

Key Points
Activated platelets facilitate plasma TAFI activation in a soluble- and/or platelet-derived thrombomodulin-dependent manner.
TAFIa inhibitor enhances accumulation of plasminogen and its propagation from activated platelets to periphery along with fibrinolysis.
Abstract
Our previous real-time imaging studies directly demonstrated the spatiotemporal regulation of clot formation and lysis by activated platelets. In addition to their procoagulant functions, platelets enhanced profibrinolytic potential by augmenting the accumulation of tissue-type plasminogen activator (tPA) and plasminogen, in vivo in a murine microthrombus model, and in vitro in a platelet-containing plasma clot model. To clarify the role of thrombin-activatable fibrinolysis inhibitor (TAFI), which regulates coagulation-dependent anti-fibrinolytic potential, we analyzed tPA-induced clot lysis times in platelet-containing plasma. Platelets prolonged clot lysis times in a concentration-dependent manner, which were successfully abolished by a thrombomodulin-neutralizing antibody or an activated TAFI inhibitor (TAFIaI). The results obtained using TAFI- or factor XIII–deficient plasma suggested that TAFI in plasma, but not in platelets, was essential for this prolongation, though its cross-linkage with fibrin was not necessary. Confocal laser scanning microscopy revealed that fluorescence-labeled plasminogen accumulated on activated platelet surfaces and propagated to the periphery, similar to the propagation of fibrinolysis. Plasminogen accumulation and propagation were both enhanced by TAFIaI, but only accumulation was enhanced by thrombomodulin-neutralizing antibody. Labeled TAFI also accumulated on both fibrin fibers and activated platelet surfaces, which were Lys-binding-site-dependent and Lys-binding-site-independent, respectively. Finally, TAFIaI significantly prolonged the occlusion times of tPA-containing whole blood in a microchip-based flow chamber system, suggesting that TAFI attenuated the tPA-dependent prolongation of clot formation under flow. Thus, activated platelet surfaces are targeted by plasma TAFI, to attenuate plasminogen accumulation and fibrinolysis, which may contribute to thrombogenicity under flow.
Visual Abstract

Introduction
In the vasculature, thrombus formation and subsequent dissolution are well-coordinated to prevent blood loss and maintain vascular patency. Platelets play a crucial role in the hemostatic response1 and also play anti-fibrinolytic roles by secreting fibrinolytic inhibitors, such as α2-antiplasmin (α2AP),2 plasminogen activator inhibitor-1 (PAI-1),3 and thrombin-activatable fibrinolysis inhibitor (TAFI),4 which are all contained within α granules at high concentrations. These inhibitors regulate fibrinolysis differently at different stages.
Plasmin is the key enzyme associated with intravascular fibrinolysis, responsible for enzymatically degrading fibrin into soluble fibrin degradation products. The activation of Glu1-plasminogen, the native form of plasminogen, is greatly enhanced after binding to the COOH-terminal lysines (C-ter Lys) of fibrin because of a conformational change in Glu1-plasminogen, from a tight to a relaxed form and the formation of a ternary complex on fibrin with tissue-type plasminogen activator (tPA), which is a key circulatory plasminogen activator.5,6 C-ter Lys, generated by the plasmin-catalyzed cleavage of fibrin, are essential for amplifying bindings of plasminogen and tPA via lysine-binding sites found on their kringle domains.7 Therefore, modifying the amounts and localizations of C-ter Lys within the fibrin thrombus can drastically alter the binding efficiency of plasminogen and the efficacy of fibrinolysis.
TAFI, a zymogen form of metallocarboxypeptidase, has been identified by several different groups and assigned multiple names, including procarboxypeptidase unstable,8 arginine procarboxypeptidase,9 plasma procarboxypeptidase B,10 procarboxypeptidase B2,11 and TAFI.12 The fundamental function of activated TAFI (TAFIa) is the removal of C-ter Lys or Arg from bioactive peptides and proteins. The removal of C-ter Lys from partially degraded fibrin reduces the binding and activation efficacy of plasminogen. Generation of TAFIa during the coagulation process therefore naturally leads to a decrease in plasminogen binding to partially degraded fibrin and to inhibit lysis.13 The half-maximal suppression of tPA-induced fibrinolysis in a purified system was achieved at a concentration of 1.0 nM TAFIa, which is equivalent to ∼2% of the total plasma TAFI concentration.14 TAFI is primarily activated by thrombin, via cleavage at Arg92-Ala93, and the catalytic efficiency of thrombin is enhanced by 3 orders of magnitude in the presence of thrombomodulin (TM),14 which is an endothelial cell transmembrane glycoprotein known to modulate the direction of thrombin activity from procoagulant to anticoagulant. TM alters the conformation of TAFI, making it more accessible to cleavage.15 Plasmin is another candidate for the activation of TAFI, and the catalytic efficiency of plasmin is enhanced by glycosaminoglycans and polyphosphates, although the catalytic efficiency of plasmin remains 10-fold lower than that of the thrombin-TM complex.16,17 Both the thrombin-TM complex and plasmin are believed to play roles in TAFI activation under both physiological and pathological conditions.15,18
Although epidemiologic studies have indicated a causative relationship between TAFI antigen/activity levels and the risk of venous thrombosis, ischemic stroke, and cardiovascular disease, the indispensable function of TAFI has not been established in a mouse model.18 The spatiotemporal regulations associated with TAFI activation and TAFIa activity remain poorly understood. The efficacy and location of thrombin generation appears to regulate TAFI activation. Thus, thrombin generation deficiencies in hemophilic patients and anticoagulants therapy for prothrombotic patients have been demonstrated to result in defective TAFI activation and the enhancement of fibrinolysis, resulting in poor clot stability and premature clot lysis, based on the results in vitro experiments.19-21
Our previous real-time imaging analyses, both in vivo, in a murine experimental microthrombus model,22,23 and in vitro, in a fibrin clot model,24,25 directly demonstrated the spatiotemporal regulation of coagulation and fibrinolysis, as follows. (1) Platelets that expressed phosphatidylserine (PS) on their surfaces following fibrin formation were present only in the core of the microthrombus that was formed after laser irradiation.22 (2) Glu1-plasminogen also accumulated in the center of the microthrombus, similar to fibrin deposition during the early stages of thrombus formation, in an endogenously generated plasmin activity-dependent manner.23 (3) Tissue factor-triggered clot formation in human platelet-containing plasma showed uneven, dense, fibrin network structures, containing PS-expressing, activated platelets.25 (4) Activated platelets displayed profibrinolytic activity by binding both plasminogen and tPA on their surfaces, initiating fibrinolysis, which was accompanied by the accumulation of plasminogen to the lytic edge, in an lysine-binding site-dependent manner.25
Based on these findings, we further analyzed the spatiotemporal regulatory mechanisms of coagulation and fibrinolysis using confocal laser scanning microscopy and a microchip-based flow chamber system, focusing on the role played by TAFI.
Materials and methods
Additional details can be found in the supplemental Information.
