

Abstract
Aging leads to functional decline of the hematopoietic system, manifested by an increased incidence of hematological disease in the elderly. Deterioration of hematopoietic integrity with age originates in part from the degraded functionality of hematopoietic stem cells (HSCs). Here, we review recent findings identifying changes in metabolic programs and loss of epigenetic identity as major drivers of old HSC dysfunction and their role in promoting leukemia onset in the context of age-related clonal hematopoiesis (ARCH). We discuss how inflammatory and growth signals from the aged bone marrow (BM) microenvironment contribute to cell-intrinsic HSC aging phenotypes and favor leukemia development. Finally, we address how metabolic, epigenetic, and inflammatory pathways could be targeted to enhance old HSC fitness and prevent leukemic transformation.
The Connection between Aging and Cancer in the Hematopoietic System
HSCs are responsible for the life-long maintenance of blood production. With age, HSCs lose their regenerative capacity, leading to typical features of blood aging, including immunosenescence, anemia, and unbalanced myeloid cell production [1,2]. These features, in turn, drive an increased risk of autoimmunity and hematological malignancies [3]. In this perspective, we review drivers of age-associated HSC dysfunction and their potential contribution to HSC clonal expansion (see Glossary) and transformation. Specifically, we discuss the intimate connection between aberrant metabolic activation, epigenetic drift, and an inflamed microenvironment in establishing these features of aging. We further highlight the existing parallels between old HSCs and leukemic stem cells (LSCs), which are important for understanding how age-related changes in the cellular and molecular fidelity of HSCs could be causal determinants in LSC formation. Finally, we address how the preservation of HSC and BM niche functionality in aging could prevent leukemia development. Our goal is to focus the conversation on the outstanding biological questions in the field of HSC aging and leukemic transformation that have the potential to be transformative for the development of novel anti-aging therapies.
Hallmarks of Aged HSCs
The hematopoietic system faces tremendous demands to produce 1011–1012 mature cells per day in humans, satisfied by a small population of BM-resident HSCs. Our understanding of HSC biology is mainly based on studies in mice, and unless indicated these reports are the primary focus of the present review. HSCs are defined by their ability to self-renew to maintain lifelong potential and to differentiate to produce all lineages of the blood and immune systems [4]. In mice, this is demonstrated functionally by the capacity of HSCs for stable engraftment and multilineage blood reconstitution on transplantation into preconditioned recipients. HSCs primarily remain quiescent during unperturbed steady-state hematopoiesis and activate en masse only in response to severe hematopoietic challenge such as infection, irradiation, or cytotoxic chemotherapy. Recent studies support a model whereby, at steady state, <1% [4] or up to 3–8% [5] of phenotypic long-term repopulating HSCs in adult mice enter the cell cycle per day to replenish the blood system. Maintenance and regulation of the HSC pool is also ensured by the specialized BM microenvironment, or niche, where HSCs reside inside the bone cavity [6].
The cellular features of mouse HSC aging are well characterized, with 18–30-month-old animals considered as having an aged hematopoietic system compared with their 6–12-week-old young counterparts. They include the paradoxical age-dependent expansion of the HSC pool, with decreased homing capacity and reduced ability to repopulate transplanted recipients, skewed balance of myeloid to lymphoid cell production and a perturbed state of quiescence of old HSCs characterized by an increase in stress-response signaling [2]. By contrast, the molecular mechanisms of HSC aging are less understood. Studies over the past decade highlight a number of interconnected cell-intrinsic and cell-extrinsic pathways contributing to HSC functional decline. Evidence for cell-intrinsic dysfunction of old HSCs include features associated with genomic instability, such as the accumulation of DNA damage, deficiency in DNA repair, and age-associated replication stress [7]. Mitochondrial and metabolic deregulation of old HSCs is also well described, including an increase in oxidative metabolism and reactive oxygen species (ROS) production [8], impaired mitochondrial function [9,10], and aberrant mechanistic target of rapamycin (mTOR) activation [11]. Loss of cell polarity and epigenetic drift are additional prominent cell-intrinsic features [1,12,13]. Cell-extrinsic mediators include the development of a proinflammatory milieu and decreased HSC-supportive function of the old BM niche [14–18]. An exploration of the interdependence of these features of HSC aging, how they could be targeted to modulate the rate of decline in HSC molecular and cellular integrity, and their contribution to leukemia are the subjects of this perspective.
Metabolic Derangement and Epigenetic Drift as the Basis of Cell-Intrinsic HSC Aging
Metabolism and epigenetics are tightly linked in their regulation of HSC function [19] and are significantly affected by aging. The cellular features of HSC dysfunction in aging primarily manifest as a failure to maintain appropriate mitochondrial and metabolic regulation [19–21] (Figure 1). In this section, we review the basis of the metabolic and epigenetic control of HSC function, provide an overview of how these mechanisms are perturbed over the course of aging, and finally discuss aspects of old HSC metabolism and epigenetics that are co-opted by or unique to LSCs.
Figure 1. Changes in Metabolism and Epigenetics during Hematopoietic Stem Cell (HSC) Activation and Aging.
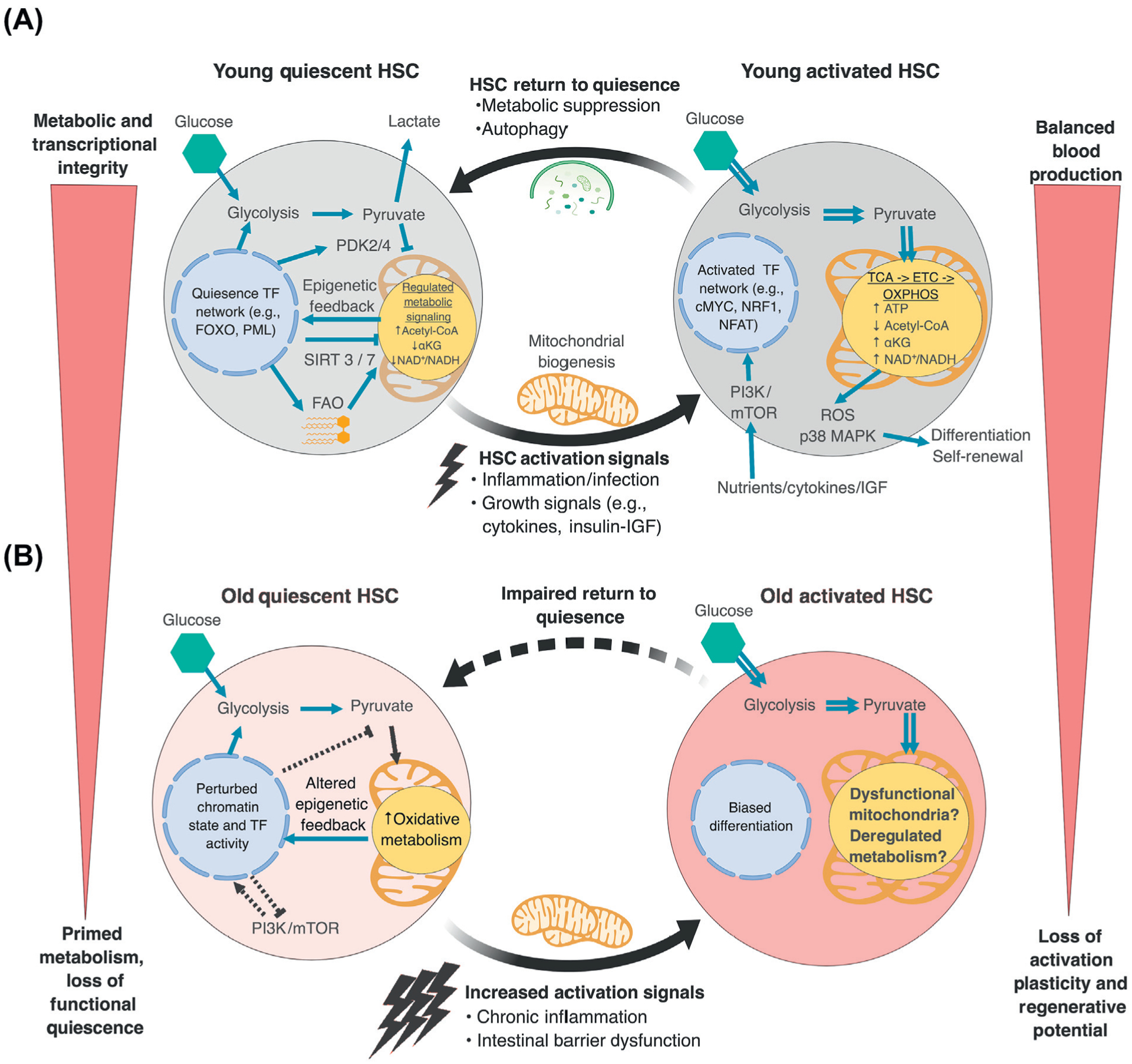
(A) In young HSCs, quiescence is maintained by a hardwired transcriptional regulatory network promoting anaerobic glycolysis and limiting the entry of pyruvate into the tricarboxylic acid (TCA) cycle. Low aerobic flux and fatty acid oxidation (FAO) also provide important substrates that participate in metabolic and epigenetic regulation and help to maintain quiescence. In response to activation signals, HSCs engage oxidative phosphorylation (OXPHOS), which is coupled with mitochondrial biogenesis, mTOR activation, and transcriptional reprogramming. This metabolic switch promotes reactive oxygen species (ROS) production and MAPK activation and stimulates HSC differentiation. When the needs of the hematopoietic system are met, HSCs engage autophagy to clear metabolically activated mitochondria and suppress oxidative metabolism to return to quiescence. (B) With age, old HSCs face challenges with the suppression of basal oxidative metabolism and maintenance of effective quiescence as a result of altered transcriptional regulatory networks and changes to cell-surface signaling, including exposure to higher levels of proinflammatory cytokines. This promotes stem cell exhaustion and blood aging phenotypes. ETC, electron transport chain.
