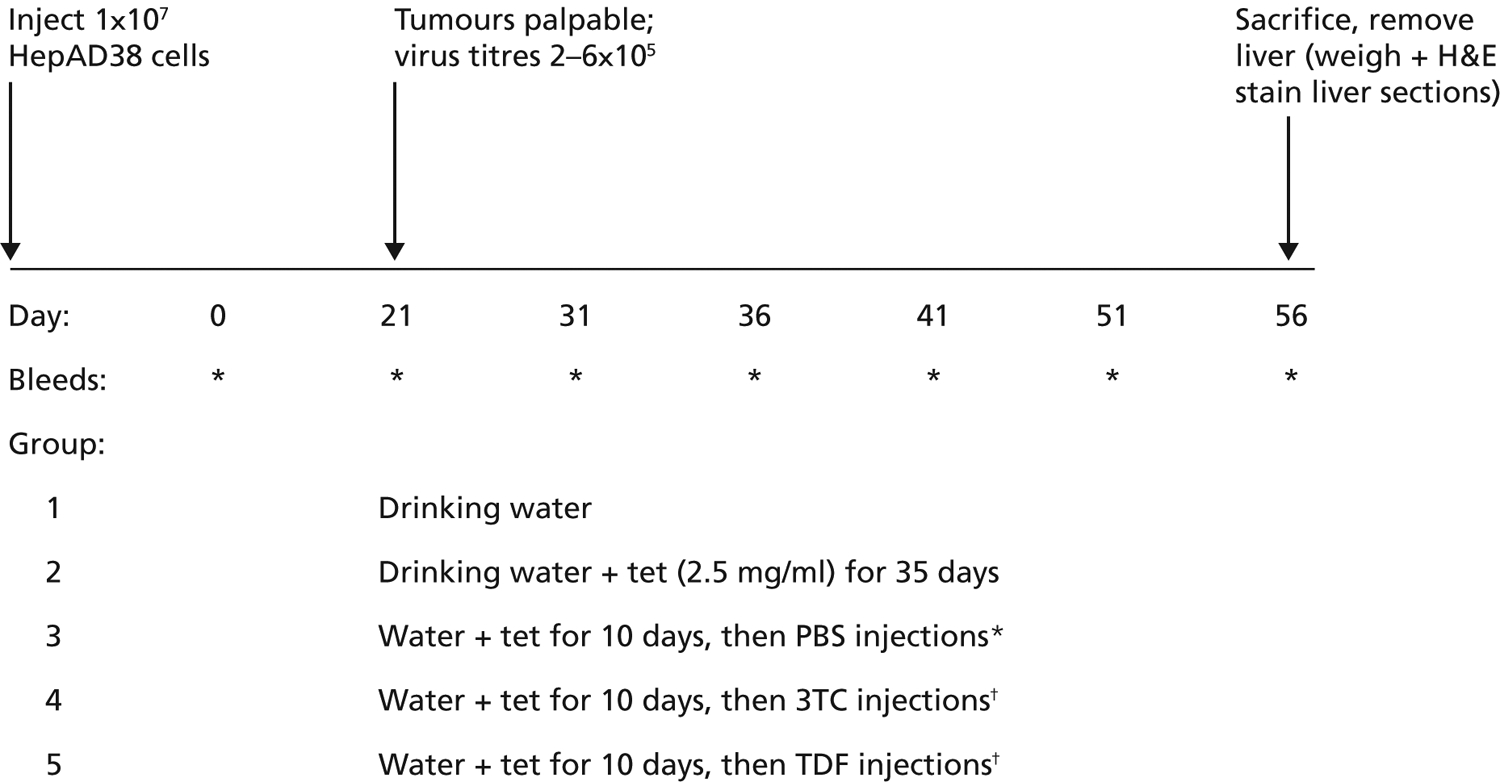
Protocol used for testing drugs in nude mice transplanted with AD38 cells
*Each injection was given daily intraperitoneally from days 31–36. †Each injection was given intraperitoneally at 100 mg/kg per day from days 31–36. H&E, hematoxylin & eosin; PBS, phosphate buffered saline; TDF, tenofovir dipivoxil fumarate; tet, tetracycline; 3TC, lamivudine.
To generate dose-response curves for single or combinations of drugs over time, the protocol outlined above was used, but different groups of mice were injected daily for 6 days (on days 31–36, inclusive) with different drug doses. The drugs used were 3TC, TDF or CLV. Mice in group A were injected with 300 mg/kg per day on each of the 6 consecutive days, group B mice were injected with 100/mg/kg per day, group C mice with 33.3 mg/kg per day, group D mice with 11.1 mg/kg per day, and group E mice with 3.7 mg/kg per day. All mice were then kept on a normal diet until the time of sacrifice (day 56). ALT values were checked as outlined above and in Figure 1. At the time of sacrifice, tumours were removed for weighing, and then formalin-fixed, paraffin-embedded sections were prepared for H&E staining.
Virus DNA determinations
To measure drug efficacy, the levels of HBV DNA in the blood of mice were quantified by real-time PCR as described elsewhere (Abe et al., 1999). Briefly, DNA was extracted from serum using the QIAamp DNA Blood Mini Kit (QIAGEN, Valencia, CA, USA). For analysis, 100 μl of mouse serum was digested with 1 μl of RNase-free DNase I (10–50 units/μl; Roche, Indianapolis, IN, USA) at 37°C for 30 min, and the DNA recovered from a minispin column in 200 μl of eluant. For real-time PCR, 10 μl of eluant containing extracted DNA was added to Ready-To-Go PCR Beads (Amersham Pharmacia Biotech, Piscataway, NJ, USA) in a total volume of 25 μl, which also contained the primers HBSF1 (5′ACATCAGGATTCCTAGGACC3′, spanning nucleotide residues 168–188), HBSR1 (5′GGTGAGTGATTGGAGGTTG3′ spanning residues 341–323) and the TaqMan probe, HBSP1 (5′FAM-CAGAGTCTAGACTCGTGGTGGACTTC-TAMRA3′, spanning residues 247–270; all from Applied Biosystems, Foster City, CA, USA), each at a concentration of 200 nmol/l. These sequences were derived from the plasmid pTKHH2, a head-to-tail dimer of HBV DNA (GenBank accession no. V01460 J02203; Galibert et al., 1979). The reaction mixture also contained manganese acetate at a final concentration of 3.5 mmol/l. PCR was performed in Smart Cycler (Cepheid, Sunnyvale, CA, USA). The protocol consisted of an initial step of 2 min at 95°C, followed by 35 cycles (15 sec at 95°C and 30 sec at 60°C). The final titre was normalized to copies of HBV DNA/ml of serum based on a standard curve generated by known amounts of pTKHH2 (Will et al., 1982) or, independently, by a surface antigen gene template provided by Biotronics, Inc (Lowell, MA, USA).
Statistical analysis
The mean difference in virus titres between groups of treated and control mice at the various time points were determined by Student’s t test, which recognized a significant difference when P<0.05.