Proteins and reagents
The following reagents were donated by each company: human recombinant tPA (TD2061; Toyobo Co., Ltd., Osaka, Japan), TAFIa inhibitor (TAFIaI, biosimilar compound of DS-104026; Daiichi Sankyo Company Limited, Tokyo, Japan), and human recombinant soluble TM (Asahi Kasei Pharma Corporation, Tokyo, Japan). Monoclonal antibody against human TM (CD141; TM-neutralizing antibody [TM-Ab]) was purchased from Hycult Biotech (Uden, The Netherlands). Human Glu-plasminogen was purified from fresh-frozen human plasma, by affinity chromatography, on sepharose-lysine.27
Subjects
The study was conducted in accordance with the Declaration of Helsinki principles and was approved by the Ethics Committee at Hamamatsu University School of Medicine. All of the participants provided written informed consent. Blood from healthy adult subjects was collected and all samples were used within 6 hours of blood collection.
Turbidimetric assay for pCLT
The turbidimetric assay was performed to determine plasma clot lysis time (pCLT). Plasma samples, either with or without platelets, were diluted to 50% at a final concentration (the concentrations shown through the manuscript represent the final concentrations), and 1.0, 1.5, or 2 nM tPA was added to initiate fibrinolysis.
ELISA for TM
The concentraion of TM in the plasma was determined using the Quantikine® Enzyme-linked immunosorbent assay (ELISA) Human TM/BDCA-3 (R&D Systems, Minneapolis, MN).
Fluorescence imaging analysis
We used a confocal laser scanning microscope, TCS SP8 (Leica Microsystems GmbH, Wetzlar, Germany). All experiments were conducted at 37°C. A total of 40 × 109 platelets/L, in half-diluted plasma, were supplemented with tPA (1.5 nM), trace amounts of Alexa Fluor-488 labeled fibrinogen (fbg-488), Alexa Fluor-568 labeled plasminogen (plg-568), and Alexa Fluor 647-labeled TAFI (TAFI-647). Fluorescence intensity was analyzed by Image J.
Microchip-based flow chamber analysis
A microchip-based flow chamber system (Total Thrombus-formation Analysis System [T-TAS]; Fujimori Kogyo Co., Ltd., Tokyo, Japan)27 was used to monitor the thrombogenicity under flow conditions. The perfusion system used was as described in the original article.28 Several modifications were implemented to the original procedure. The process of thrombus formation, at 240 seconds−1 shear rate, was monitored by recording flow pressure changes. The following parameters were analyzed to evaluate thrombogenicity: (1) time to 10 kPa (T10) is the lag time necessary for the flow pressure to increase by 10 kPa because of the partial occlusion of the capillary, as an indicator of the onset of thrombus formation; (2) occlusion time (OT) is the lag time for the flow pressure to increase by 80 kPa because of the near-complete occlusion of the capillary; and (3) area under the curve for 30 minutes (AUC30) refers to the area under the flow pressure curve for 30 minutes after the start of the assay.
Statistical analysis
Data were statistically analyzed by Statcel 3, an add-in software for Excel (OMS Publishing Inc., Saitama, Japan). The precise methods are indicated in the figure legends. All data were judged to have normal distribution and equal variance.
Results
TAFI activation contributed to the prolongation of pCLT in a platelet concentration-dependent manner
We first assessed the effect of TAFI activation on tPA-induced, platelet-containing pCLT by turbidimetric assay. pCLT was significantly prolonged with increased platelet counts (80 or 160 × 109 platelets/L) (Figure 1A). Both TAFIaI and TM-Ab effectively attenuated the prolongation of pCLT, to similar extents, in both platelet-free and platelet-containing plasma (Figure 1B). Because the soluble TM concentration in platelet-free plasma was 4.29 ± 0.98 ng/mL (average ± standard deviation [SD], from 15 healthy volunteers), such a low concentration of soluble TM appeared enough for TAFI activation in plasma. Supplementation with soluble recombinant TM, to activate TAFI, potently prolonged pCLT, which was most prominently observed at the highest platelet count (160 × 109 platelets/L). The prolongation induced by soluble TM could be completely canceled by TAFIaI (Figure 1B).
Figure 1.

Platelet-dependent prolongation of plasma clot lysis time (pCLT) determined by the turbidimetric method. A final concentration of 2 nM (A-B) or 1.5 nM/1.0 nM (C) tissue-type plasminogen activator (tPA). pCLTs are shown as the average ± standard deviation. (A) pCLT values at different concentrations of platelet-containing plasma. The number of samples is as follows; 0, 20, 40, 80, and 160 (× 109/L) platelets, n = 18, n = 14, n = 6, n = 17, and n = 10, respectively (9 independent experiments from 4 donors). (B) The involvement of the thrombomodulin (TM)/thrombin-activatable fibrinolysis inhibitor (TAFI) system in pCLT. Red (control), 0, 80, and 160 (× 109/L) platelets, n = 18, n = 17, and n = 10, respectively; blue (activated TAFI inhibitor [TAFIaI]), 0, 80, and 160 (× 109/L) platelets, n = 11, n = 11, and n = 8 respectively; and green (neutralizing antibody of TM [TM-Ab]), 0, 80, and 160 (× 109/L) platelets, n = 7, n = 7, and n = 6, respectively. The effects of TAFIaI were determined in the presence of 5 nM soluble TM, which potently activates TAFI, at different platelet concentrations. Red bars (control): 0, 80, and 160 (× 109/L) platelets, n = 8, n = 7, and n = 3, respectively. Blue bars (TAFIaI): 0, 80, and 160 (× 109/L) platelets, n = 5, n = 6, and n = 3, respectively. (C) Requirement for TAFI in plasma for the platelet-dependent prolongation of pCLT. The number of samples was the same in each group; normal plasma and TAFI-deficient plasma (tPA 1.5 nM, tPA 1.0 nM), n = 5, n = 4, and n = 7, respectively. Red bars are control and blue bars are TAFIaI. Data were analyzed as follows: Williams multiple comparison test: *P < .05 (TM), **P < .01, Welch’s t test: #P < .005 vs control, ##P < .001 vs control; Student t test: +P < .005, ++P < .001 vs control.
TAFI is present in both the plasma and in the platelet granule; therefore, we analyzed the participation of TAFI secreted by activated platelets during the prolongation of pCLT. Normal washed platelets in normal plasma successfully activated TAFI and prolonged pCLT. However, normal washed platelets did not prolong pCLT when TAFI-deficient plasma was used (Figure 1C). This was also true when larger number of platelets were used and when a lower concentration of tPA was used to prolong pCLT (Figure 1C). Collectively, these results suggested that physiological concentrations of platelets attenuated pCLT through the activation of TAFI, which was derived from plasma but not from platelets.
Real-time imaging analysis of clot lysis enabled us to estimate the spatiotemporal regulation of TAFI activation
We visualized platelet-containing plasma clot formation and lysis, using confocal laser scanning microscopy, to evaluate the spatiotemporal regulation of the TAFI system. In the present study, we used a lower concentration of tPA (1.5 nM) than was used in a previous study25 to achieve slower clot lysis. Representative sequential images of both Alexa Fluor 488-labeled fibrinogen (green; fbg-488) and Alexa Fluor 568-labeled plasminogen (red; plg-568) are displayed in Figure 2A. The accumulation of plg-568 in the control sample began in the dense fibrin network regions (high magnification images in supplemental Figure 1), where activated platelets with exposed PS can be found (supplemental Figure 2), and propagated to the periphery. Larger amounts of accumulation were obtained by the addition of either TAFIaI or TM-Ab, especially in the dense fibrin regions, suggesting that the influence of TAFIa is more prominent in the dense fibrin regions than at the periphery.