Dynamics of HSC Metabolism and Epigenetic Regulation
At steady state, HSCs utilize glycolysis and actively suppress mitochondrial respiration, a metabolic state that appears to be transcriptionally hardwired [19,22,23] and may be supported by the low oxygen tension of the BM niche as a result of increased activity of hypoxia inducible factor 1 alpha (HIF1α) [22,24]. HIF1α is thought to promote glycolysis in HSCs by upregulating pyruvate dehydrogenase kinase (PDK) and lactate dehydrogenase A (LDHA), thereby limiting mitochondrial pyruvate import and flux through the tricarboxylic acid (TCA) cycle and instead favoring anaerobic catabolism to lactate. The role of hypoxia and HIF1α in HSC quiescence is reviewed in detail elsewhere [22,25]. During activation and differentiation, HSCs undergo a metabolic switch to oxidative phosphorylation (OXPHOS) and produce elevated levels of ATP and ROS to enter the cell cycle. Genetic loss of HIF1α [24], siRNA knockdown of its transcriptional activator Meis1 [26], or genetic knockout of the PDK enzymes that block pyruvate transit into the TCA cycle [27,28], as well as aberrant OXPHOS activation via deletion of the negative regulator mitochondrial carrier homolog 2 (MTCH2) [29], prevent the maintenance of HSC quiescence and promote stem cell exhaustion in mice, thus mimicking aging phenotypes. Complete inhibition of OXPHOS conversely leads to the accumulation of dysfunctional HSCs incapable of differentiation [30]. Low levels of basal respiration also appear to have important roles in HSC quiescence and self-renewal activity [31,32], hence also suggesting critical functions for the TCA cycle and respiratory metabolic intermediates in HSC biology.
The profound metabolic changes that occur when HSCs transition from quiescence to proliferation are accompanied by substantial epigenetic remodeling. DNA demethylation is observed on metabolic activation of HSCs [8] and during differentiation as measured in direct downstream multipotent progenitors [33]. In this context, mitochondria may serve as key epigenetic signaling hubs in HSCs, with metabolic flux controlling the abundance of metabolites acting as cofactors and substrates for enzymes with epigenetic modification activity [23,34]. The sirtuin (SIRT) family of NAD+-dependent deacetylases (Box 1), acetyl-CoA-dependent histone acetyl transferases (HATs), and alpha ketoglutarate (αKG)-dependent dioxygenases are prominent examples with documented histone and DNA modulating activity that is regulated by the metabolic state of the cell (Table 1). For instance, OXPHOS regenerates NAD+ pools that keep the TCA cycle running, and αKG is in turn generated by the TCA cycle [35]. The αKG-dependent methylcytosine dioxygenase 2 (TET2) promotes DNA demethylation by catalyzing the oxidation of 5-methylcytosine to 5-hydroxymethylcytosine, eventually leading to DNA demethylation and regulating HSC differentiation [36,37]. The αKG cofactor ascorbate has been shown to preferentially accumulate in HSCs and promote TET2 function [38] and loss of Tet2 leads to aberrant self-renewal activity of HSCs in mice [36,37]. DNA methyltransferases such as DNA methyltransferase 3 alpha (DNMT3A) require the product of folate metabolism, S-adenosyl methionine (SAM), and are responsible for the establishment and maintenance of methylated nucleotides. Loss of Dnmt3a promotes self-renewal at the expense of differentiation [39] and immortalizes HSCs in mice [40]. It is unclear how loss of enzymes that methylate and demethylate DNA both lead to HSC differentiation defects, but these results point to an essential role for the dynamic regulation of DNA methylation and demethylation in controlling HSC function [41]. Hence, the reliance on glycolysis by quiescent HSCs may help to preserve their genomic methylation in part by limiting αKG and TET activity, whereas in activated HSCs the increase in OXPHOS drives αKG upregulation and may contribute to their altered chromatin acetylation and DNA methylation profile [19,41].
Box 1. SIRTs at the Interplay between Metabolism and Epigenetics in HSC Aging.
SIRTs are a family of seven NAD-dependent proteins that function as deacetylases and/or as ADP-ribosyltransferases. SIRT3, SIRT4, and SIRT5 are localized in the mitochondria, SIRT1, SIRT6, and SIRT7 are predominantly nuclear, and SIRT2 is mostly found in the cytosol. SIRTs play complex protective roles along the mitochondrial-metabolic-nuclear axis in HSCs by modulating the acetylation status and activity of multiple targets including histones. For instance, SIRT1 supports FOXO3 activity, whose activity is carefully regulated in HSCs to maintain quiesence and self-renewal capacity [49,50], and its loss leads to aberrant aging-like HSC phenotypes [51]. SIRT6 has been shown to regulate HSC homeostasis through the epigenetic repression of Wnt signaling via its nuclear histone 3 deacetylase activity [52]. The mitochondrial SIRT3 directly suppresses ROS and its overexpression improves the functionality of old HSCs [10]. SIRT7 suppresses translation and the mitochondrial respiratory machinery in part through inhibition of the respiratory transcription factor NRF1 and activates a proteostatic mitochondrial unfolded protein response pathway important in metabolic suppression during aging [9]. SIRT2 has also been implicated in supporting the resolution of mitochondrial protein folding stress in old HSCs [53]. These mitochondrial suppressive activities are specifically required in aged HSCs, and since SIRT2, SIRT3, and SIRT7 levels decline in old HSCs they have been suggested as important regenerative targets.
Table 1.
HSC Metabolites and Their Epigenetic Targets
| Name | Metabolic function and route of synthesis | Regulated enzyme | Effect on HSC function | Changes in hematopoiesis with age | Changes with leukemia |
|---|---|---|---|---|---|
| αKG | Derived from glucose or glutamine; generated by the TCA cycle | αKG dependent dioxygenases (e.g., TETs) and Jumanji c domain histone demethylases (JHDMs) | αKG increases in activated HSCs along with OXPHOS [19]; TET2 is essential for proper regulation of HSC differentiation [36,37] | Unclear whether αKG levels change in HSCs with age or whether deregulation primarily occurs via loss of cofactors or target epigenetic enzyme activity | TET2 frequently mutated in ARCH and subsequently in MDS and AML [128]; IDH1/2 mutations convert αKG to the oncometabolite D-2-hydroxyglutarate, which competitively inhibits αKG function [74]; BCAT1 enzyme upregulation in AML suppresses αKG and TET2 activity [79,80] |
| Succinate | TCA cycle metabolite; feeds into complex II of electron transport chain (succinate dehydrogenase) | Succinate accumulation results in negative regulation of αKG-dependent enzyme activity; also generated as product of JHDM activity [46] | Unclear, but negatively regulates αKG and the activity of JHDMs and TETs in human pluripotent stem cells [173] | No direct evidence; succinate accumulation could phenocopy TET inhibition [174] | Mutations in succinate dehydrogenase, which result in succinate accumulation, occur in several nonhematopoietic cancers [175]; no direct evidence for tumor-promoting role of succinate accumulation, but could possibly act by the same mechanism as TET inhibition [174] |
| Ascorbate (vitamin C) | Derived from the diet in humans; generated by the gulonolactone oxidase enzyme in mice | αKG-dependent dioxygenases use ascorbate as a cofactor; also regulates JHDMs [46] | TET2 activity is particularly sensitive to ascorbate levels in HSCs; ascorbate accumulates in HSCs and negatively regulates HSC self-renewal and myelopoiesis through Tet2-dependent and -independent mechanisms [38] | Ascorbate levels may decline in human HSCs in elderly patients with vitamin C deficiency, promoting loss of TET2 function | Ascorbate depletion accelerates leukemogenesis while repletion delays leukemogenesis and increases survival in Flt3 internal tandem duplication (Flt3ITD) mouse models [38] |
| NAD+ | NAD+: NADH ratio is essential for mitochondrial function, with pools of mitochondrial NAD+ regenerated by OXPHOS | Required cofactor for the HDAC function of SIRTs | SIRT1 supports FOXO3 activity [51], SIRT2 prevents NLRP3 inflammasome and caspase 1 activation [53], SIRT3 suppresses ROS [10], SIRT6 regulates Wnt signaling [52], SIRT7 suppresses NRF1 activation and promotes response to mitochondrial protein folding stress [9] | Loss ofSIRT2, SIRT3, and SIRT7 associated with old HSC oxidative and mitochondrial stress-related dysfunction; NAD+ precursor nicotinamide riboside supplementation improves muscle, neural, and melanocyte stem cell function in old mice [176] | Unclear role in leukemia |
| Acetyl-CoA | Main function is to deliver acetyl group to TCA cycle for oxidation; generated by mitochondrial oxidation or recycling of acetate groups | Acetyl-CoA-dependent HATs | Quiescent HSCs maintain high acetyl-CoA levels; several HATs genetically required for HSC function: CREB [42], MOZ [43], and HAT cofactor TRAPP [44] | Unclear whether levels change with age | Unclear role in leukemia |