Results
Regulation of HBV titre in vivo by tet
The HepAD38 cell line was constructed so that HBV replication was under the control of tet (Ladner et al., 1997). Previous work showed an inverse relationship between the presence of tet and the levels of HBV DNA in the cell culture supernatant (Ladner et al., 1997; Zhou et al., 2006). To determine whether this happens in vivo, the protocol in Figure 1 was used. Accordingly, 1×107 HepAD38 cells were injected subcutaneously into a single site on the backs of nu/nu mice. Most mice developed visible, palpable tumours by about day 21 and, at that time, about 60–70% of the injected mice had HBV DNA titres in the range of 2–6×105 virus genome equivalents/ml of serum. When five mice were fed a normal diet, which included autoclaved water (group 1), the mean virus titre rose to >109, suggesting a >4 log increase in the levels of virus within a 35 day period (Figure 2, curve 1). When another five mice were fed a normal diet that included 2.5 mg/ml of tet, which was consistently present in the drinking water (group 2), mean virus titres decreased slightly over the same time period (Figure 2, curve 2). Groups 1 and 2 differed by a mean of almost 1.2 logs by day 36 (P<0.01), 2.1 logs by day 41 (P<0.005), 2.6 logs by day 46 (P<0.002), nearly 4 logs by day 51 (P<0.001) and >4.5 logs on day 56 (P<0.001). These results indicate that a transplantable small animal model could make high titres of virus within a few weeks of injection and that virus levels could be regulated by tet in the drinking water.
Figure 2.
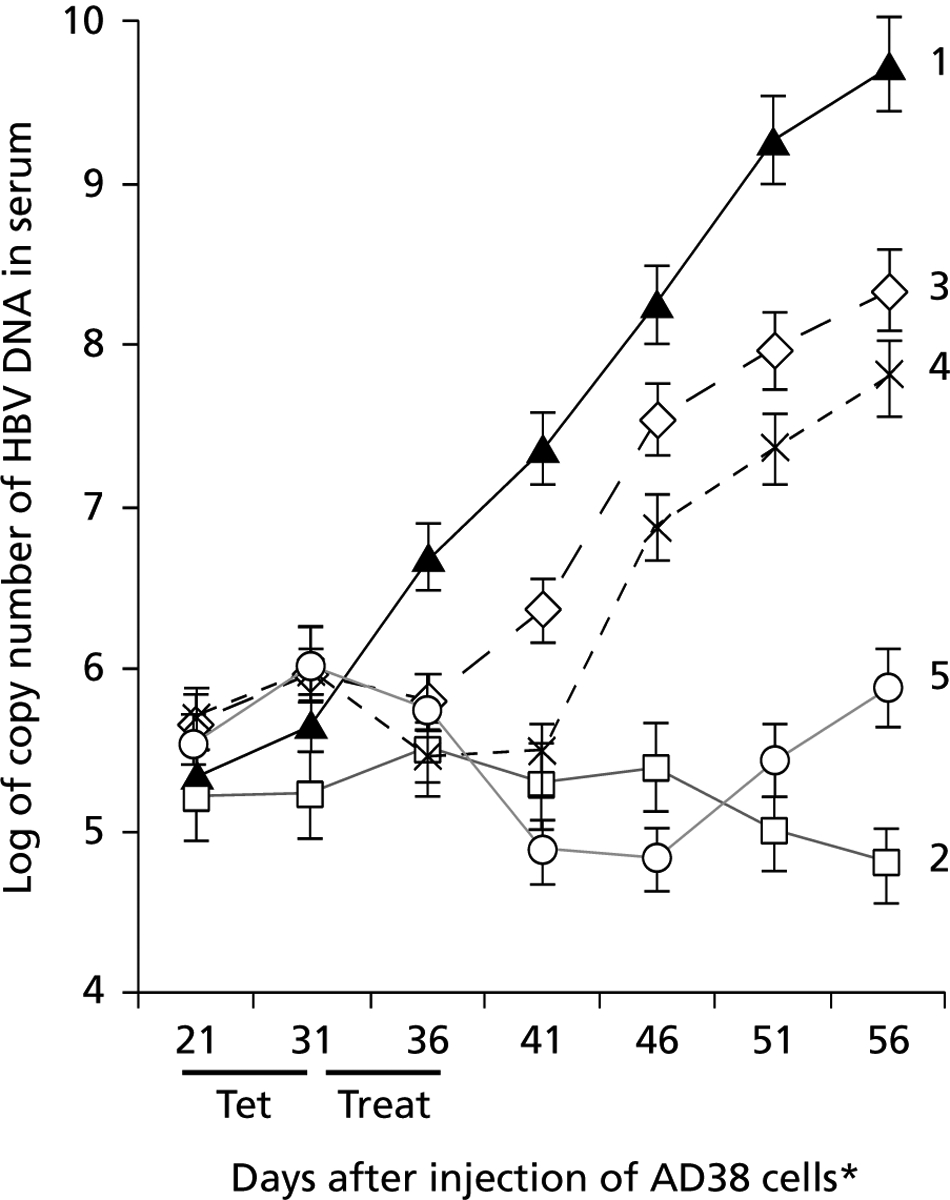
HBV DNA titres (assayed by real-time PCR) in serum samples from nude mice injected subcutaneously with AD38 cells
Tumours and virus in the blood were detected by day 21 after injection of AD38 cells. Mice were on drinking water for 35 days (curve 1), on tetracycline (tet; 2.5 mg/ml) for 35 days (curve 2), or on tet for 10 days (in their drinking water for days 21–31), and then injected daily for 6 days with phosphate buffered saline (curve 3), lamivudine (3TC; curve 4), or tenofovir dipivoxil fumarate (TDF; curve 5). 3TC and TDF were used at doses of 100 mg/kg per day. Each point is the mean titre of HBV DNA from five mice in each group. *Day 21 is the first day of treatment. Treat, treatment period.
To see whether the effects of tet were reversible, another five mice were treated with tet for 10 days (days 21–31), injected daily for a further 6 days (days 31–36) with PBS (group 3), and then observed for 20 days (day 56). By day 41, which was 10 days after the end of tet treatment, there was a log-fold increase in virus titre in group 3 mice compared with group 2 mice (P<0.01). This difference increased to >2 logs by day 46 (P<0.004), to almost 3 logs by day 51 (P<0.002) and to 3.3 logs by day 56 (P<0.001). Interestingly, the slope of the curve for virus titre in group 3 mice was similar to that in group 1 mice, suggesting that once the inhibitory effects of tet wore off, virus titres increased at nearly the same rate as mice that never had tet in their drinking water (Figure 2, curves 1 and 3). These combined results suggest that virus production was under the control of tet in vivo depending upon whether tet was added to the drinking water. This was similar to that observed earlier in cell culture (Ladner et al., 1997).