Figure 2.

Real-time imaging analysis of the effect of the thrombomodulin (TM)/thrombin-activatable fibrinolysis inhibitor (TAFI) system on the accumulation of labeled plasminogen. (A) Representative sequential overlaid images of Alexa Fluor 488-labeled fibrinogen (fbg-488; green) and Alexa Fluor 568-labeled plasminogen (plg-568; red). The dense fibrin network formed at the activated platelets indicates the heterogeneity of the fibrin network structure. Tissue-type plasminogen activator, at 1.5 nM, was added to initiate plg-568 accumulation at the activated platelets and on the fibrin fibers. (Upper) Control (2, 15, 30, 45, and 60 minutes from the start of video capture). (Middle) TAFIa inhibitor (TAFIaI; 2, 10, 15, 20, and 25 minutes from the start of video capture). (Lower) Neutralizing antibody of TM (TM-Ab; 2, 10, 15, 20, and 25 minutes from the start of video capture). (B) Average fluorescence intensities of fbg-488 (blue circles) and plg-568 (red circles) in the dense fibrin region, in 30-pixel (∼34-μm) circular regions of interest, are plotted over time for the control. Plasminogen accumulation (Plg. accum.) time (double-headed arrow) was measured as the period from the appearance of the fibrin fiber structure to the achievement of maximal intensity for plg-568 fluorescence. (C) Plg. accum. time in control (n = 31, median 45 [minutes], interquartile range [IQR] 30.5-48), TAFIaI (n = 22, median 16 [minutes], IQR 14.3-16), and TM-Ab (n = 22, median 17 [minutes], IQR 12.3-17). Statistical analysis was performed using the Mann-Whitney U test (#P < .001 vs control).
The average fbg-488 fluorescence intensities in dense fibrin network regions within a 30-pixel circular region of interest (ROI) were plotted over time, for control samples (Figure 2B, open circles). The simultaneous measurement of plg-568 fluorescence intensities within the same ROI (Figure 2B, closed circles) showed an increase during the early phase, when fbg-488 fluorescence intensities increased, from fibrin network formation. Almost 10 minutes after an initiation of clot formation, plg-568 fluorescence intensity ceased to increase and rather decreased, which was followed by another gradual increase in plg-568 fluorescence to a maximum level. Finally, the fluorescence of both fbg-488 and plg-568 steadily decreased during clot lysis. We measured the time from the appearance of the fibrin fiber structure to the achievement of maximal plg-568 fluorescence intensity and defined this time as the plasminogen accumulation time (Figure 2B, double-headed arrow). Because the accumulation of plg-568 is dependent on the C-ter-Lys residues on fibrin fibers, we assumed that the plasminogen accumulation time would be TAFI-dependent. The plasminogen accumulation time in control samples was widely distributed, suggesting that TAFI activation likely differed among differentially activated platelets (Figure 2C). The plasminogen accumulation times as well as plasma clot lysis times were substantially shortened in the presence of either TAFIaI or TM-Ab (Figure 2C).
To further analyze the involvement of TAFI activation on the platelet surface, we precisely analyzed how plasminogen accumulation propagated from the activated platelets to the periphery, using concentric-circle ROIs (Figure 3A). A time delay in the maximum plasminogen accumulation was observed, from the center of the circle to outside the circle, in control platelets (Figure 3Ba,Ca), which was inhibited by TAFIaI (Figure 3Bb,Cb). Though TM-Ab also shortened the plasminogen accumulation time on average to a similar extent (Figure 2C), their shortening patterns were different. In the case of TM-Ab, the delay was essentially canceled in one-third of platelets (Figure 3Bd); two-thirds showed similar delays as those observed in the control (Figure 3Bc,Cc). These results may suggest that in the propagation process of the plasminogen accumulation to the periphery, other mechanism is also involved in the activation of TAFI than the thrombin/TM-dependent mechanism. The insufficient neutralization of TM by the antibody used in the present study, however, must be considered.
Figure 3.
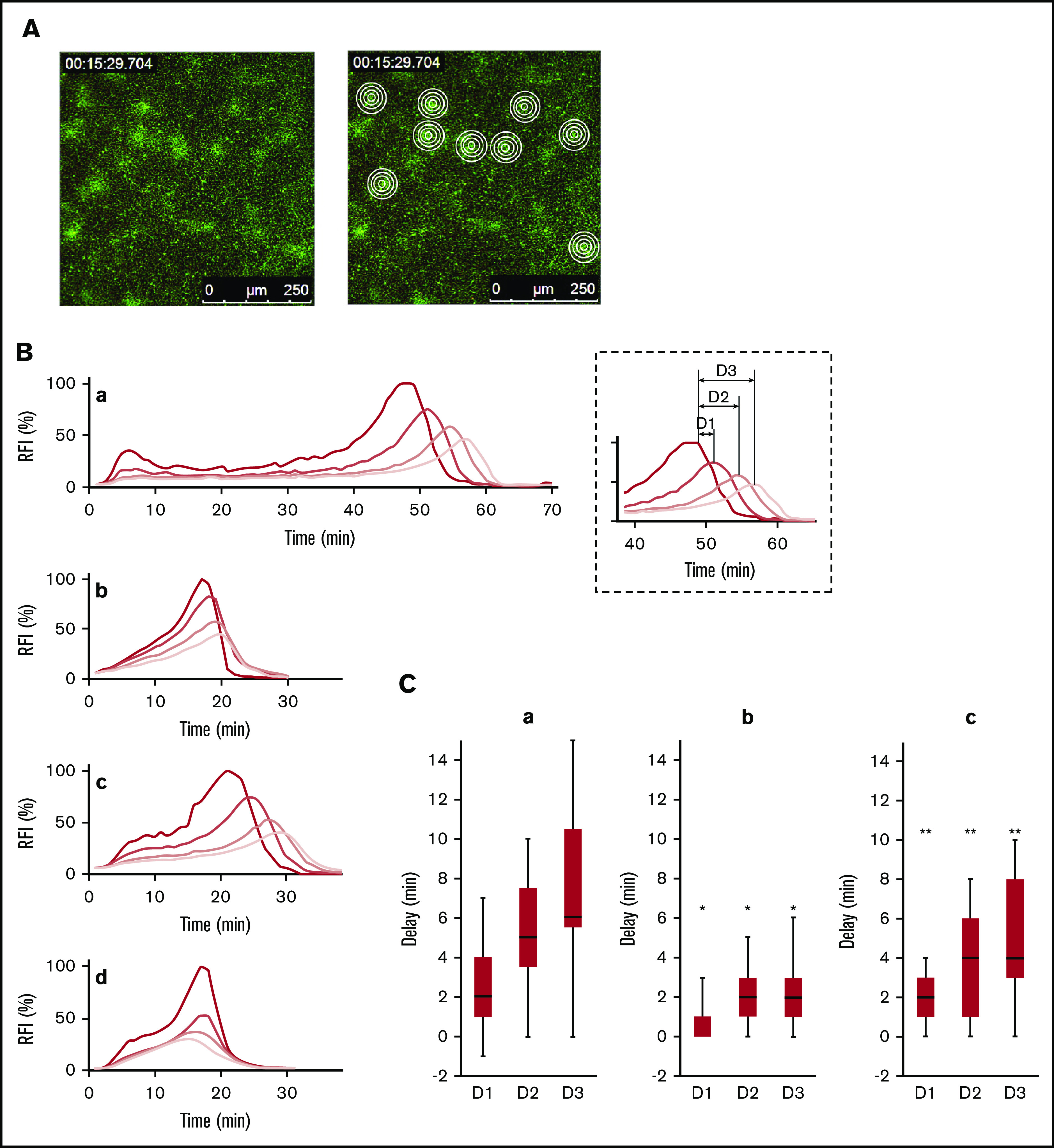