| SAM | Derived from mitochondrial threonine catabolism and the methionine cycle | Required methyl donor for all methylation reactions, including lysine methyltransferases (MLL/KMT2) and cytosine DNA methyltransferases (DNMT1, DNMT3a, DNMT3b) | Unclear how SAM levels are regulated in HSCs; MLL is essential for hematopoiesis; DNMT1 and DNMT3a have nonredundant functions in regulating HSC self-renewal and differentiation [39,177] | No changes to SAM levels reported in HSC aging | KTM2A/MLL1 translocations drive a subset of infant and adult leukemia [178]; DNMT3a mutations occur in ARCH and MDS/AML |
| FAD | Redox-active coenzyme; biochemical source is the vitamin riboflavin | Lysine-specific demethylase 1 (LSD1) requires FAD as a cofactor; LSD1 removes methyl groups from lysine 4 on histone H3 (H3K4) | LSD1 essential for hematopoiesis [179], but unclear how FAD levels are regulated in HSCs; loss of LSD1 reduces HSC self-renewal and generation of myeloid and lymphoid progenitors; LSD1 decommissions specific Hox genes through epigenetic repression to allow differentiation [179] | Unclear, but indirect evidence that LSD1 inhibition reverses upregulation of genes in OXPHOS pathway and downregulation ofgenes in glycolysis pathway in induced pluripotent stem cells [180] | Heightened expression of LSD1 in AML, with antileukemic effects of LSD1 inhibition observed in mouse models of human leukemia [179] |
Glycolysis-derived pyruvate is also the predominant source of acetyl-CoA for HATs, and the histone deacetylase (HDAC) function of nuclear SIRTs is heavily regulated by the nuclear NAD+: NADH ratio. Quiescent HSCs have a high-acetyl-CoA to low-NAD+ ratio, which maintains acetylation and is potentially important for their self-renewal activity [13,33]. This is consistent with the genetic requirement for HATs such as CREB-binding protein (CBP) [42] and the monocytic leukemia zinc-finger protein MOZ [43], as well as the HAT cofactor TRRAP [44], in the preservation of HSC self-renewal activity and maintenance of the hematopoietic compartment in mice [19,41]. The biochemistry of these metabolite-dependent chromatin modifiers, as well as their molecular biological consequences for gene expression, are reviewed in detail elsewhere [45,46], although direct understanding of their consequences for HSC gene regulation remains limited. To summarize, the metabolic switch from HSC glycolytic quiescence to oxidative activation is accompanied by substantial epigenetic remodeling, which is likely to be supported in part by changes in the abundance of mitochondrial metabolic flux-derived metabolites. Precise epigenetic control appears necessary to regulate prodifferentiation and proself-renewal transcription factor regulatory networks. Further establishment of how the bioavailability of metabolic intermediates is regulated by HSC metabolism at quiescence and on activation, and how this regulates the activity of epigenetic modifying enzymes, will greatly improve our understanding of these processes.
How Changes to Metabolism and Epigenetics Promote HSC Aging Phenotypes
While limited direct connections have been made between the metabolism and epigenetics of aged stem cells in model organisms, it is logical that metabolic changes resulting from age-associated stress could induce epigenetic alterations in the chromatin of HSCs and, by the same token, that epigenetic alterations could promote changes to transcriptional programs controlling HSC metabolism. An initial step towards the connection of epigenetics, gene expression, and metabolism arose from a study integrating transcriptome, DNA methylome, and histone modification profiling of purified HSCs isolated from young and 24-month-old mice. While exclusively correlative, this work reported a broad increase in H3K4me3 histone marks on ChIP-seq and a modest increase in global DNA hypermethylation on reduced-representation bisulfite sequencing (RRBS) during aging. More interestingly, they found that targeted hypomethylation of HSC signature genes correlated with increased expression and enrichment in pathways supporting self-renewal, myeloid skewing, and ribosomal activation in old HSCs by RNA-seq [12]. It is possible that these epigenetic changes result from the failure of HSCs to suppress aerobic metabolism in aging or that, conversely, they promote gene expression changes leading to old HSC metabolic phenotypes. The combination of these chromatin remodeling activities may also be partially responsible for activation-associated epigenetic drift in old HSCs. However, other groups have described a more limited correlation between DNA methylation changes and transcriptional alterations associated with HSC aging, instead suggesting that these epigenetic changes predominantly affect gene expression in downstream progeny through cellular inheritance of epigenetic marks [41,47]. These contradictory findings may be explained by the limitations of promoter-proximal or gene-body methylation and gene expression correlation, as epigenetic regulation of distal enhancers may reveal a more direct regulatory role of DNA methylation changes in old HSCs [33,41]. Collectively, these findings point to the importance of the HSC epigenetic landscape not only in regulating transcriptional programs but also in stem cell differentiation potential toward distinct effector lineages via heritable transmission of epigenetic marks. Mechanistic approaches are necessary to validate the functional significance of age-associated histone and DNA methylation changes. However, the observation of increased DNA hypermethylation at CpG dinucleotide islands in human peripheral blood samples from old individuals [48], independent of changes in mature blood cell type composition, potentially reflects aging-associated DNA methylation changes in the stem cell compartment and supports the relevance of these mouse model findings to human aging.
Studies of the function of SIRTs have provided the most direct evidence for the interplay between metabolism and epigenetics in the context of HSC aging. SIRTs are well recognized for their antioxidant functions and role in longevity [35] and play complex protective roles along the mitochondrial-metabolic-nuclear axis in HSCs (Box 1). Although we still have an incomplete mechanistic picture, mitochondrial biogenesis, metabolism, and the protein folding stress response appear to be coordinated by SIRTs in a cell-protective manner in young HSCs and have damaging consequences for HSC function as the expression of specific SIRT proteins declines with age. Mitochondrial protein folding stress in old HSCs has recently been linked to activation of the NLRP3 inflammasome and caspase 1 activity. Old HSC engraftment and reconstitution can be improved by direct genetic inhibition of these inflammatory mediators or inhibition of their activity by SIRT2 overexpression [53]. This link suggests that the functional decline of metabolically stressed HSCs could also result from the activation of inflammatory signaling pathways and that the activation of stress response pathways in aging is increasingly necessary for functional preservation of HSCs.
Replicative History Promotes HSC Epigenetic Drift in Aging
Age-associated changes to the epigenetic state and functional fitness of HSCs may relate to the replicative history and serial metabolic activation of HSCs. Inducible H2B-GFP pulse-chase labeling of HSCs in transgenic mice has been used to associate HSC replicative history with fitness over the mouse lifespan [54]. HSCs lose the H2B-GFP label as it dilutes in dividing daughter cells, but approximately 3% of HSCs can retain the label for 10–22 months. Importantly, functional engraftment capacity from old HSCs is limited to this label-retaining compartment. In support of this model, HSC behavior following inflammatory stress and genotoxic irradiation stress has been shown to be predetermined by epigenetic configuration, suggesting that HSCs retain epigenetic memory of their activation context and subsequently become biased in their lineage potentiality [55]. The implication of these observations is that with successive cell divisions HSCs become incapable of maintaining their cellular identity and pan-hematopoietic lineage potential, which may chiefly result from the changes to chromatin accessibility occurring from modifications to DNA and histones that accompany serial cell cycling from quiescence to activation and back, which we refer to as epigenetic drift. As a consequence, the organism has evolved a capacity to maintain a fraction of HSCs in a dormant state to limit epigenetic drift and preserve lifelong hematopoiesis (Figure 2).
Figure 2. Molecular Mechanisms Underlying Loss of Hematopoietic Stem Cell (HSC) Fitness with Age.