Validation of the model with nucleoside analogues
Studies were also conducted to assess the efficacy of 3TC and TDF against HBV replication in vivo. A group of mice was treated with tet for 10 days (days 21–31) and then injected daily for 6 days with 3TC (days 31–36; Figure 1, group 4), which resulted in virus suppression until day 41 (Figure 2, curve 4). Rebound virus titre among group 4 mice was 1.5 logs (P<0.01), 2.4 logs (P<0.004) and 3 logs (P<0.002) higher than group 2 mice on day 46, 51 and 56, respectively. Finally, when TDF was substituted for 3TC in a separate set of mice (Figure 1, group 5), sustained virus suppression was observed for at least 10 days after the end of therapy (day 46; Figure 2, curve 5). In TDF-treated compared with PBS-treated mice (groups 5 and 3, respectively), TDF suppressed virus by about 1.5 logs on day 41 (P<0.01), by approximately 2.7 logs on day 46 (P<0.002), by 2.5 logs on day 51 (P<0.002) and 2.4 logs on day 56 (P<0.002). Hence, the antiviral effects of TDF were sustained compared with those of 3TC evaluated under the same conditions.
Additional experiments using the protocol above were conducted in the absence of tet, which means that the mice had higher virus loads at day 31 when 3TC or TDF treatments were initiated. Here, too, the virus titres were depressed during nucleoside analogue treatment, although the mean titres were 3–5× higher in these mice during and after treatment compared with the corresponding curves in Figure 2 for each drug (data not shown). Hence, nucleoside analogues known to be effective against HBV in human infections are also effective against HBV replication in this small animal model.
Further work was conducted to determine whether the decreased virus titres were associated with alterations in tumour growth or toxicity. At 56 days after injection of HepAD38 cells, all mice were sacrificed. The tumour in each animal was excised and wet weights determined. As shown in Figure 3A, there were no significant differences in the mean tumour size among mice in any of the groups, indicating that the drugs did not reduce virus titre by inhibiting HepAD38 growth. In addition, when human ALT values were determined in serum samples from each mouse, no increases >1.5 fold were noted, suggesting that the drugs were not toxic. The ALT levels at day 56 (Figure 3B) were about the same as in the earlier time points for each animal (data not shown). Toxicity to HepG2 cells may have otherwise resulted in the release of intracellular virus DNA, which would have been detected by PCR. H&E staining of tumour tissue from each animal did not show any signs of necrosis (data not shown), confirming that the compounds were not toxic as used. Furthermore, there were no changes in water consumption in any of the mice, and no significant changes in mouse weight or behaviour during treatment, again suggesting that these compounds were non-toxic and implying that the decreased virus titres were likely to be due to the antiviral activity of the nucleoside analogues. These results validate the use of this small animal model as a way to assess antiviral activities of drug candidates.
Figure 3.
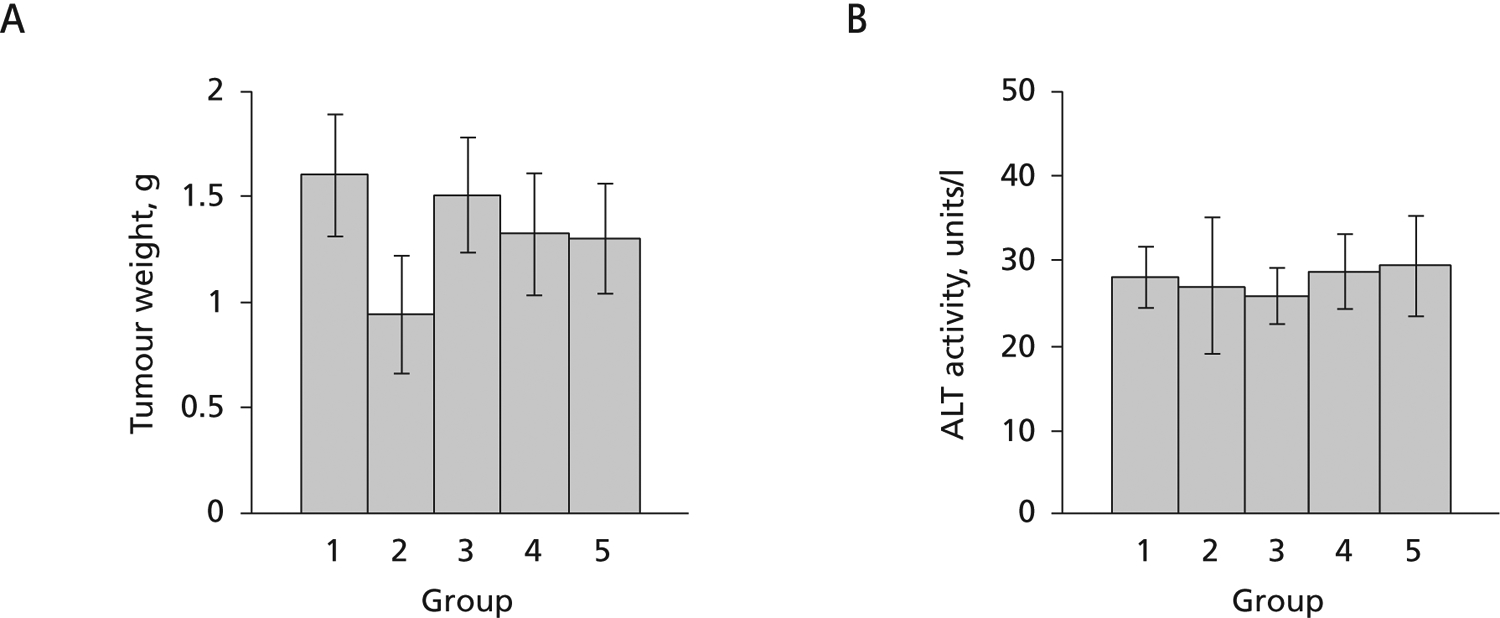
Tumour weights and ALT levels from nude mice injected subcutaneously with AD38 cells at day 56
(A) Tumour weights. (B) Alanine aminotransferase (ALT). From the left, mice were treated with water only (group 1), water plus tetracycline (tet; 2.5 mg/ml) for 35 days (group 2), or water plus tet for 10 days, then phosphate buffered saline (group 3), lamivudine (3TC; group 4) or tenofovir dipivoxil fumarate (TDF; group 5). 3TC and TDF were used at doses of 100 mg/kg per day.
Dose–response curves for 3TC, TDF and CLV in vivo
To use this model for assessing the efficacy of combination therapy, dose-response curves over time were generated with 3TC, TDF and CLV, using the protocol shown in Figure 1. In all cases, the concentration ranges tested were within those used in the clinic (Kuo et al., 2004; Hui & Lau, 2005; Yurdaydin et al., 2005; Korba et al., 2006; Yan et al., 2006). The results of the dose-response curves for each of these drugs, presented in Figures 4A–C, demonstrated an inverse relationship between drug dose and the levels of virus DNA in the blood. At high doses of CLV (300 and 100 mg/kg per day), virus DNA titres remained repressed for at least 10 days after the end of drug treatment. Compared with the dose-response curve for the least amount of drug (3.7 mg/kg per day), the two highest drug doses suppressed virus by ≥1.3 logs on day 41 (P<0.01) and by >2 logs on day 46 (P<0.005). After that time, virus rebound was observed at all doses of CLV used (Figure 4A). In comparison, all doses of 3TC suppressed virus during the treatment period (Figure 4B). At day 41, suppression of approximately 1 log was still observed with the three highest doses of 3TC compared with the lowest dose (P<0.01), but this suppression was sustained through to day 46 only among mice treated with the highest dose of 3TC (P<0.01). In contrast, dose-response curves for TDF resembled those of CLV: there was a sustained 1–1.5 log suppression of virus titre among mice treated with the three highest doses of drug (Figure 4C; P<0.01). Comparing the dose-response curves of these three drugs with virus titres in mice fed diets containing water or water plus tet for days 21–31 (Figure 4D) indicate that the highest doses of these drugs suppress virus replication during treatment and for 6–10 days after the end of treatment by ≥2 logs (P<0.005). Hence, three different nucleoside analogues that are effective against HBV and suppress virus titre in this model also demonstrate a dose-dependent suppression of virus titre up to 10 days after the end of treatment. The absence of increased ALT throughout the observation period, combined with the fact that these drugs do not impact upon tumour growth, further suggests that the differences seen were not due to toxicity, but to the antiviral activities of these compounds.