Activated thrombin-activatable fibrinolysis inhibitor (TAFIa)-associated propagation of plasminogen accumulation initiated from the activated platelet-surfaces. (A) The left panel is a 15-minute “control” panel. Four concentric circles in the dense fibrin network region were analyzed based on Alexa Fluor 488-labeled fibrinogen fluorescence; green (right). Concentric circles have diameters of 15, 30, 45, and 60 pixels (17, 34, 51, and 68 µm, respectively). (B) Fluorescence intensities (FI) of Alexa Fluor 568-labeled plasminogen (plg-568) were measured in 4 concentric circles in the dense fibrin network region, and the average FI for the center and the 3 donut-shaped areas were determined. Representative FI changes for plg-568 are shown relative to the maximum FI at the center (RFI %). The shading of the lines is from the center (ie, activated platelets to the outer [light red line to gray lines]). Control (a), TAFIa inhibitor (TAFIaI) (b), neutralizing antibody for thrombomodulin (TM-Ab) (c-d). (C) Propagation of plasminogen accumulation is represented as the time delay in peak plasminogen FI from the center to each outer donut-shaped area (definition is indicated in the dotted square areas, as D1, D2, and D3). D1, D2, and D3 are presented as the median and interquartile range. Control (n = 23) (a), TAFIaI (n = 21) (b), TM-Ab (n = 17) (c). Significance was analyzed by the Mann-Whitney U test (*P < .01 vs control, **P < .01 vs TAFIaI).
Fluorescence-labeled TAFI localized to both C-ter-Lys-exposed fibrin fibers and the surfaces of activated platelets
We examined the localization of Alexa Fluor 647-labeled TAFI (TAFI-647) during clot formation and lysis. When lysis occurred both during the early phase (Figure 4A, plg-568a and plg-568b; upper left and lower left angles) and during the later phase (Figure 4A, plg-568d and plg-568e; upper right), the localization of TAFI-647 (Figure 4A, TAFI-647) was similar to the localization of plg-568, which was observed at both C-ter-Lys-exposed fibrin fibers and the surfaces of activated platelets. TAFI-647 fluorescence on activated platelet surfaces increased in a time-dependent manner, even before the initiation of clot lysis (Figure 4A; TAFI-647). Further, ε-aminocaproic acid, a lysine analog, completely prevented plasminogen accumulation (Figure 4B; supplemental Figure 3; plg-568), but only partially prevented TAFI accumulation on the activated platelet surfaces, and some amounts of TAFI-647 fluorescence was unexpectedly detectable (Figure 4B; supplemental Figure 3; TAFI-647). These results suggested that TAFI could localize to both lysing fibrin fibers and to activated platelet surfaces, and that the latter is possibly through a C-ter-Lys-independent mechanism.
Figure 4.

Labeled thrombin-activatable fibrinolysis inhibitor (TAFI) localized to both C-terminal lysine exposed fibrin fibers and the surfaces of activated platelets. (A) Representative sequential (a-e at the indicated times) high magnification images (63× objective) of Alexa Fluor 488-labeled fibrinogen (fbg-488, top, green), Alexa Fluor 568-labeled plasminogen (plg-568, second from top, red), and Alexa Fluor 647-labeled TAFI (TAFI-647; third from top, blue). (B) Images of fbg-488, plg-568, TAFI-647, and overlay at 20 minutes, 40 seconds from the start of video capture, in the presence of 1 mM ε-aminocaproic acid, a lysine analog.
Effects of FXIII on the activation and accumulation of TAFI
Because the localization of TAFI on activated platelet surfaces appeared to be C-ter-Lys-independent, we analyzed the role played by both plasma factor XIII (FXIII) and cellular FXIII catalytic A subunit (FXIII-A), the latter of which is exposed on activated platelets and crosslinks TAFI with the Aα chain of fibrin(ogen). Both R283, an irreversible inhibitor of FXIIIa that inhibits the crosslinking of α2AP to fibrin fibers, and TAFIaI effectively shortened the pCLT in platelet-free normal plasma (Figure 5A, left columns; normal), and an additive effect was obtained following treatment with both inhibitors. These effects were more obvious in the presence of platelets (Figure 5A; normal). FXIII-deficient plasma containing normal platelets, however, did not show any changes in the presence of the FXIIIa inhibitor, even though TAFIaI potently facilitated clot lysis (Figure 5A, right columns; FXIII-deficient). The distribution and accumulation of TAFI-647 on both fibrin fibers and activated platelets were similarly in both FXIII-deficient plasma (Figure 5B) and normal plasma (Figure 4A) in the presence of normal platelets. Collectively, neither plasma-derived FXIII nor platelet-derived FXIII-A affected the effective activation of TAFI, under the condition used in the present study.
Figure 5.