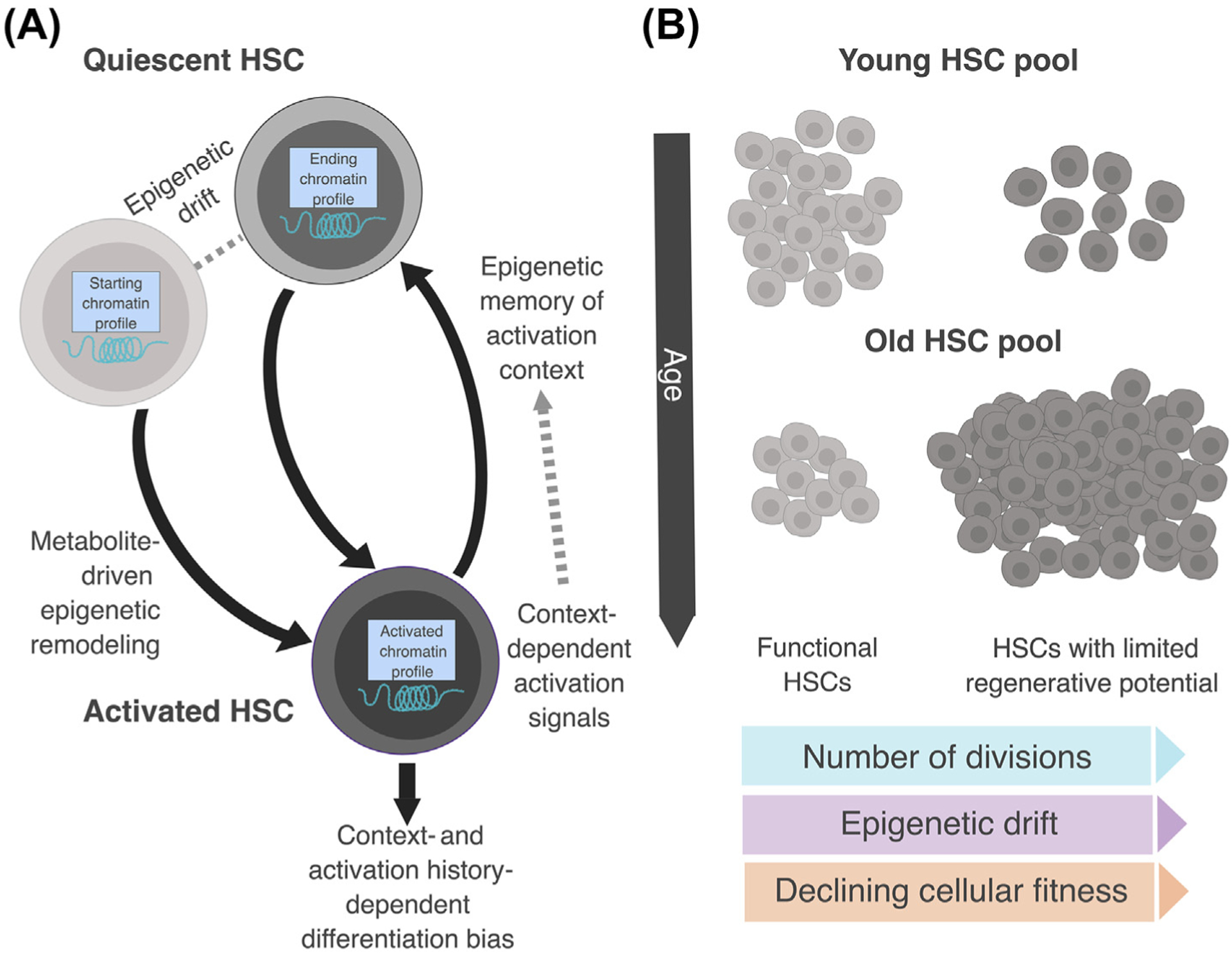
(A) Model of HSC cell cycle and environmental activation signals driving context-dependent and age-related epigenetic drift. (B) Aging is associated with an expanded HSC compartment but a decrease in the number of cells meeting the ‘functional’ HSC definition (i.e., cells with serial pan-lineage hematopoietic repopulation activity following transplantation). Evidence suggests that only HSCs with limited divisional history can fulfill this functional criterion. Repeated cell division may promote changes to the chromatin accessibility profile of HSCs, biasing them via epigenetic drift towards differentiation programs that they have been signaled to promote. Cellular activation also perturbs HSC quiescence and thus may contribute to declining cellular fitness by altering HSC proteostasis or metabolic homeostasis. While the number of HSCs expands with age, the number of HSCs with limited divisional history declines, potentially explaining this paradox. Light gray, functional HSCs; dark gray, nonfunctional HSCs.
The idea that HSC function is linked to mitotic history is supported by several additional studies showing that HSCs lose regenerative capacity with progressive divisions and that several aspects of aged hematopoiesis can be observed by enforcing HSC proliferation by repeated monthly injections of the myeloablative chemotherapy 5-fluorouracil (5-FU) [47,56,57]. Serial replicative challenges induced by 5-FU in HSCs trigger deregulation in the methylation state of DNA, affecting genomic loci associated with hematopoietic lineage potential and genes expressed in downstream progenitors [58]. Additionally, targeted DNA hypermethylation was associated with loss of expression of polycomb repressive complex 2 (PRC2) in replication-induced and physiological HSC aging [12, 58,59]. Hypermethylation of PRC2 target genes is associated with the silencing of its enzymatic component Ezh2 [12,58], a histone methyltransferase whose loss of expression promotes therapy resistance in acute myeloid leukemia (AML). Age-related epigenetic changes are also linked to DNA damage and the activation of the DNA damage response (DDR) in old HSCs (Box 2). Taken together, these studies indicate that replicative history and aging promote significant epigenetic changes in HSCs and that such epigenetic drift might be causal to their functional decline.
Box 2. DNA Damage, DDR Activation, and Epigenetic Drift in Aged HSCs.
While DNA damage and mutation accumulation have long been considered major contributors to HSC aging, recent data provide alternative interpretations. The well-established DNA damage theory of HSC aging is based on the combined observations of elevated levels of the canonical DNA damage marker γH2AX and higher comet-assay tail movements in old HSCs [58,160], increased reliance on the error-prone nonhomologous end-joining DNA repair pathway in quiescent HSCs [160], the ability of genotoxic agents to promote HSC dysfunction [161,162], and the striking inherited BM-failure phenotypes in patients with mutations in DNA repair factors or HSCs devoid of DNA repair capacity [163–166]. Early transcriptome profiling of young versus old mouse HSCs indicate that DDR pathways are among the most significantly downregulated in aging [13]. Attenuated DDR activation has also recently been reported in old HSCs exposed to genotoxic agents, which promote their survival by decreasing apoptotic priming [167]. However, other recent evidence suggests that the DDR process might not be intrinsically perturbed in old HSCs [168]. The overall mutation level in old HSCs has been estimated to be roughly 2–3-times higher than in young HSCs [168] and only modest DNA damage accrual has been reported in the HSPC compartment of elderly humans [150]. Experiments using mitochondrial DNA ‘mutator’ mice have also resulted in hematopoietic-dysfunction phenotypes that significantly diverged from physiological HSC aging [169]. The constant rate of somatic genetic decay over the short lifespan of a mouse (~2 years) compared with the long lifespan of a human (~80 years), but conservation in HSC aging phenotypes, also makes difficult to reduce aging to a direct consequence of mutation accumulation. However, it is plausible that repeated environmental exposures and activation of the DDR process promote epigenetic drift in old HSCs. Old HSCs undergo significant amounts of replication stress, which is a key contributor to their functional decline [7]. Cycling old HSCs have elevated levels of γH2AX foci and replication fork defects, resulting from decreased expression of minichromosome maintenance (MCM) helicase components. Interestingly, replication stress and γH2AX foci are observed in the absence of DDR pathway activation and persist on re-entry into quiescence. The suggestion emerging from this work is that γH2AX foci may instead function as a histone modification associated with ribosomal DNA gene silencing in old HSCs. Collectively, these studies indicate that we still have an imperfect understanding of the mutagenic versus epigenetic consequences of DDR activation and replication stress in driving physiological HSC aging.
Cellular Responses to Metabolic Perturbation in Aging
HSCs are poised to respond to activating signals from their environment when the hematopoietic system is challenged, which becomes more frequent during aging. Nutrient-sensing metabolic pathways that modulate the rate of organismal aging also control the glycolytic/oxidative respiration axis in HSCs [19,60]. Activation of PI3K/AKT, mTOR, and p38-MAPK, drive OXPHOS engagement, ROS production, and HSC differentiation [19]. mTORC1 controls an adaptive cell cycle transition from a quiescent G0 state to a G-alert primed cell-cycle activation state in response to injury or inflammation in muscle stem cells and HSCs [61]. While mTOR activity is critical for effective hematopoietic activation in response to stress, chronic PI3K-mTOR pathway overactivation increases mitochondrial biogenesis and metabolic deregulation in quiescent HSCs, producing phenotypes similar to those of glycolytic impairment or overactive OXPHOS [19,62,63]. Conversely, AKT1/2-deficient HSCs are arrested in quiescence, resembling HSCs with inhibited OXPHOS [19]. While induction of ROS is similarly important for the activation of HSCs, aberrantly high ROS levels can cause DNA damage over time and may accelerate HSC exhaustion and aging downstream of chronic p38-MAPK activation [19].
The catabolic activity of the FOXO family of transcription factors plays important roles in mediating HSC metabolism and oxidative stress response. Loss of FOXO3 in HSCs, or suppression of FOXO3 by AKT signaling, results in elevated mitochondrial membrane potential, increased ROS levels, and aberrant AKT and ERK activation, together causing HSC exhaustion and loss of long-term repopulation activity [19]. Conversely, genetic ablation of Id1 has recently been suggested to protect HSCs from exhaustion and aging by interfering with the HSC stress activation response [64]. The proteostasis mechanism of autophagy plays complex roles in protecting HSC function at steady state, during aging, and on transformation, as reviewed in Box 3. Nonselective macroautophagy allows HSCs to cope with metabolic challenges and promote their return to quiescence following metabolic activation [8,49], while selective mitophagy acts only under stress conditions and contributes to LSC survival [65,66].