Figure 4.
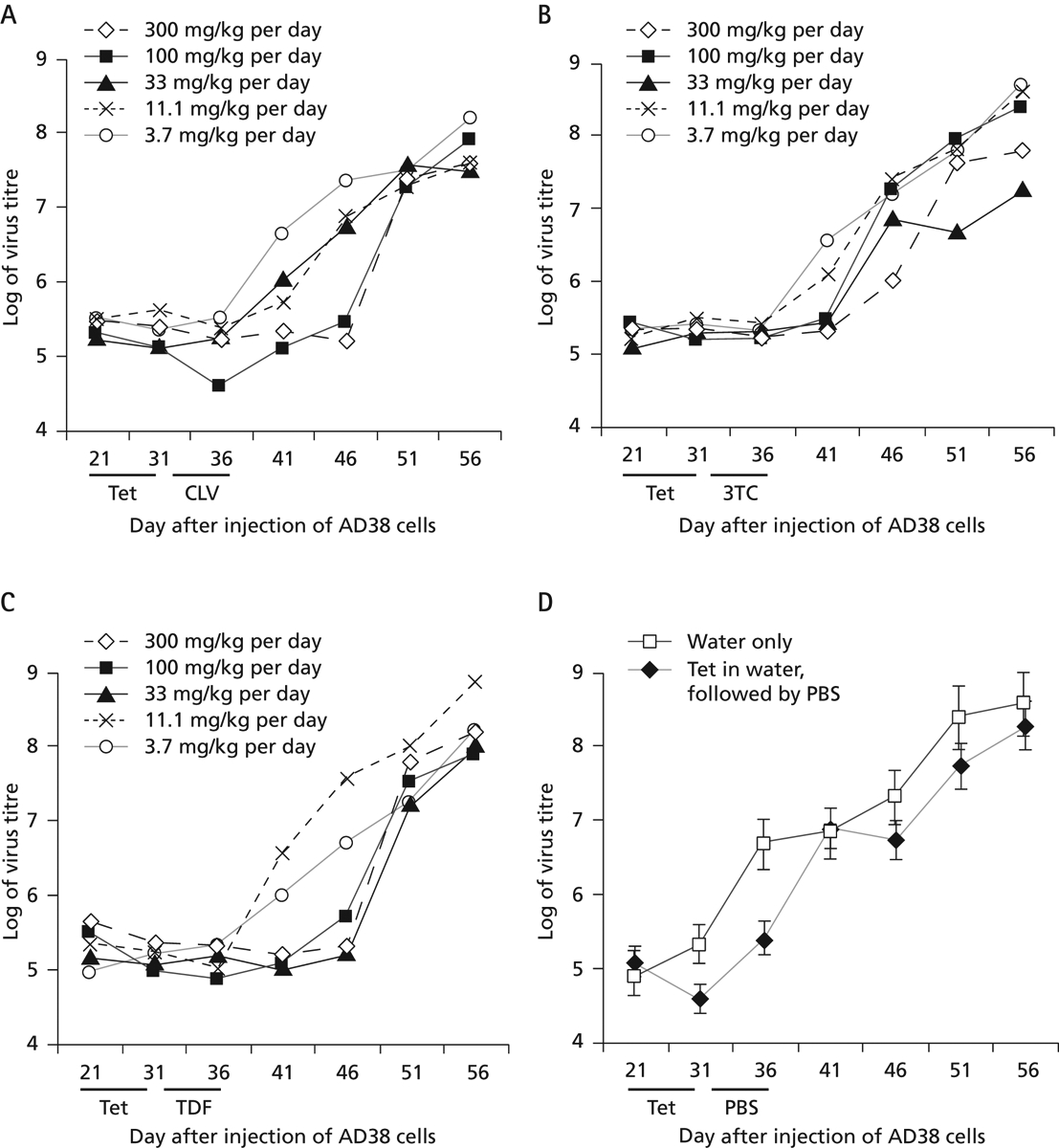
Dose–response curves for CLV-, 3TC- and TDF-treated mice and for control mice
(A) Clevudine (CLV), (B) lamivudine (3TC), (C) tenofovir dipivoxil fumarate (TDF) and (D) control. In (D), mice were fed with water only or with tetracycline (tet) in water, followed by phosphate buffered saline (PBS). Each point represents the average value from five mice in each group. In panels A–C, the average values shown varied by no more than ±6%.
Combination therapy with TDF plus CLV
With the recent availability of multiple reverse transcriptase inhibitors in the clinic, opportunities now exist for the development of combination therapies that are highly efficacious against chronic HBV infection, but do not give rise to drug resistance, even after many years of treatment. Hence, experiments were designed to ask whether CLV and TDF work together in vivo in an additive or synergistic way. Accordingly, the protocol in Figure 1 was used to evaluate the dose-response curves of CLV, TDF, and the combination of these drugs (Figure 5). The molar ratio of the two drugs remained constant (1:1) for each group of mice, although the dosages used for treatment were diluted threefold in each panel. For example, in panel A, each of the five mice were fed tet for 10 days (days 21–31), given 300 mg/kg per day of each drug separately or in combination therapy for days 31–36 inclusive, and then the mice were followed for rebound for another 20 days. In panel B, the mice were given a dose of 100 mg/kg per day in the same protocol, and so on, with lower doses in panels C–E.
Figure 5.