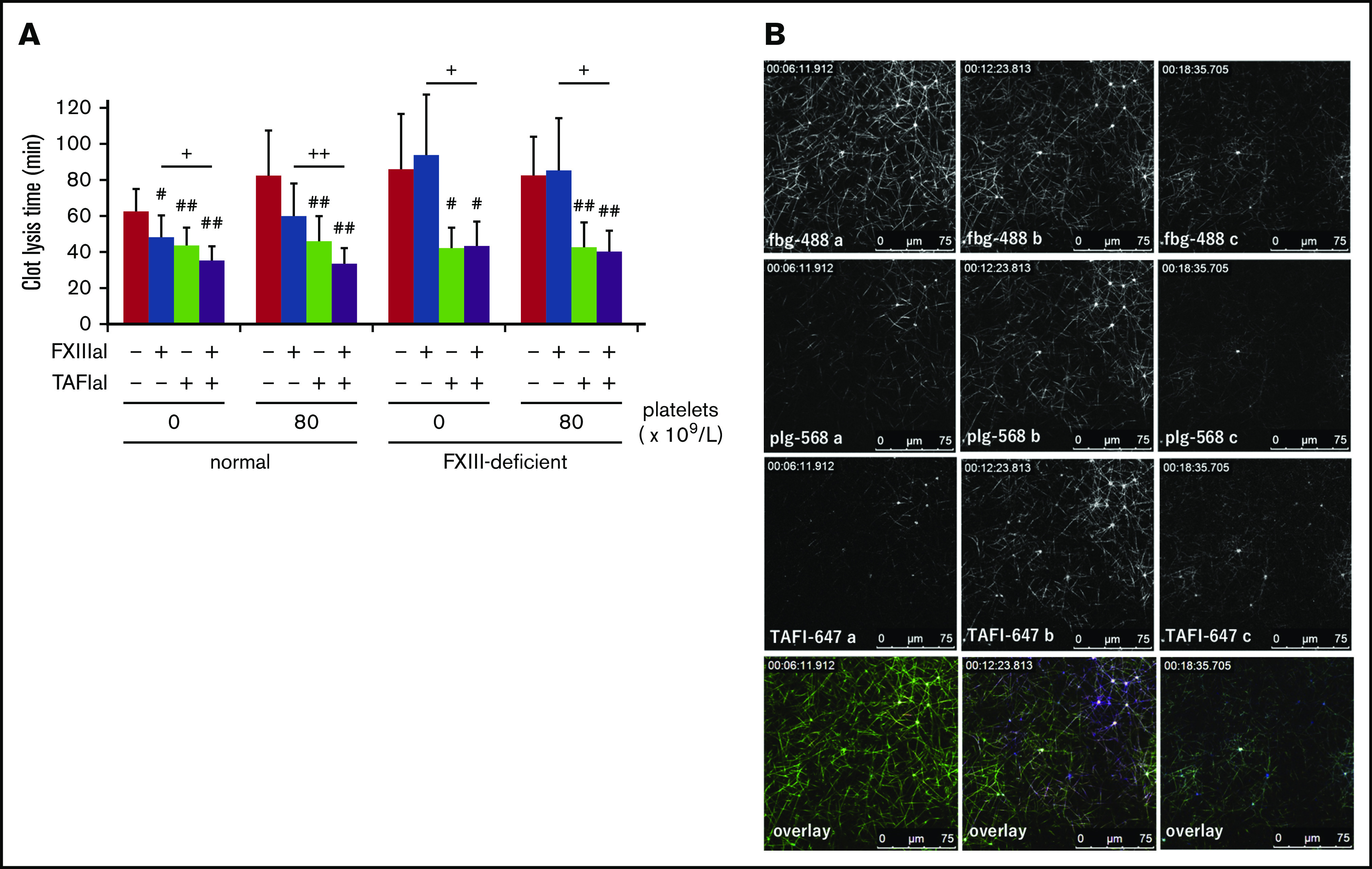
Cross-linkage between thrombin-activatable fibrinolysis inhibitor (TAFI) and fibrin is not necessary for TAFI function. (A) Activated factor XIII inhibitor (FXIIIaI; R283, an irreversible inhibitor against FXIIIa), at 1 mM, and/or activated TAFI inhibitor (TAFIaI), at 5 µM, were added to normal (n = 7) or FXIII-deficient plasma (n = 5), with or without washed normal platelets. Tissue-type plasminogen activator (tPA, 1.5 nM)-induced plasma clot lysis times were determined and are presented as the average ± SD. Significant differences were analyzed using Student t test (#P < .05, ##P < .01 vs control, +P < .05, ++P < .01 R283 vs R283 and TAFIaI). (B) Representative sequential high magnification (63× objective) images of Alexa Fluor 488-labeled fibrinogen (fbg-488, top, green), Alexa Fluor 568-labeled plasminogen (plg-568, second from top, red), and Alexa Fluor 647-labeled TAFI (TAFI-647, third from top, blue) in normal platelet-containing FXIII-deficient plasma. Clot lysis was initiated by the addition of tPA (2 nM).
TAFI activation and the modification of tPA-dependent resistance during thrombogenicity under flow conditions
To evaluate the physiological role played by TAFI during thrombogenicity, we assessed the effects of TAFI activation on thrombolysis under flow conditions (low shear rate; approximately at 240 seconds−1), using a microchip-based flow chamber system (T-TAS). We slightly modified the original protocol to determine the thrombogenicity of whole blood (WB), because the occlusion of the microchip capillary, which had a channel cross-section of 300 µm wide and 80 µm depth, was largely affected by the concentration of platelets during the preliminary experiment, as described in the following section.
First, we evaluated the analytical parameters for thrombogenicity, T10, OT, and AUC30, for different platelet counts. Representative pressure curves in normal (Figure 6A; blue line) and platelet-rich (Figure 6A; pink line) WB were similar, in contrast to those in platelet-poor WB, which showed either nonocclusion (Figure 6A; red line) or prolonged occlusion (Figure 6A; orange line) patterns. When equal amounts of normal and platelet-poor WB were mixed (platelet-half WB), the pressure curve showed an intermediate pattern (Figure 6A; green line). The average of platelet counts, T10, OT, and AUC30 values for each category are summarized in Figure 6B. Based on these results, we chose platelet-half WB to limit the size-dependent efficacy of occlusion in our experimental condition.
Figure 6.

Activation of thrombin-activatable fibrinolysis inhibitor (TAFI) under flow conditions. (A) Thrombogenicity of different platelet concentrations of whole blood (WB), supplemented with 5 nM tissue-type plasminogen activator was examined using Total Thrombus formation Analysis System (T-TAS). Representative pressure changes are indicated as color lines: pink, platelet-rich WB; blue, normal WB; green, 50% normal and 50% platelet-poor (platelet-half) WB; orange and red, platelet-poor WB. (B) Thrombogenicity-related parameters (T10 [red bars]: time to 10-kPa increase, OT [blue bars]: occlusion time, AUC30 [green line]: area under the curve for 30 minutes) in platelet-poor (80-kPa increase achieved in just 1 sample [74 × 109/L platelets]; the other 2 samples did not achieve an 80-kPa increase within 30 minutes), platelet-half (n = 4, 162 ± 16 [average ± SD] × 109/L platelets), normal (n = 4, 293 ± 7.8 [average ± SD] × 109/L platelets), and platelet-rich (n = 7, 376 ± 65 [average ± SD] × 109/L platelets) WB are displayed. Statistical analysis was performed using Williams multiple comparison (*P < .01 against platelet-half WB). (C-E) Thrombogenicity in platelet-half WB, with or without 5 nM tissue-type plasminogen activator (tPA) or 10 μM activated TAFI inhibitor (TAFIaI), was examined at a shear rate of 240 seconds−1. Sample numbers are as follows (tPA/TAFIaI): −/−, +/−, −/+, and +/+, n = 6, n = 6, n = 3, and n = 6, respectively. (C) T10 is shown as the average ± SD. No significant change was determined by Student t test. OT (D) and AUC30 (E) are shown as the median and interquartile range and were analyzed using the Mann-Whitney U test (#P < .01 vs tPA−/TAFIaI−, +P < .05). Two samples treated with both tPA and TAFIaI, which did not achieve an 80-kPa increase within 1800 seconds, are represented as closed 2 circles in panel D. Four individuals donated blood to these experiments.
Although the onset of thrombus formation, T10, was not modified by the presence of tPA and TAFIaI (Figure 6C), we successfully detected the tPA-induced prolongation of OT, which was further prolonged by the simultaneous use of tPA and TAFIaI; 2 samples did not reach 80 kPa above baseline values (Figure 6D). Greater differences between this group and others were observed in the AUC30 values (Figure 6E). These results suggested that TAFI may regulate the thrombogenicity under physiological flow conditions by regulating tPA-induced fibrinolysis.