Box 3. The Paradox of Autophagy and Mitophagy in HSC Aging and Leukemogenesis.
The proteostasis mechanism of nonselective macroautophagy (autophagy) is a central integrator of the metabolic and cellular activity that protects HSC function [49]. Genetic inhibition of autophagy in HSCs results in metabolic activation and stem cell exhaustion [8,170]. It was recently demonstrated that a small subset of old HSCs with high levels of autophagy exhibit long-term regeneration potential, a quiescent metabolic state, and low replication stress similar to young HSCs, while most old HSCs with low autophagy levels fail to suppress oxidative respiration and display all of the classical features of age-related functional decline [8]. These results suggest that autophagy is necessary for the metabolic quiescence and functional activity of HSCs in the context of age-associate stress. While loss of autophagy leads to significant changes in DNA methylation in young HSCs [8], it remains to be determined why a subset of old HSCs engage autophagy and whether this has epigenetic or leukemogenic consequences.
Activation of the PINK1–PARKIN mitophagy pathway supports the enhanced regenerative potential of HSCs with damaged mitochondria [67]. Other groups have failed to identify a specific requirement for mitophagy in healthy HSCs at steady state [8,171], suggesting that engagement in mitophagy may be important only under stress conditions. In line with this view, unchecked mitophagy in HSCs results in aberrant self-renewal and loss of BM cellularity, leading to anemia, lymphopenia, and decreased survival [172]. LSCs appear to hijack the mitophagy pathway to promote their self-renewal and metabolic stress resistance [66] in part through AMPK signaling, previously demonstrated to be important for LSC fitness [65]. In summary, while nonselective autophagy appears to play important roles in the functional and metabolic maintenance of HSCs at steady state with consequences for HSC aging, LSCs may appropriate the mitophagy stress response pathway to promote their survival under conditions of metabolic or cytotoxic challenge.
Less well-studied, additional metabolic pathways are important in HSC functional maintenance. The promyelocytic leukemia (PML) nuclear protein activates a peroxisome proliferator-activated receptor delta (PPARδ)-mediated mitochondrial fatty acid oxidation (FAO) program, which preserves HSC stemness and prevents exhaustion through a pathway that also converges on the regulation of ROS balance [67,68]. FAO promotes an increase in global histone acetylation by providing metabolic sources of acetyl-CoA [69], suggesting that HSC epigenetic poising may be affected by fatty acid metabolism in addition to glucose metabolism. FAO was also demonstrated to be essential for HSC polarity and asymmetric cell division [68]. Polarity defects arise in old HSCs as a result of elevated activity of the small Rho GTPase CDC42, leading to a random nuclear distribution of acetylated H4K16 [70]. Further epigenetic profiling by single-cell ATAC-seq suggested that aged, apolar HSCs lose the capacity to pass on asymmetric epigenetic information to their downstream progenitors [71]. While it remains to be demonstrated that FAO is perturbed in old HSCs, these converging findings suggest that the epigenetic consequences of age-associated loss of polarity could be mediated in part by metabolic aberration. Collectively, these discoveries reinforce the tight relationship between the metabolic phenotype of HSCs and their transcriptional regulatory networks, implicating dysregulated growth signaling pathways in old HSC functional decline.
Leukemic Stem Cell Metabolism and Epigenetic Consequences
The connection between the metabolism and epigenetics of old HSCs and that of LSCs remains underexplored and a ripe area for new research. Importantly, disruption of either glycolysis or respiration impairs leukemogenesis, supporting conserved sensitivity to glycolytic/respiratory metabolic balance [72,73]. The first approved metabolic agents for AML, ivosidenib and enasidenib, inhibit mutant forms of isocitrate dehydrogenase 1 and 2 (IDH1/2) that convert αKG from the TCA cycle to the oncometabolite R-2-hydroxyglutarate. R-2-Hydroxyglutarate competitively inhibits αKG-dependent dioxygenases, resulting in aberrant epigenetic poising downstream of reduced TET2 and KDM2a histone-modifying enzyme activity, blocking differentiation and promoting self-renewal [74]. More recently, the combination of the BCL-2 inhibitor venetoclax with the hypomethylating agent 5-azacitidine has demonstrated high therapeutic activity in AML, with overall survival and standard of care implications [75]. Intriguingly, the depth and durability of remissions observed with this combination are believed to result from the selective targeting of LSC oxidative metabolism. Specifically, venetoclax together with 5-azacitidine has been shown to decrease OXPHOS by disrupting the electron transport chain complex II downstream of a reduction in cellular glutathione levels [76]. This reduction in glutathione appears to be related to a block in amino acid uptake, with these results highlighting a connection with the increased amino acid dependency required for LSCs to effectively use OXPHOS [77]. Regulation of amino acid levels is similarly important for HSC maintenance, with depletion of the essential amino acid valine leading to HSC depletion [78]. However, LSCs appear to have a greater reliance on amino acid metabolism that can be therapeutically exploited. The cytosolic aminotransferase for branched-chain amino acids, BCAT1, is upregulated in both patients and mouse models of CML and AML and promotes LSC survival by depleting αKG [81,82]. Elevated levels of BCAT1 thereby phenocopy Idh-mutant suppression of TET activity or direct loss of Tet2, leading to DNA hypermethylation. These findings provide an interesting example of nongenetic convergence on an important epigenetic pathway for LSC fitness by a metabolic enzyme. Dihydroorotate dehydrogenase (DHODH), which functions in pyrimidine synthesis, is another attractive target for leukemia therapy, as its inhibition induces differentiation of LSCs thus sensitizing leukemic cells to chemotherapy [81].
Intriguingly, a potential LSC resistance mechanism following amino acid deprivation resulting from venetoclax/5-azacitidine combination therapy was mediated by the upregulation of fatty acid metabolism to rescue OXPHOS in LSCs [77]. These results highlight the metabolic adaptation of LSCs and the potential synergy of targeting multiple metabolic pathways in concert. In support of the metabolic dependency of LSCs on fatty acid metabolism, inhibition of carnitine palmitoyl transferase 1a (CPT1a), the rate-limiting catalytic enzyme in the FAO pathway, has cytotoxic activity in AML cells [82]. Inhibition of fatty acid-binding protein 4 (FABP4) or the fatty acid transporter CD36 may similarly be active in targeting LSCs [83], potentially via a conserved mechanism. The specific epigenetic mechanisms by which metabolic enzyme or metabolite uptake inhibition affect LSC fate decision merits further investigation. Collectively, these results illustrate how the metabolic vulnerability of LSCs can be therapeutically targeted by perturbing the epigenetic basis of LSC self-renewal and differentiation blockade.
Deteriorating HSC Niche as Driver of Aging and Blood Cancer
While the majority of studies on HSC aging and transformation focus on intrinsic changes, the BM niche where HSCs reside also contributes to these processes (Figure 3). In this section, we review the role of the BM niche in HSC function, provide an overview of the changes in the BM microenvironment occurring during aging, and finally summarize how functional alterations of the niche can contribute to the development and propagation of leukemia.
Figure 3. The Dysregulated Aging Bone Marrow (BM) Niche Microenvironment.