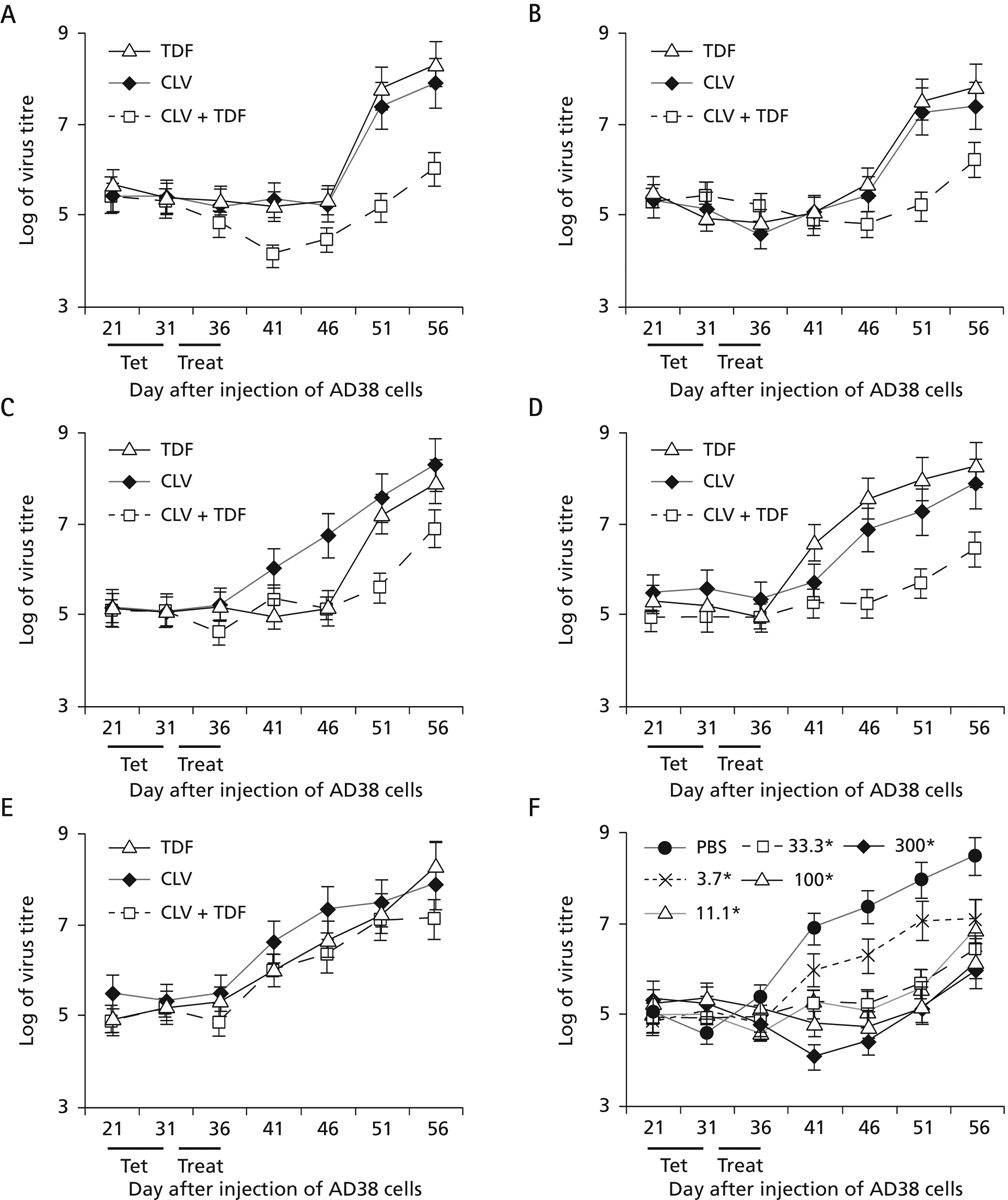
Treatment of mice with different doses of TDF, CLV or with TDF + CLV combination therapy
The protocol outlined in Figure 1 was followed. Five mice per group were treated with tenofovir dipivoxil fumarate (TDF), clevudine (CLV) or with TDF + CLV combination therapy. Doses were administered at (A) 300 mg/kg per day, (B) 100 mg/kg per day, (C) 33.3 mg/kg per day, (D) 11.1 mg/kg per day or (E) 3.7 mg/kg per day for 6 consecutive days (days 31–36). Mice were then bled over the next 30 days as indicated. (F) Curves reflecting virus DNA titres in serum resulting from different concentrations of combination therapy shown in panels A–E were compared with each other and to phosphate buffered saline (PBS)-fed mice. *All concentrations in F are measured as mg/kg per day. Treat, treatment period.
In Figure 5A–E, virus titres were very similar during days 21–31 when the mice were fed tet, suggesting that tet was inhibiting the accumulation of virus in blood (compared with the ‘PBS’ curve in panel F), even though the tumours were growing. At the highest dose of drugs (panel A), there was a further decrease in virus titre with combination therapy compared with monotherapy (see day 41). After day 36, mice were no longer treated with drugs. Those previously treated with CLV or TDF after day 46 had a strong rebound, whereas combination CLV plus TDF therapy demonstrated a much weaker rebound, with virus levels 2.2–2.6 logs below the monotherapy values on days 51 and 56 (panel A; P<0.004). In panel B, combination therapy continued to suppress virus on days 51 and 56 by 1.6–2.3 logs compared to monotherapy with either drug (P<0.01). In panel C, virus remained suppressed in combination therapy 1.4–2.0 logs below that achieved with either drug alone on days 51 and 56 (P<0.01). Similar results were obtained in panel D, where virus levels in mice treated with combination therapy were 1.5–2.0 logs below that achieved with either drug alone (P<0.01). Only at the lowest dose of drugs tested were there no differences in the virus titres among mice treated with one drug compared with mice treated with combination therapy (panel E). In panel F, the curves for combination therapies at each dose (from panels A–E) were plotted and compared to the curve of virus titre produced by the placebo treatment (the ‘PBS’ curve). Note that the curves for combination therapies at doses ranging from 300 down to 11.1 mg/kg per day were similar (they had overlapping error bars) and that only the curve for the lowest dose (3.7 mg/kg per day) showed significant rebound as soon as treatment ended. In other words, combination therapy at 11.1 mg/kg per day (for each drug; panel D) suppressed virus by nearly 2 logs compared with monotherapy at concentrations up to 27-fold higher (as in panel A). This may not be so surprising, as TDF is a member of the acyclic phosphates, whereas CLV is an l-nucleoside analogue; therefore, both drugs should inhibit HBV replication by complementary mechanisms. Hence, two potent compounds working together inhibit virus at significantly lower concentrations than either drug used alone.
Discussion
This study demonstrates that a small animal model was capable of assessing the antiviral efficacy of compounds known to be active against HBV. This was determined by the extent to which virus DNA suppression was observed in drug-treated compared with PBS-treated mice, and by the length of time virus DNA levels remained suppressed after the end of therapy (Figure 2). These data provide a system to evaluate combination therapies with different nucleoside analogues (Figure 5) and to evaluate their efficacy with compounds directed against other targets within the virus or host cell that support virus replication. For example, compounds that target hepatitis B core antigen synthesis and/or nucleocapsid assembly would block encapsidation of pregenomic RNA during virus replication (Lin et al., 2001) and could be assayed in vivo with the model described herein. In addition, there is increasing evidence that the hepatitis B X antigen stimulates virus gene expression and replication (Bouchard et al., 2001), correlates with the pathogenesis of chronic liver disease (Jin et al., 2001), and contributes centrally to the development of HCC (Feitelson et al., 2005), suggesting that any lead compounds against X antigen could be further characterized in this system.