Discussion
In addition to our previous findings, which showed that activated platelets promoted fibrinolysis via the progressive surface accumulation of both tPA and plasminogen at coagulation start sites,25 here, we provided evidence of the opposite effect, demonstrating that activated platelets suppressed fibrinolysis by diminishing the accumulation and slowing the propagation of fluorescent plasminogen. This effect was attributed to the activation of TAFI in plasma in a soluble and/or platelet-derived TM-dependent manner. Thus, the surfaces of the activated platelets appeared to be essential loci for the regulation of the coagulation-dependent initiation of fibrinolysis by TAFI. TAFI activation during thrombus formation was also demonstrated under flow conditions, which suggested the physiological relevance of TAFI activity for thrombus stabilization.
Not only supplemented TM, as was shown in our previous study,25 but also endogenously present soluble TM in the plasma from healthy volunteers effectively activated TAFI following thrombin generation in our in vitro experimental conditions. TM was initially demonstrated to be predominantly expressed on vascular and lymphatic endothelial cells.29 Later, small amounts of TM were also identified on platelets, megakaryocytes, monocytes, and neutrophils.30,31 Soluble TM is released from endothelial cells and can be accelerated during various disease states by proteolytic activity, generated either in circulation or on the surfaces of endothelial cells.32-34 Based on the observed shortening of pCLT in the presence of TM-Ab and TAFIaI (Figure 1B-C), these limited amounts of soluble TM in plasma appeared sufficient for TAFI activation, even under platelet-free conditions. However, further TAFI activation may have been induced in the presence of activated platelets because of the augmented thrombin generation and the presence of TM on activated platelets.
In vitro findings reported by our25 and other35,36 laboratories suggested that plasminogen accumulated on activated platelet surfaces, where dense fibrin networks form, further supporting the profibrinolytic potential of activated platelets. This mechanism appears to underly the coagulation-associated enhancement of fibrinolysis. Therefore, these loci appear to be targets for the TAFI-mediated regulation of plasminogen accumulation and fibrinolysis. We successfully semiquantified TAFI activity as a decrease in fluorescence intensity of plasminogen and a prolongation of plasminogen accumulation time at the dense fibrin network regions. We used a lower concentration of tPA (below 2 nM) in our in vitro tPA-triggered pCLT assay to detect platelet-dependent TAFI activation constantly, as the maximum activity of TAFI has been reported to occur within 10 minutes after fibrin clot formation.37 A small increase of plasminogen (Figures 2B, closed circles and Figure 3Ba) at initial phase (within 10 minutes after fibrin network formation) was followed by its decrease because of the initiation of TAFI activation at the site. The TAFI system appeared to regulate the profibrinolytic properties of activated platelets by modulating the amounts of accumulated plasminogen on their surfaces. In addition to a threshold-dependent TAFIa mechanism for preventing clot lysis,38 a temporal limitation on the thrombin-dependent activation of TAFI appeared to be critical under high-plasminogen-activation conditions.
A detailed analysis of the propagation of plasminogen (red) signals, initiated from the activated platelets to the peripheral fibrin network, revealed the existence of a finely regulated mechanism for TAFI activation. TAFIa, produced by thrombin on the activated platelet surfaces, appears to propagate to the periphery, along with fibrin network formation. When TAFIaI was used, the maximum plasminogen accumulation quickly propagated and its delay at the periphery disappeared (Figure 3Bab,Cab). This phenomenon fits the threshold-dependent mechanism of TAFIa activity, in which the cleavage of C-ter-Lys and the inhibition of plasminogen binding do not occur when TAFIa activity falls below a specific threshold.38 When TM-Ab was used, however, the propagation pattern observed was similar to that for the control, even though the lysis occurred earlier than in the control. These results suggest that TAFIa is still generated by other mechanism than thrombin/TM system and participates the regulations of both plasminogen accumulation and fibrinolysis.
Plasmin may also be involved in the activation of TAFI. Previously, the biphasic pattern of TAFI activation during in vitro, platelet-free, plasma clot lysis was described. The first peak of TAFIa activity appeared after the initiation of the coagulation phase, and a second rise in TAFIa activity was observed during the clot lysis phase.37 The latter phase, however, had no apparent effect on clot lysis time. In our present study, although we did not observe a clear biphasic activation of TAFI, the plasmin-dependent activation of TAFI may be involved, especially during the clot lysis phase, which may contribute to the observed delay of propagation in plasminogen accumulation from the activated platelets to the periphery in the case of platelet-rich thrombus. A monoclonal antibody that selectively inhibits the plasmin-mediated human TAFI activation was demonstrated to have effectively facilitated the lysis of whole blood model thrombi.39 In addition to the coagulation-associated activation phase of TAFI,19,20,40-42 the physiological importance of plasmin-dependent TAFI activation should be identified.
Using a microchip-based flow chamber system, T-TAS (Figure 6), we also demonstrated the relevant role of TAFIa in thrombogenesis of WB under flow conditions. During our previous in vivo study,23 we demonstrated that fibrinolysis proceeded concurrently with thrombus formation and that both processes were regulated by platelet-secreted PAI-1 using an in vitro flow chamber model.43 Based on these findings, we tried to verify whether TAFIa also modifies thrombogenicity under flow conditions. We successfully demonstrated, in the present study, that the platelet-dependent stabilization of thrombi was attenuated by TAFIaI under flow conditions in the presence of tPA. These results contrasted with a report by Leenarerts et al, in which no statistically significant effects of TAFIa inhibitor were observed on the tPA-mediated fibrinolysis of thrombi formed under arterial shear flow.44 Differences in the flow rate, channel size, and platelet count may underlie these different results.
As reviewed by Whyte et al,45 the crucial roles played by platelets during hemostatic responses are well-established. Their roles in the fibrinolysis, however, are much more diverse, and consensus has not yet been reached. In contrast with the profibrinolytic potential of procoagulant platelets, the anti-fibrinolytic effects of platelets have previously been reported. All 3 inhibitors of fibrinolysis, PAI-1, α2AP, and TAFIa, have been demonstrated to attenuate fibrinolytic activity.46 Platelet-derived TAFI, in conjunction with the TAFI present in plasma, has been demonstrated to enhance the attenuation of fibrinolysis,47 as was speculated by Nesheim et al.48 Our results and those of other groups,41 however, have clarified that activated platelets increased TAFIa activity and prolonged clot lysis time only when sufficient amounts of TAFI were available in the plasma. Although TAFI secreted from platelets during platelet-rich thrombus formation may affect the platelet-mediated resistance to fibrinolysis, plasma-derived TAFI appeared to be essential for the attenuation of fibrinolysis.
In platelet-containing plasma clots, labeled TAFI accumulated on both activated platelet surfaces and the progressive lytic edges of fibrin fibers, in 2 distinct manners (Figure 4A-B). Because TAFI is well-known to coexist with plasminogen in plasma,10 the similar distributions between TAFI and plasminogen at these 2 sites is reasonable. A recently identified new C-ter Lys residue, Lys 556 on α-chain of fibrinogen, which is generated by thrombin on the activated platelet surfaces, is also a cleavage target by TAFIa.36 A certain amount of TAFI, however, was not dissociated by the use of a lysine analog. Regardless of TAFI activation, therefore, C-ter-Lys-independent binding with activated platelets occurs, which may be due to platelet-expressing proteins, including TM30 and FXIII-A.49 Although a functional role for FXIII-A during the TAFI activation was not demonstrated in our experimental conditions (Figure 5A), FXIII-A was shown to be exposed at the protruding caps of the activated platelet surface and to exert anti-fibrinolytic functions by crosslinking α2AP to fibrin.50 Further imaging studies, at higher resolutions, should be performed to visualize both activated platelet-specific cap structures and the localization of functional molecules, such as TM.