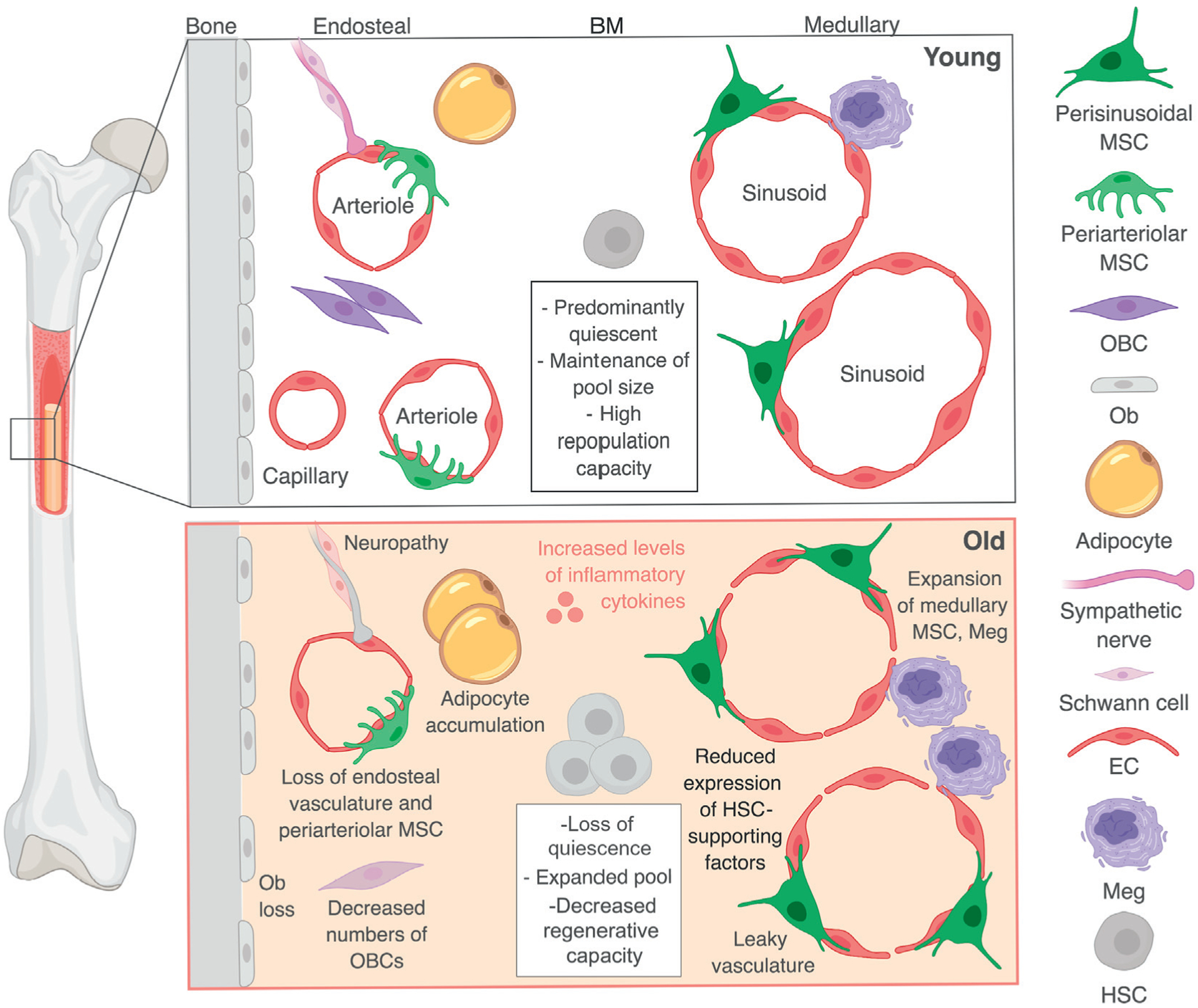
In the BM, endosteal regions are rich in arterioles, capillaries, periarteriolar mesenchymal stem cells (MSCs), and their osteoprogenitor derivatives [i.e., osteoblastic lineage cells (OBCs), osteoblasts (Ob)], while medullary regions are enriched in sinusoids and perisinusoidal MSCs. Sympathetic nerves track alongside the vasculature, with predominantly periarteriolar localization. Megakaryocytes (Meg) are located next to BM vessels and adipocytes are found throughout the BM cavity. Various cell types instruct hematopoietic stem cells (HSCs) to stay dormant, cycle, or differentiate through direct interactions and cytokine secretion. Aging leads to remodeling of the BM microenvironment, with increased levels of inflammatory cytokines, loss of endosteal populations, accumulation of adipocytes, and functional deterioration of the central marrow, leading to decreased HSC-supportive activity and promotion of HSC aging features.
Microenvironmental Regulation of HSC Fate
The BM niche regulates HSC fate decisions through direct cell-cell interactions and the secretion of cytokines and chemokines [7]. While the exact localization of HSCs within the marrow cavity and the precise function of distinct BM niche cells remain a matter of investigation, several cell types have emerged as essential players. Stromal cells including mesenchymal stromal cells (MSCs), osteoblasts and their osteoprogenitor cells (OBCs), endothelial cells (ECs), adipocytes, sympathetic nerves, and nonmyelinating Schwann cells, as well as mature hematopoietic cells such as megakaryocytes, macrophages, and T cells, all play interdependent roles in controlling HSC homeostasis [6,84]. Imaging of HSCs in the BM cavity demonstrated that the majority of HSCs reside in close proximity to sinusoidal vessels [54], although others argue that this association could be random due to the high density of the sinusoidal network and that other cell types, such as arterioles and megakaryocytes, are more important in instructing HSC fates [85–87]. The functions of the HSC niches include maintaining the HSC pool [88,89], preserving HSC quiescence [85–87,90], regulating HSC differentiation behavior [88,91–94], and supporting HSC regeneration during stress hematopoiesis [95] as detailed in Table 2. The function and composition of these niche elements differs based on their location in the marrow cavity, with major anatomical differences observed between niche cells lining the bone surface (endosteal) versus in the central cavity (medullary) locations (Figure 3).
Table 2.
Changes in HSC Niche Cells in Mouse Models of Aging and Leukemia
| Niche component | Main location in the BM | Role at steady state | Change with age | Change in leukemia | Potential for intervention |
|---|---|---|---|---|---|
| MSCs | Periarteriolar MSCs - mostly endosteal; perisinusoidal MSCs-mainly in the central marrow | Ensure HSC maintenance, produce HSC-supporting factors including CXCL12, SCF, ANGPT1 [6,84], contribute to pan-lineage differentiation [181] | Preservation [17,99] or expansion of central marrow perisinusoidal MSCs with decreased expression of Cxcl12, Scf, and Angptl [15]; lossof endosteal NG2+ periarteriolar MSCs [15,18] | Expansion of central marrow perisinusoidal Nestin-GFP+ MSCs with decreased expression of HSC maintenance factors, loss of NG2+ MSCs, and differentiation block towards mature osteoblasts in MLL-AF9 AML model [119]; loss of Nestin-GFP+ MSCs in JAK2V167F MPN model [121] | Functional improvement could be achieved by blocking the underlying cause (e.g., blocking IL-1 signaling helped to mitigate apoptosis-induced death of perisinusoidal MSCs in JAK2V167F MPN model [121], restoration of innervation helped to restore the functional impairment of aged MSCs [15]) |
| OBCs | Preferentially located at the endosteum | Important for HSC maintenance and lymphoid differentiation | Decreased numbers of OBCs [15,17] with decreased production of osteopontin [17] | Expanded population of proinflammatory myelofibrosis OBCs in BCR/ABL CML model [118]; loss of osteoprogenitors in Notch-driven T-ALL model [117] | Functional improvement of OBCs could be achieved by reestablishment of HIF1α stability and consequent increased expression of OBC-supporting growth factors in ECs [182] |
| Osteoblasts | Endosteal location | Indirect role in HSC maintenance, support erythroid and lymphoid differentiation | Decreased numbers in middle-aged and old mice [96] | Loss of mature osteoblasts in Notch-driven T-ALL and MLL-AF9 AML models [117,120] | Clearance of senescent cells and inhibition of inflammatory signaling prevented age-related bone loss [183] |
| Sympathetic nerves and nonmyelinating Schwann cells | Endosteal and central marrow location, frequently associated with arterioles | Critical for maintenance of HSC dormancy and regulation of HSC trafficking, produce noradrenaline, regulate circadian rhythm, activate latent TGFβ [6,84] | Neuropathy, loss of sympathetic nerves leading to loss of adrenergic signaling, expansion of medullary MSCs with decreased HSC-supporting activity, loss of arterioles, and increased HSC numbers [15] | Neuropathy [119,121] contributing to loss of MSCs [121] in MLL-AF9 AML and JAK2V167F MPN models | Functional improvement could be achieved by blocking the underlying cause; for example, blocking IL-1 signaling decreased apoptosis of Schwann cells in JAK2V167F MPN model [121] and administration of β2-adrenoceptor agonist (clenbuterol) helped to reduce LSC expansion in MLL-AF9 AML model [119] and improve HSC repopulation capacity in aged mice [15] |
| ECs | Enriched at the endosteum for arterioles and H-type capillaries and in the central marrow for sinusoids | Ensure HSC maintenance, produce HSC-supporting factors including CXCL12 and SCF, regulate lineage differentiation via Notch ligands [6,84] | Loss of H-type capillaries [18] and arterioles [15]; decreased [16], unchanged [17,99,182], or expanded [15] EC pool with increased vascular leakiness [16]; decreased expression of HSC-supporting CXCL12 and SCF [16] | Decreased numbers of endosteal, but not central marrow, blood vessels leading to decreased survival of normal HSCs in MLL-AF9 AML model [120] | Functional improvement could be achieved through restoration of Notch signaling in aged ECs with recovered numbers of capillaries and arterioles, but failed to rescue aged HSC function [18]; stimulation of ECs at endosteum by deferoxamine, with improved survival of normal HSCs in model of MLL-AF9-driven AML [120]; transplantation of young ECs, with improved regeneration potential of old HSCs [16] |
| Adipocytes | Throughout BM cavity | Contradictory: either HSC antagonistic [184] or HSC supportive through SCF production [185] | Increased numbers in steady-state aging [186] and during irradiation-induced regeneration [15] | Potential contribution to leukemia propagation and resistance through secretion of inflammatory factors and leptin, no direct evidence in mouse models [187] | No intervention in aging described yet |
| Megakaryocytes | In proximity to BM vasculature | Maintain HSC quiescence and differentiation by secretion of TGFβ, TPO, CXCL4, FGF1 [6,84] | Increased numbers [15,188] with associated increase in TGFβ production [188] | Decreased numbers and expression of Cxcl4 in BCR/ABLCML model, decreased numbers in JunB MPN model [95] | No intervention described yet |
The Aged Microenvironment Compromises HSC Function
Major changes in the bone and vasculature are observed in both old mice and elderly humans, suggesting remodeling of the entire niche microenvironment during aging. Pronounced thinning, impaired regeneration potential, and reduced hormonal function of the bone manifest by mid-life [96], preceding age-associated hematopoietic dysfunction. Systemic vascular aging manifests by thickening of arterial walls and loss of vasodilation in larger vessels, leading to increased incidence and progression of vascular disease [97]. The deterioration of endosteal and medullary niche elements in aging have distinct effects on HSC maintenance (Table 2). In the endosteum, aging is accompanied by the attrition of bone-forming osteoblasts [98] and OBCs [15,17] as well as the loss of endosteal vasculature including H-type capillaries [18], arterioles [15], and closely associated sympathetic nerves [15]. As the endosteal niche is implicated in the maintenance of a subset of quiescent HSCs and in the regulation of lymphoid and erythroid differentiation [6], age-related loss of these cells could directly contribute to the myeloid skewing and loss of quiescence of aged HSCs. Consistent with this idea, genetic ablation of sympathetic nerves in the BM promotes age-related HSC dysfunction, while the administration of β3-adrenoceptor agonist restores aspects of HSC function in old mice [16]. In the central marrow, MSC numbers are either preserved [17,99] or expanded [15] with age, but have downregulated expression of key HSC-supporting factors including SCF, CXCL12, and