This mouse model has the following advantages: it requires relatively small amounts of drug for in vivo assessment, it can be used for the preclinical evaluation of HBV-specific therapies (that cannot be tested against HBV-like viruses in other animals), it will provide rapid information on rebound viraemia and is cost effective. The model is also simple to construct with HepAD38 cells in hand, as it does not require the time or expense of breeding woodchucks or transgenic mice. The model could also be used to assess drug toxicity in the transplanted tumour, the liver, or other organs (Figure 3), and to conduct limited pharmacokinetic studies with new drug candidates. Although this could also be done in transgenic mice, infected woodchucks and chimps, larger numbers of HepAD38-transplanted nude mice could be used to increase the statistical power of the studies performed. Furthermore, the recent finding that HBeAg may be a surrogate marker for covalently closed circular (ccc) DNA levels in HepAD38 cells (Zhou et al., 2006) may extend the utility of this model in evaluating compounds that may target cccDNA in the viral minichromosome. Although cccDNA is detectable in ducks and woodchucks infected with their respective viruses (Zhu et al., 2001; Zhang et al., 2003), the model herein represents the first time that compounds could be evaluated against HBV-associated cccDNA. In addition, the recent construction of HepG2 cell lines stably replicating selected drug-resistant mutants of HBV, and/or mutants that arise during chronic infection (Ladner et al., 1998; Ying et al., 2000), provide opportunities to evaluate additional compounds that may be effective against these mutants in vitro and to further evaluate their effectiveness in vivo. Single or multiple compounds with strong antiviral efficacy and low toxicity will be prime candidates for further assessment against chronic liver disease in the HBV-transgenic SCID mouse model (Larkin et al., 1999). Hence, the model described herein is a robust system that fills an important gap in the development of HBV-specific therapeutic approaches that bridge compound discovery and characterization in tissue culture with only the best candidates going for human clinical trials.
Acknowledgements
This work was supported by NIH grants CA48656, CA66971 and CA104025 to MAF; by NIH grants 5R37-AI-41980 and 5R37-AI-25899; support from the Emory Center for AIDS Research 5P30-AI-50409; and from the US Department of Veterans Affairs (to RFS). A portion of this work was presented at the HepDART 2005 meeting, held on the Kohala Coast of the Big Island of Hawaii on December 11–15, 2005.
References
- Abe A, Inoue K, Tanaka T, Kato J, Kajiyama N, Kawaguchi R, Tanaka S, Yoshiba M & Kohara M (1999) Quantitation of hepatitis B virus genomic DNA by real-time detection PCR. Journal of Clinical Microbiology 37:2899–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguesse-Germon S, Liu SH, Chevallier M, Pichoud C, Jamard C, Borel C, Chu CK, Trepo C, Cheng YC & Zoulim F (1998) Inhibitory effect of 2′-fluoro-5-methyl-β-l-arabinofuranosyluracil on duck hepatitis B virus replication. Antimicrobial Agents & Chemotherapy 42:369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AL, Banks KE, Pontoglio M, Yaniv M & McLachlan A (2005) Alpha/beta interferon differentially modulates the clearance of cytoplasmic encapsidated replication intermediates and nuclear covalently closed circular hepatitis B virus (HBV) DNA from the livers of hepatocyte nuclear factor 1alpha-null HBV transgenic mice. Journal of Virology 79:11045–11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijsterbosch MK, Ying C, de Vrueh RL, de Clercq E, Biessen EA, Neyts J & van Berkel TJ (2001) Carrier-mediated delivery improves the efficacy of 9-(2-phosphonylmethoxyethyl)adenine against hepatitis B virus. Molecular Pharmacology 60:521–527. [PubMed] [Google Scholar]
- Bouchard MJ, Wang LH & Schneider RJ (2001) Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 294:2376–2378. [DOI] [PubMed] [Google Scholar]
- Butt AA (2006) Tenofovir for chronic hepatitis B virus infection in HIV-coinfected patients. The AIDS Reader 16:219–222. [PubMed] [Google Scholar]
- Chang CN, Doong S, Zhou J, Beach J, Jeong L, Chu C, Tsai C, Cheng Y, Liotta D & Schinazi RF (1992) Deoxycytidine deaminase-resistant stereoisomer is the active form of (+/−)2′,3′-dideoxy 3′-thiacytidine in the inhibition of hepatitis B virus replication. Journal of Biological Chemisry 267:13938–13942. [PubMed] [Google Scholar]
- Chu CK, Boudinot FD, Peek SF, Hong JH, Choi Y, Korba BE, Gerin JL, Cote PJ, Tennant BC & Cheng YC (1998). Preclinical investigation of L-FMAU as an anti-hepatitis B virus agent. Antiviral Therapy 3 Suppl 3:113–121. [PubMed] [Google Scholar]
- Chu CK, Ma T, Shanmuganathan K, Wang C, Xiang Y, Pai SB, Yao GQ, Sommadossi JP & Cheng YC (1995) Use of 2′-fluoro-5-methyl-β-l-arabinofuranosyluracil as a novel antiviral agent for hepatitis B virus and Epstein-Barr virus. Antimicrobial Agents & Chemotherapy 39:979–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Schinazi RF, Gosselin G, Imbach JL, Chu CK, Rando RF, Revankar GR & Sommadossi JP (1996) Effect of beta-enantiomeric and racemic nucleoside analogues on mitochrondrial functions in HepG2 cells. Implications for predicting drug hepatotoxicity. Biochemical Pharmacology 52:1577–1584. [DOI] [PubMed] [Google Scholar]
- Cullen JM, Smith SL, Davis MG, Dunn SE, Botteron C, Cecchi A, Linsey D, Linzey D, Frick L, Paff MT, Goulding A & Biron K (1997) In vivo antiviral activity and pharmacokinetics of (−)-cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine in woodchuck hepatitis virus infected woodchucks. Antimicrobial Agents & Chemotherapy 41:2076–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandri M, Burda MR, Gocht A, Torok E, Pollok JM, Rogler CE, Will H & Petersen J (2001) Woodchuck hepatocytes remain permissive for hepadnavirus infection and mouse liver repopulation after cryopreservation. Hepatology 34:824–833. [DOI] [PubMed] [Google Scholar]
- Dandri M, Burda MR, Zuckerman DM, Wursthorn K, Matschl U, Pollok JM, Rogiers X, Gocht A, Kock J, Blum HE, von Weizsacker F & Petersen J (2005) Chronic infection with hepatitis B viruses and antiviral drug evaluation in uPA mice after liver repopulation with tupaia hepatocytes. Journal of Hepatology 42:54–60. [DOI] [PubMed] [Google Scholar]
- Dienstag JL, Schiff ER, Mitchell M, Casey DE Jr, Gitlin N, Lissoos T, Gelb LD, Condreay L, Crowther L, Rubin M & Brown N (1999) Extended lamivudine retreatment for chronic hepatitis B: maintenance of viral suppression after discontinuation of therapy. Hepatology 30:1082–1087. [DOI] [PubMed] [Google Scholar]