As Hoffman et al proposed in a cell-based model of hemostasis,1 fibrinolysis appeared to be regulated by both cell- and coagulation-based mechanisms. Here, we demonstrated platelet-based pro- and anti-fibrinolytic mechanisms. How these mechanisms are balanced and spatiotemporally regulated in normal subjects and become dysregulated during pathological states remain to be determined.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by grants from the JSPS KAKENHI (JP19K08577) (Y.S.) and the Smoking Research Foundation (T.U.). Daiichi Sankyo Company Limited (Tokyo, Japan) and Asahi Kasei Pharma Corporation (Tokyo, Japan) kindly provided chemical compounds. The T-TAS experiment was supported by Fujimori Kogyo Co., Ltd. (Tokyo, Japan). The fluorescence imaging experiment was performed at the Advanced Research Facilities & Services, Hamamatsu University School of Medicine.
Footnotes
For original data, please contact the corresponding author (seigan@hama-med.ac.jp).
Authorship
Contribution: Y.S. designed the study, performed the experiments, analyzed and interpreted the data, and wrote the paper; H.S. and N.H. discussed the results; L.M. performed a part of the turbidimetric assays; T.U. conceptualized the study and wrote the paper; and all authors read and approved the paper.
Conflict-of-interest disclosure: T.U. received research funding from Daiichi Sankyo Company Limited. The remaining authors declare no competing financial interests.
Correspondence: Yuko Suzuki, Department of Medical Physiology, Hamamatsu University School of Medicine, 1-20-1, Handa-yama, Higashi-ku, Hamamatsu 431-3192, Japan; e-mail: seigan@hama-med.ac.jp.
References
- 1.Hoffman M, Monroe DM III. A cell-based model of hemostasis. Thromb Haemost. 2001;85(6):958-965. [PubMed] [Google Scholar]
- 2.Plow EF, Collen D. The presence and release of alpha 2-antiplasmin from human platelets. Blood. 1981;58(6):1069-1074. [PubMed] [Google Scholar]
- 3.Booth NA, Simpson AJ, Croll A, Bennett B, MacGregor IR. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol. 1988;70(3):327-333. [DOI] [PubMed] [Google Scholar]
- 4.Mosnier LO, Buijtenhuijs P, Marx PF, Meijers JC, Bouma BN. Identification of thrombin activatable fibrinolysis inhibitor (TAFI) in human platelets. Blood. 2003;101(12):4844-4846. [DOI] [PubMed] [Google Scholar]
- 5.Rijken DC, Lijnen HR. New insights into the molecular mechanisms of the fibrinolytic system. J Thromb Haemost. 2009;7(1):4-13. [DOI] [PubMed] [Google Scholar]
- 6.Urano T, Castellino FJ, Suzuki Y. Regulation of plasminogen activation on cell surfaces and fibrin. J Thromb Haemost. 2018;16(8):1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorsen S. The mechanism of plasminogen activation and the variability of the fibrin effector during tissue-type plasminogen activator-mediated fibrinolysis. Ann N Y Acad Sci. 1992;667(1 Plasminogen A):52-63. [DOI] [PubMed] [Google Scholar]
- 8.Hendriks D, Scharpé S, van Sande M, Lommaert MP. Characterisation of a carboxypeptidase in human serum distinct from carboxypeptidase N. J Clin Chem Clin Biochem. 1989;27(5):277-285. [DOI] [PubMed] [Google Scholar]
- 9.Campbell W, Okada H. An arginine specific carboxypeptidase generated in blood during coagulation or inflammation which is unrelated to carboxypeptidase N or its subunits. Biochem Biophys Res Commun. 1989;162(3):933-939. [DOI] [PubMed] [Google Scholar]
- 10.Eaton DL, Malloy BE, Tsai SP, Henzel W, Drayna D. Isolation, molecular cloning, and partial characterization of a novel carboxypeptidase B from human plasma. J Biol Chem. 1991;266(32):21833-21838. [PubMed] [Google Scholar]
- 11.Vanhoof G, Wauters J, Schatteman K, et al. The gene for human carboxypeptidase U (CPU)–a proposed novel regulator of plasminogen activation–maps to 13q14.11. Genomics. 1996;38(3):454-455. [DOI] [PubMed] [Google Scholar]
- 12.Bajzar L, Manuel R, Nesheim ME. Purification and characterization of TAFI, a thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1995;270(24):14477-14484. [DOI] [PubMed] [Google Scholar]
- 13.Sakharov DV, Plow EF, Rijken DC. On the mechanism of the antifibrinolytic activity of plasma carboxypeptidase B. J Biol Chem. 1997;272(22):14477-14482. [DOI] [PubMed] [Google Scholar]
- 14.Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271(28):16603-16608. [DOI] [PubMed] [Google Scholar]
- 15.Plug T, Meijers JC. Structure-function relationships in thrombin-activatable fibrinolysis inhibitor. J Thromb Haemost. 2016;14(4):633-644. [DOI] [PubMed] [Google Scholar]
- 16.Mao SS, Cooper CM, Wood T, Shafer JA, Gardell SJ. Characterization of plasmin-mediated activation of plasma procarboxypeptidase B. Modulation by glycosaminoglycans. J Biol Chem. 1999;274(49):35046-35052. [DOI] [PubMed] [Google Scholar]
- 17.Leung LLK, Morser J. Carboxypeptidase B2 and carboxypeptidase N in the crosstalk between coagulation, thrombosis, inflammation, and innate immunity. J Thromb Haemost. 2018;16(8):1474-1486. [DOI] [PubMed] [Google Scholar]
- 18.Foley JH, Kim PY, Mutch NJ, Gils A. Insights into thrombin activatable fibrinolysis inhibitor function and regulation. J Thromb Haemost. 2013;11(suppl 1):306-315. [DOI] [PubMed] [Google Scholar]
- 19.Broze GJ Jr., Higuchi DA. Coagulation-dependent inhibition of fibrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood. 1996;88(10):3815-3823. [PubMed] [Google Scholar]
- 20.Mosnier LO, Meijers JC, Bouma BN. Regulation of fibrinolysis in plasma by TAFI and protein C is dependent on the concentration of thrombomodulin. Thromb Haemost. 2001;85(1):5-11. [PubMed] [Google Scholar]
- 21.Semeraro F, Incampo F, Ammollo CT, et al. Dabigatran but not rivaroxaban or apixaban treatment decreases fibrinolytic resistance in patients with atrial fibrillation. Thromb Res. 2016;138:22-29. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi T, Mogami H, Murakami Y, et al. Real-time analysis of platelet aggregation and procoagulant activity during thrombus formation in vivo. Pflugers Arch. 2008;456(6):1239-1251. [DOI] [PubMed] [Google Scholar]