ANGPT1 [15]. Aging also impairs the function of ECs and increases vascular leakiness throughout the BM cavity. Aged ECs are sufficient to drive hematopoietic aging phenotypes in young HSCs, as demonstrated in mouse ex vivo co-culture and in vivo EC infusion assays [16]. These changes in the microenvironment occur alongside and are likely to be promoted by increased inflammatory signaling in the old BM niche. Low-level chronic upregulation of circulating proinflammatory cytokines with age, referred to as ‘inflammaging’, is a well-documented phenomenon that contributes to many aspects of organismal aging [100–102]. For example, two- to sixfold upregulation of MIP1α/β and RANTES levels is observed in the BM fluid of old mice at steady state [14,17]. While the exact cellular source of these cytokines is currently unknown, an increase in chronic cytokine exposure may contribute to the loss of physiological integrity of the aged microenvironment, including bone loss [103] and vascular leakiness [104,105], and age-associated hematopoietic dysfunction [102,106]. Deregulated inflammatory cytokine signaling is known to perturb HSC quiescence and functional maintenance [102] and is a prominent molecular signature of old HSCs [13]. In keeping with these findings, recent evidence in a murine model suggests that myeloid-biased HSCs preferentially expand in response to lipopolysaccharide (LPS)-induced inflammation, which suggests the increased myeloid output observed in old mice may be the consequence of increased exposure to inflammatory stimuli [107]. In summary, the increase in inflammatory signaling and loss of niche-supportive function in aging are two critical aspects of age-associated HSC dysfunction.
BM Microenvironment Remodeling in Leukemia
Changes in the aged BM stroma not only lead to loss of HSC supportive capacity but may also provide the permissive environment that enables the initial expansion and further propagation of leukemic clones. The activity of BM stromal cells is frequently dysregulated in hematological malignancies, with stroma acting as both initiators and reinforcers of leukemia [6,108]. Pioneering evidence for the role of the niche in promoting leukemia came from the discovery that deficiency in transcriptional regulator retinoic acid receptor gamma (RARγ) drives the development of myeloproliferative neoplasms (MPNs) in a nonhematopoietic cell-mediated manner, resulting primarily from an increase in inflammatory cytokine production by BM stromal cells [109]. Many leukemia-associated mutations induced solely in stromal cells, like osterix (Osx)-expressing osteoprogenitors [110,111], collagen type 1 alpha 1 (Col1a1)-positive osteoblasts [112], angiopoietin receptor (Tie2+)-positive ECs [113], and MSCs [114], have since been shown to lead to the development of MPN-like or AML-like pathology and have been associated with promoting mutagenesis in hematopoietic cells [110,111]. Mechanisms affected in stromal cells in these niche-initiated leukemia models include changes in Notch and Wnt signaling, ribosome biogenesis, and RNA processing, which lead to the remodeling of the BM stroma and promote a deregulated inflammatory milieu [110–114].
Experimental and clinical observations also suggest that leukemic cells can directly remodel the BM stroma to promote their preferential survival over healthy HSCs [6]. This can occur by perturbation of the physiological BM niche, leading to decreased production of HSC maintenance factors, changes in the niche structure, and the activation of inflammatory signaling that exhausts normal HSCs and enhances LSC survival (Table 2). Remodeling of the BM microenvironment by leukemic clones is reported in many leukemia types, including B cell acute lymphoblastic leukemia (B-ALL) [115], multiple myeloma (MM) [116], T-ALL [117], CML [118], AML [119,120], and MPN [121]. Leukemias can also result in the death of Schwann cells, the promotion of neuropathy, and the subsequent loss of essential Nestin+ MSCs [119,121]. Leukemias can also modulate the differentiation capacity of MSCs, leading to decreased levels of HSC maintenance factors and HSC depletion [115,118,119,121]. In addition, several blood malignancies lead to the destruction of endosteal niches, reflected in the loss of OBCs and development of myelofibrosis [117,118,120,122] and, in the case of AML, changes in endosteal vasculature that preferentially decrease the engraftment and survival of healthy HSCs [120]. A common thread in these leukemic niche remodeling events is the role of inflammation, with increased secretion of factors such as IL-1α/β [118,121], TNFα [113,120,123], MIP1α [115,119], and CXCL2 [120]. Inflammatory cytokine signaling promotes cell death and the degeneration of normal niches, allowing leukemia to co-opt the BM microenvironment. IL1RAP overexpression and STAT3 activation in AML have also emerged as central pathways by which leukemic stem cells use inflammation to support oncogenic signaling pathways [124,125].
Interestingly, hematopoietic-specific deletion of Tet2 is associated with defective intestinal barrier maintenance, which contributes to the development of preleukemic myeloproliferation in mice [126]. While the mechanisms leading to intestinal permeability are not understood, the resultant bacterial translocation into the circulation and innate immune response activation promote systemic IL-6 signaling, which subsequently drives critical myeloproliferative signaling events. This provides an interesting case study of how subclinical hematopoietic abnormalities can lead to intestinal dysfunction that promotes malignancy. This concept is supported by additional evidence that enhanced clonal competitiveness of Tet2-KO HSPCs occurs as a result of increased autocrine IL-6 signaling in response to acute inflammatory insults [127]. It is interesting in this context to also consider the necessity of systemic inflammatory defects that arise independently of hematopoietic dysfunction in leukemic transformation. In summary, changes to the BM microenvironment during aging and as a result of remodeling by malignancies can contribute to leukemic transformation and disease progression.
ARCH and Preleukemic Disposition in Humans
In humans, aging is frequently accompanied by the development of clonal hematopoiesis, in approximately 10% of individuals greater than 70 years old [128,129]. The full extent of the clinical relevance of ARCH was realized only recently, when copy number variation (CNV) and next-generation sequencing analysis demonstrated that ARCH is characterized by the presence of leukemia-associated mutations in expanded, nonleukemic clones of blood cells in the elderly [130–132]. Clonal expansion of mutant ARCH clones is likely to result from a combination of cell-intrinsic fitness advantages over normal HSCs and preferential survival in the dysfunctional aged microenvironment. Intriguingly, more that 90% of ARCH mutations identified so far occur in genes encoding epigenetic modification enzymes, including DNMT3A, TET2, and additional sex combs-like transcriptional regulator 1 (ASXL1) [128,129,133], hence reinforcing the relevance of epigenetic drift in the promotion of HSC aging phenotypes. The impact of ARCH extends far beyond leukemia predisposition, with strong associations now observed with chronic inflammation and cardiovascular disease [134], poor solid tumor prognosis [135], and increased overall mortality risk [136], suggesting a systemic detrimental effect of altered hematopoiesis in aging.
Furthermore, recent studies show that cancer-associated mutations also accumulate and accompany aging in solid organs without obvious disease development [137]. While the mutation profiles are organ dependent, these observations suggest that age-related clonal selection is a universal aspect of human aging [137], and one that is at present difficult to simulate in model organisms.
ARCH is assumed to represent the expansion of mutated old HSC clones, although no direct experimental evidence is currently available. Retrospective analysis of HSC clones from patients who developed AML provides support for this model and demonstrates that mutations in epigenetic modification enzymes can frequently be found in HSCs and precede mutations in driver genes found in LSCs [138,139]. This suggest that preleukemic mutation accumulation in long-lived stem and progenitor cells could be both deterministic and predictive. Two recent studies provide strong evidence for this theory by showing that the likelihood of ARCH developing into AML can be predicted by a high-risk mutational signature in the peripheral blood, years prior to diagnosis [140,141]. In particular, significantly higher frequencies of specific mutations, including tumor protein p53 (TP53) and IDH2, were strongly associated with AML development. This suggests that monitoring of peripheral blood variant allele frequencies could help to predict cancer development and may change clinical management by allowing earlier therapeutic intervention.