- Doong S, Tsai C, Schinazi RF, Liotta D & Cheng Y (1991) Inhibition of the replication of hepatitis B virus in vitro by 2′,3′-dideoxy-3′thiacytidine and related analogues. Proceedings of the National Academy of Sciences U S A 88:8495–8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feitelson MA, Reis H, Liu J, Lian Z & Pan J (2005) Hepatitis B virus X antigen (HBxAg) and cell cycle control in chronic infection and hepatocarcinogenesis In Frontiers in Bioscience. Viral Infection and Cell Cycle Control; pp.1558–1572. Edited by Zhao R. Alberton, NY: Frontiers in Bioscience. [DOI] [PubMed] [Google Scholar]
- Foster WK, Miller DS, Scougall CA, Kotlarski I, Colonno RJ & Jilbert AR (2005) Effect of antiviral treatment with entecavir on age- and dose-related outcomes of duck hepatitis B virus infection. Journal of Virology 79:5819–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman PA, Davis M, Liotta DC, Paff M, Frick LW, Nelson DJ, Dornsife RE, Wurster JA, Wilson LJ, Fyfe JA, Tuttle JV, Condreay L, Averett DR, Schinazi RF, & Painter GR (1992) The anti-hepatitis B virus activities, cytotoxicities, and anabolic profiles of the (−) and (+) enantiomers of cis-5-fluoro-1-(2-[hydroxymethyl]-1,3-oxythiolan-5-yl)-cytosine (FTC). Antimicrobial Agents & Chemotherapy 36:2686–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman PA, Jeffrey J, Kieffer L, Feng JY, Anderson KS, Borroto-Esoda K, Hill E, Copeland WC, Chu CK, Sommadossi JP, Liberman I, Schinazi RF & Painter GR (2001) Mechanism of action of 1-β-d-2,6-diaminopurine dioxolane, a prodrug of the human immunodeficiency virus type 1 inhibitor 1-β-d-dioxolane guanosine. Antimicrobial Agents & Chemotherapy 45:158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galibert F, Mandart E, Fitoussi F, Tiollais P & Charnay P (1979) Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281:646–650. [DOI] [PubMed] [Google Scholar]
- Gilson RJC, Chopra KB, Newell AM, Murray-Lyon IM, Nelson MR, Rice SJ, Tedder RS, Toole J, Jaffe HS & Weller IVD (1999) A placebo-controlled phase I/II study of adefovir dipivoxil in patients with chronic hepatitis B virus infection. Journal of Viral Hepatitis 6:387–395. [DOI] [PubMed] [Google Scholar]
- Han SH (2005) Telbivudine: a new nucleoside analogue for the treatment of chronic hepatitis B. Expert Opinion on Investigational Drugs 14:511–519. [DOI] [PubMed] [Google Scholar]
- Hoofnagle JH, Doo E, Liang TJ, Fleischer R, & Lok AS (2007) Management of hepatitis B: summary of a clinical research workshop. Hepatology 45:1056–1075. [DOI] [PubMed] [Google Scholar]
- Hui CK & Lau GK (2005) Clevudine for the treatment of chronic hepatitis B virus infection. Expert Opinion on Investigational Drugs 14:1277–1284. [DOI] [PubMed] [Google Scholar]
- Hunt CM, Brown NA & Rubin M (1999) Lamivudine therapy of chronic hepatitis B. Advances in Experimental Medicine and Biology 458:11–21. [DOI] [PubMed] [Google Scholar]
- Iyer RP, Roland A, Jin Y, Mounir S, Korba B, Julander JG & Morrey JD (2004) Anti-hepatitis B virus activity of ORI-9020, a novel phosphorothioate dinucleotide, in a transgenic mouse model. Antimicrobial Agents & Chemotherapy 48:2318–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob JR, Korba BE, Cote PJ, Toshkov I, Delaney WE 4th, Gerin JL & Tennant BC (2004) Suppression of lamivudine-resistant B-domain mutants by adefovir dipivoxil in the woodchuck hepatitis virus model. Antiviral Research 63:115–121. [DOI] [PubMed] [Google Scholar]
- Jacquard AC, Nassal N, Pichoud C, Ren S, Schultz U, Guerret S, Chevallier M, Werle B, Peyrol S, Jamard C, Rimsky LT, Trepo C & Zoulim F (2004) Effect of a combination of clevudine and emtricitabine and adenovirus-mediated delivery of gamma interferon in the woodchuck model of hepatitis B virus infections. Antimicrobial Agents & Chemotherapy 48:2683–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YM, Yun C, Park C, Wang HJ & Cho H (2001) Expression of hepatitis B virus X protein is closely correlated with the high periportal inflammatory activity of liver diseases. Journal of Viral Hepatitis 8:322–330. [DOI] [PubMed] [Google Scholar]
- Julander JG, Colonno RJ, Sidwell RW & Morrey JD (2003) Characterization of antiviral activity of entecavir in transgenic mice expressing hepatitis B virus. Antiviral Research 59:155–161. [DOI] [PubMed] [Google Scholar]
- Korba BE (1996) In vitro evaluation of combination therapies against hepatitis B virus replication. Antiviral Research 29:49–51. [DOI] [PubMed] [Google Scholar]
- Korba BE, Cote P, Hornbuckle W, Schinazi R, Gerin JL & Tennant BC (2000) Enhanced antiviral benefit of combination therapy with lamivudine and famciclovir against WHV replication in chronic WHV carrier woodchucks. Antiviral Research 45:19–32. [DOI] [PubMed] [Google Scholar]
- Korba BE. Furman PA. & Otto MJ (2006) Clevudine: a potent inhibitor of hepatitis B virus in vitro and in vivo. Expert Review of Antiinfective Therapy 4:549–561. [DOI] [PubMed] [Google Scholar]
- Korba BE, Xie H, Wright KN, Hornbuckle WE, Gerin JL, Tennant BC & Hostetler KY (1996) Liver targeted antiviral nucleosides: enhanced antiviral activity of phosphatidyl-dideoxyguanosine versus dideoxyguanosine in woodchuck hepatitis in vivo. Hepatology 23:958–963. [DOI] [PubMed] [Google Scholar]
- Kuo A, Dienstag JL & Chung RT (2004) Tenofovir disoproxil fumarate for the treatment of lamivudine-resistant hepatitis B. Clinical Gastroenterology & Hepatology 2:266–272. [DOI] [PubMed] [Google Scholar]
- Ladner SK, Miller TJ, Otto MJ & King RW. (1998) The hepatitis B virus M539V polymerase variation responsible for 3TC resistance also confers cross-resistance to other nucleoside analogues. Antiviral Chemistry & Chemotherapy 9:65–72. [PubMed] [Google Scholar]
- Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C & King RW (1997) Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrobial Agents & Chemotherapy 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Clayton MM, Sun B, Perchonock CE, Morgan JL, Siracusa LD, Michaels F & Feitelson MA (1999) Hepatitis B virus transgenic SCID mouse model of chronic liver disease. Nature Medicine 5:907–912. [DOI] [PubMed] [Google Scholar]
- Li D, Xu DZ, Choi BC, Men K, Zhang JX, Lei XY & Yan YP (2005) Preliminary study on the efficacy and safety of lamivudine and interferon alpha therapy in decreasing serum HBV DNA level in HBV positive transgenic mice during pregnancy. Journal of Medical Virology 76:203–207. [DOI] [PubMed] [Google Scholar]