- 23.Brzoska T, Tanaka-Murakami A, Suzuki Y, Sano H, Kanayama N, Urano T. Endogenously generated plasmin at the vascular wall injury site amplifies lysine binding site-dependent plasminogen accumulation in microthrombi. PLoS One. 2015;10(3):e0122196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki Y, Yasui H, Brzoska T, Mogami H, Urano T. Surface-retained tPA is essential for effective fibrinolysis on vascular endothelial cells. Blood. 2011;118(11):3182-3185. [DOI] [PubMed] [Google Scholar]
- 25.Brzoska T, Suzuki Y, Sano H, et al. Imaging analyses of coagulation-dependent initiation of fibrinolysis on activated platelets and its modification by thrombin-activatable fibrinolysis inhibitor. Thromb Haemost. 2017;117(4):682-690. [DOI] [PubMed] [Google Scholar]
- 26.Zhou J, Kochan J, Yin O, et al. A first-in-human study of DS-1040, an inhibitor of the activated form of thrombin-activatable fibrinolysis inhibitor, in healthy subjects. J Thromb Haemost. 2017;15(5):961-971. [DOI] [PubMed] [Google Scholar]
- 27.Powell JR, Castellino FJ. Activation of human neo-plasminogen-Val442 by urokinase and streptokinase and a kinetic characterization of neoplasmin-Val442. J Biol Chem. 1980;255(11):5329-5335. [PubMed] [Google Scholar]
- 28.Hosokawa K, Ohnishi T, Kondo T, et al. A novel automated microchip flow-chamber system to quantitatively evaluate thrombus formation and antithrombotic agents under blood flow conditions. J Thromb Haemost. 2011;9(10):2029-2037. [DOI] [PubMed] [Google Scholar]
- 29.Maruyama I, Bell CE, Majerus PW. Thrombomodulin is found on endothelium of arteries, veins, capillaries, and lymphatics, and on syncytiotrophoblast of human placenta. J Cell Biol. 1985;101(2):363-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki K, Nishioka J, Hayashi T, Kosaka Y. Functionally active thrombomodulin is present in human platelets. J Biochem. 1988;104(4):628-632. [DOI] [PubMed] [Google Scholar]
- 31.Dittman WA, Majerus PW. Structure and function of thrombomodulin: a natural anticoagulant. Blood. 1990;75(2):329-336. [PubMed] [Google Scholar]
- 32.Ishii H, Majerus PW. Thrombomodulin is present in human plasma and urine. J Clin Invest. 1985;76(6):2178-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takano S, Kimura S, Ohdama S, Aoki N. Plasma thrombomodulin in health and diseases. Blood. 1990;76(10):2024-2029. [PubMed] [Google Scholar]
- 34.Boehme MW, Deng Y, Raeth U, et al. Release of thrombomodulin from endothelial cells by concerted action of TNF-alpha and neutrophils: in vivo and in vitro studies. Immunology. 1996;87(1):134-140. [PMC free article] [PubMed] [Google Scholar]
- 35.Whyte CS, Swieringa F, Mastenbroek TG, et al. Plasminogen associates with phosphatidylserine-exposing platelets and contributes to thrombus lysis under flow. Blood. 2015;125(16):2568-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ni R, Neves MAD, Wu C, et al. Activated thrombin-activatable fibrinolysis inhibitor (TAFIa) attenuates fibrin-dependent plasmin generation on thrombin-activated platelets. J Thromb Haemost. 2020;18(9):2364-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leurs J, Wissing BM, Nerme V, Schatteman K, Björquist P, Hendriks D. Different mechanisms contribute to the biphasic pattern of carboxypeptidase U (TAFIa) generation during in vitro clot lysis in human plasma. Thromb Haemost. 2003;89(2):264-271. [PubMed] [Google Scholar]
- 38.Leurs J, Nerme V, Sim Y, Hendriks D. Carboxypeptidase U (TAFIa) prevents lysis from proceeding into the propagation phase through a threshold-dependent mechanism. J Thromb Haemost. 2004;2(3):416-423. [DOI] [PubMed] [Google Scholar]
- 39.Vercauteren E, Mutch NJ, Declerck PJ, Gils A. Plasmin and the thrombin-thrombomodulin complex both contribute to thrombin-activatable fibrinolysis inhibitor activation in whole blood model thrombi. J Thromb Haemost. 2013;11(1):190-192. [DOI] [PubMed] [Google Scholar]
- 40.von dem Borne PA, Meijers JC, Bouma BN. Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood. 1995;86(8):3035-3042. [PubMed] [Google Scholar]
- 41.Carrieri C, Galasso R, Semeraro F, Ammollo CT, Semeraro N, Colucci M. The role of thrombin activatable fibrinolysis inhibitor and factor XI in platelet-mediated fibrinolysis resistance: a thromboelastographic study in whole blood. J Thromb Haemost. 2011;9(1):154-162. [DOI] [PubMed] [Google Scholar]
- 42.Wyseure T, Cooke EJ, Declerck PJ, et al. Defective TAFI activation in hemophilia A mice is a major contributor to joint bleeding. Blood. 2018;132(15):1593-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hosokawa K, Ohnishi-Wada T, Sameshima-Kaneko H, et al. Plasminogen activator inhibitor type 1 in platelets induces thrombogenicity by increasing thrombolysis resistance under shear stress in an in-vitro flow chamber model. Thromb Res. 2016;146:69-75. [DOI] [PubMed] [Google Scholar]
- 44.Leenaerts D, Loyau S, Mertens JC, et al. Carboxypeptidase U (CPU, carboxypeptidase B2, activated thrombin-activatable fibrinolysis inhibitor) inhibition stimulates the fibrinolytic rate in different in vitro models. J Thromb Haemost. 2018;16(10):2057-2069. [DOI] [PubMed] [Google Scholar]
- 45.Whyte CS, Mitchell JL, Mutch NJ. Platelet-mediated modulation of fibrinolysis. Semin Thromb Hemost. 2017;43(2):115-128. [DOI] [PubMed] [Google Scholar]
- 46.Mutch NJ, Thomas L, Moore NR, Lisiak KM, Booth NA. TAFIa, PAI-1 and alpha-antiplasmin: complementary roles in regulating lysis of thrombi and plasma clots. J Thromb Haemost. 2007;5(4):812-817. [DOI] [PubMed] [Google Scholar]
- 47.Schadinger SL, Lin JH, Garand M, Boffa MB. Secretion and antifibrinolytic function of thrombin-activatable fibrinolysis inhibitor from human platelets. J Thromb Haemost. 2010;8(11):2523-2529. [DOI] [PubMed] [Google Scholar]
- 48.Nesheim M, Wang W, Boffa M, Nagashima M, Morser J, Bajzar L. Thrombin, thrombomodulin and TAFI in the molecular link between coagulation and fibrinolysis. Thromb Haemost. 1997;78(1):386-391. [PubMed] [Google Scholar]
- 49.Muszbek L, Bereczky Z, Bagoly Z, Komáromi I, Katona É. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev. 2011;91(3):931-972. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell JL, Lionikiene AS, Fraser SR, Whyte CS, Booth NA, Mutch NJ. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. 2014;124(26):3982-3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.