- Lim SG, Krastev Z, Ng TM, Mechkov G, Kotzev IA, Chan S, Mondou E, Snow A, Sorbel J & Rousseau F for the FTCB-204 Study Group (2006) Randomized, double-blinded study of emtricitabine (FTC) plus clevudine versus FTC alone in treatment of chronic hepatitis B. Antimicrobial Agents & Chemotherapy 50:1642–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Song Y, Kong X, Xie N, Wu X, Liu N, Wang N, Cao E & Jin Y (2001) Anti-viral activity of hairpin ribozyme directed against HBV core region in vitro. Journal of Tongji Medical University 21:219–221. [DOI] [PubMed] [Google Scholar]
- Lok ASF (1994) Treatment of chronic viral hepatitis. Journal of Viral Hepatitis 1:105–124. [DOI] [PubMed] [Google Scholar]
- Marion PL, Oshiro LS, Regnery DC, Scullard GH & Robinson WS (1980) A virus in Beechey ground squirrels that is related to hepatitis B virus of humans. Proceedings of the National Academy of Sciences U S A 77:2941–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason WS, Cullen J, Moraleda G, Saputelli J, Aldrich CE, Miller DS, Tennant B, Frick L, Averett D, Condreay LD & Jilbert AR (1998) Lamivudine therapy of WHV-infected woodchucks. Virology 245:18–32. [DOI] [PubMed] [Google Scholar]
- Mason WS, Seal G & Summers J (1980) Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. Journal of Virology 36:829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuleman P, Libbrecht L, De Vos R, de Hemptinne B, Gevaert K, Vandekerckhove J. Roskams T & Leroux-Roels G (2005) Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 41:847–856. [DOI] [PubMed] [Google Scholar]
- Nicoll A, Colledge D, Toole J, Angus P, Smallwood R & Locarnini S (1998) Inhibition of duck hepatitis B virus replication by 9[2-phosphonylmethoxyethyl]adenine, an acyclic phosphonate nucleoside analogue. Antimicrobial Agents & Chemotherapy 42:3130–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paff MT, Averett DR, Prus KL, Miller WH & Nelson DJ (1994) Intracellular metabolism of (−) and (+)-cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine in HepG2 derivative 2.2.15 (subclone P5A) cells. Antimicrobial Agents & Chemotherapy 38:1230–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrillo RP (1993) Antiviral therapy of chronic hepatitis B: past, present, and future. Journal of Hepatology 17 Suppl 3:S56–S63. [DOI] [PubMed] [Google Scholar]
- Schinazi RF, Ilan E, Black PL, Yao X & Dagan S (1999) Cell-based and animal models for hepatitis B and C viruses. Antiviral Chemistry & Chemotherapy 10:99–114. [DOI] [PubMed] [Google Scholar]
- Shi J, Mathew JS, Tharnish PM, Rachakonda S, Pai SB, Adams M, Grier JP, Gallagher K, Zhang H, Wu JT, Shi G, Geleziunas R, Erickson-Viitanen S, Stuyver L, Otto MJ, Watanabe KA & Schinazi RF (2003) N4-acyl-modified D-2′,3′-dideoxy-5-fluorocytidine nucleoside analogues with improved antiviral activity. Antiviral Chemistry & Chemotherapy 14:81–90. [DOI] [PubMed] [Google Scholar]
- Summers J, Smolec JM & Snyder R (1978) A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks. Proceedings of the National Academy of Sciences U S A 75:4533–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Kumada H, Ikeda K, Chayama K, Arase Y, Saitoh Y, Tsubota A, Kobayashi M, Koike M, Ogawa N & Tanikawa K (1999) Histological changes in liver biopsies after one year of lamivudine treatment in patients with chronic hepatitis B infection. Journal of Hepatology 30:743–748. [DOI] [PubMed] [Google Scholar]
- Szmuness W (1978) Hepatocellular carcinoma and hepatitis B virus: Evidence for a causal association. Progress in Medical Virology 24:40–69. [PubMed] [Google Scholar]
- Tiollais P, Pourcel C & Dejean A (1985) The hepatitis B virus. Nature 317:489–495. [DOI] [PubMed] [Google Scholar]
- Tsiquaye KN, Sutton D, Maung M & Boyd MR (1996) Antiviral activities and pharmacokinetics of penciclovir and famciclovir in Pekin ducks chroncally infected with duck hepatitis B virus. Antiviral Chemistry & Chemotherapy 7:153–159. [Google Scholar]
- Will H, Cattaneo R, Koch HG, Darai G, Schaller H, Schellekens H, van Eerd PM & Deinhardt F (1982) Cloned HBV DNA causes hepatitis in chimpanzees. Nature 299:740–742. [DOI] [PubMed] [Google Scholar]
- Xiong X, Flores C, Yang H, Tooke JJ & Gibbs CS (1999) Mutations in hepatitis B DNA polymerase associated with resistance to lamivudine do not confer resistance to adefovir in vitro. Hepatology 28:1669–1673. [DOI] [PubMed] [Google Scholar]
- Yan ML, Yan LN, Li B, Zeng Y, Wen TF, Wang WT, Yang JY, Xu MQ, Li ZH & Chen YB (2006) Intramuscular hepatitis B immune globulin combined with lamivudine in prevention of hepatitis B recurrence after liver transplantation. Hepatobiliary & Pancreatic Diseases International 5:360–363. [PubMed] [Google Scholar]
- Ying C, De Clercq E & Neyts J (2003) Selective inhibition of hepatitis B virus replication by RNA interference. Biochemical & Biophysical Research Communications 309:482–484. [DOI] [PubMed] [Google Scholar]
- Ying C, De Clercq E, Nicholson W, Furman P & Neyts J (2000) Inhibition of the replication of the DNA polymerase M550V mutation variant of human hepatitis B virus by adefovir, tenofovir, L-FMAU, DAPD, penciclovir and lobucavir. Journal of Viral Hepatitis 7:161–165. [DOI] [PubMed] [Google Scholar]
- Yurdaydin C, Bozkaya H, Cetinkaya H, Sahin T, Karaouz D, Toruner M, Erkan O, Heper AO, Erden E, Bozdayi AM & Uzunalimolu O (2005) Lamivudine vs lamivudine and interferon combination treatment of HBeAg(−) chronic hepatitis B. Journal of Viral Hepatitis 12:262–268. [DOI] [PubMed] [Google Scholar]
- Zhang YY, Zhang BH, Theele D, Litwin S, Toll E & Summers J (2003) Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proceedings of the National Academy of Sciences of the U S A 100:12372–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Guo H, Guo JT, Cuconati A, Mehta A & Block TM (2006) Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Research 72:116–124. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Yamamoto T, Cullen J, Saputelli J, Aldrich CE, Miller DS, Litwin S, Furman PA, Jilbert AR & Mason WS (2001) Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. Journal of Virology 75:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoulim F, Berthillon P, Guerhier FL, Seigneres B, Germon S, Pichoud C, Cheng C & Trepo C (2002) Animal models for the study of HBV infection and the evaluation of new anti-HBV strategies. Journal of Gastroenterology & Hepatology 17:S460–S463. [DOI] [PubMed] [Google Scholar]
- Zoulim F, Poynard T, Degos F, Slama A, El Hasnaoui A, Blin P, Mercier F, Deny P, Landais P, Parvaz P, Trepo C & The Lamivir Study Group (2006) A prospective study of the evolution of lamivudine resistance mutations in patients with chronic hepatitis B treated with lamivudine. Journal of Viral Hepatitis 13:278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]