The enhanced competitive fitness of ARCH clones in aging may be supported by or require concurrent dysfunction of the aged BM niche microenvironment. Murine studies show that p53-deficient clones preferentially expand in a stressed/irradiated niche, while hematopoietic p53 mutations do not confer a selective fitness advantage at steady state [142]. Recent high-resolution sequencing of the stem cell compartment in MDS and AML suggests that clones with sufficient oncogenic potential may exist in the HSC compartment for prolonged periods of time prior to disease onset and may lead to disease only in specific environmental contexts [143]. Furthermore, MDS and AML clones appear to evolve in a parallel rather than stepwise manner, suggesting that the changes to the niche following MDS and associated treatment may initiate AML from separate preleukemic stem and progenitor cells. Additionally, clonal analyses of murine BM cells transplanted into young and old recipients demonstrated that the aged microenvironment supported a lower number of transplanted clones, suggesting a selective pressure that could contribute to clonal expansion [144]. While ARCH has not been demonstrated in mice during physiological aging, serial transplantation of aged BM cells was recently linked to the expansion of clones with mutations of cancer-associated genes [145]. This suggests that longer chronological time and/or hematopoietic stress may be essential for the development of ARCH, which underlies the context dependency of mutant clonal expansion and transformation. Together, these findings reinforce the notion of clonal and environmental selection promoting epigenetic drift in aging HSCs, in turn leading to subsequent myeloproliferative and systemic physiological consequences in aging (Figure 4). In summary, ARCH is quickly becoming one of the most interesting predictive biomarkers of aging, given the ease with which it can be assayed using collected blood samples and its relevance to both hematopoietic and nonhematopoietic age-related chronic diseases.
Figure 4. From Aging to Leukemia.
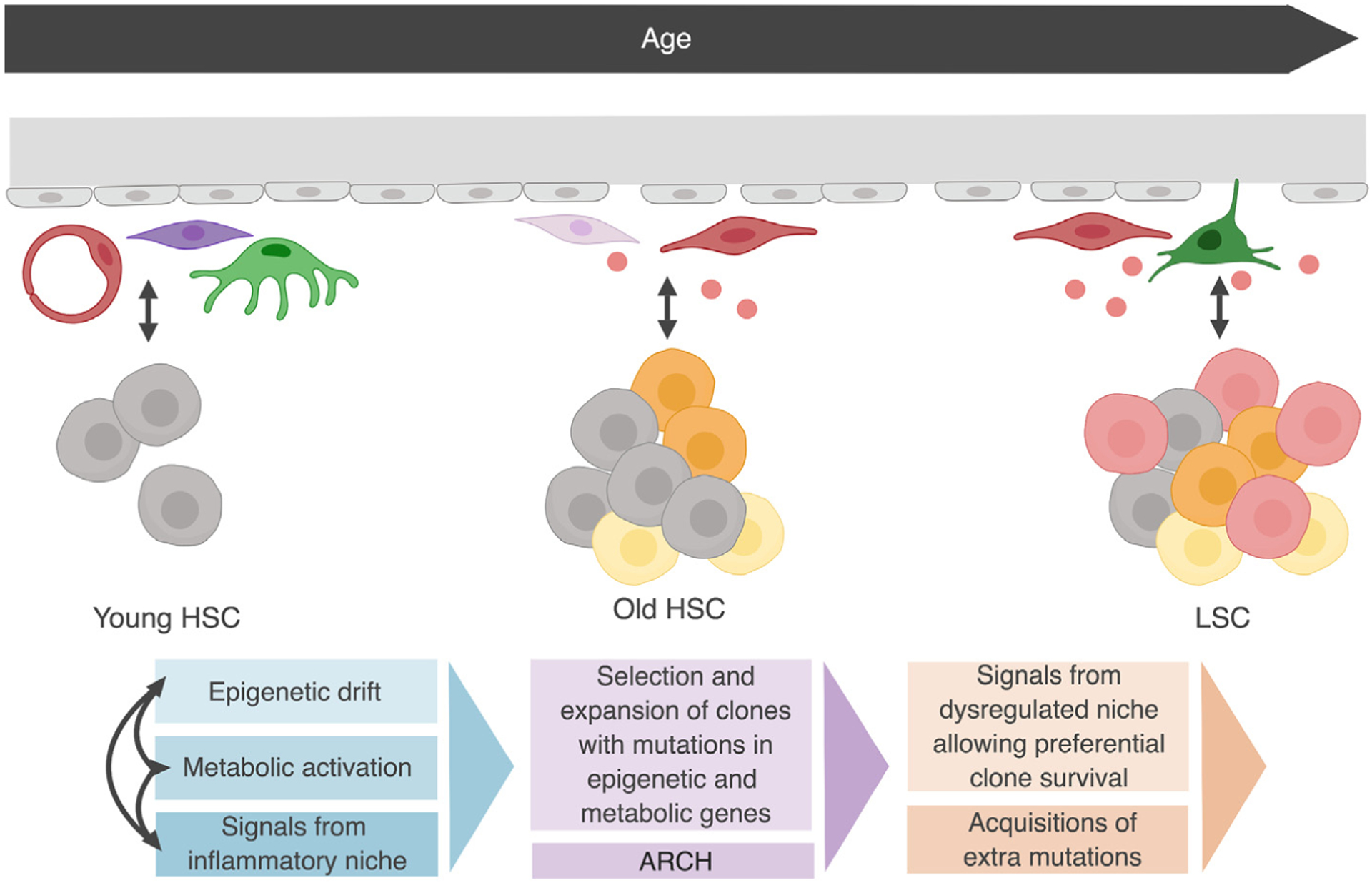
Model for how age-related changes in hematopoietic stem cell (HSC) epigenetic and metabolic state and signals from the dysregulated aging bone marrow (BM) microenvironment lead to clonal expansion and predisposition to leukemic transformation. Gray, functional HSCs; yellow/orange, HSCs that acquired mutations in epigenetic and metabolic genes; red, leukemic stem cells (LSCs). Partially based on [159].
New Insights into the Rational Design of Antiaging Interventions
Metabolism, epigenetics, and niche-mediated inflammatory signaling pathways have emerged as critical interrelated determinants of HSC aging and leukemic transformation. Several approaches aimed at restoring the metabolic and epigenetic profiles of old HSCs, and rebuilding the supportive activity of or blocking inflammatory signals from the BM microenvironment, have been recently described in mouse models of aging. Overexpression of Sirt3 [10] and Sirt7 [9] or treatment with the mTOR inhibitor rapamycin [11] and the CDC42 inhibitor CASIN [70] are proposed to restore HSC function by rejuvenating metabolic and epigenetic programs. Similarly, suppression of the inflammatory cytokine RANTES mitigates myeloid bias in HSCs from middle-aged mice [14]. Recent studies also demonstrate the potential for restored niches to alleviate aspects of HSC aging phenotypes, with improved repopulation capacity of old HSCs on young EC transplantation [16], reactivation of Notch signaling in old ECs [18], and administration of the ADRb3-selective agonist BRL3734 to restore adrenergic function and old MSC activity [15]. It is therefore likely that a combination of interventions concurrently targeting cell-intrinsic deregulation and cell-extrinsic environmental factors will be needed to develop effective therapies targeting age-related functional decline of the hematopoietic system.
An important limitation in the assessment and implementation of antiaging interventions is that specific criteria for the assessment of HSC rejuvenation have not yet been established and vary from study to study as described in Table 3. The HSC-aging community must therefore formulate standardized guidelines for assessing rejuvenation or the functional restoration of old HSCs. In this context, it is important to note that decreased HSC engraftment and myeloid-biased blood regeneration are features of aging that are also found in response to regenerative stress and inflammation [146,147] and therefore cannot be solely equated with aging. While approaches enforcing HSC replication through several rounds of chemotherapeutic treatment [47] or repeated injection of inflammatory mediators [148] are used to mimic the cellular features of aging, they have significantly different molecular consequences on HSCs than physiological aging. We suggest that long-lasting cell-intrinsic changes such as replication stress (Box 2), loss of polarity, or epigenetic or metabolic status [7] should be used instead as a hallmark of a molecularly old HSC state and as a benchmark by which to test the efficacy of rejuvenation interventions. It also remains to be seen whether criteria defined in mice are relevant for human antiaging interventions. Multiple phenotypic and functional differences between human and mouse hematopoietic systems have been previously described [149]. However, many of the features of mouse blood aging, including HSC pool expansion, reduced proliferative capacity, lineage bias, and increased number of γH2AX loci, are recapitulated in human BM samples [150–153], hence confirming the relevance of murine models for understanding human aging.
Table 3.
Methods for Assessing HSC Aging
| HSC feature | Effect of age | Method of measurement | Advantage | Limitation |
|---|---|---|---|---|
| Pool size | Expanded with age [1] | Flow cytometry-based analyses of BM populations | Quantitative | Does not reflect functionality |
| Repopulation capacity | Decreased with age [1] | Flow cytometry-based analyses of multilineage blood and BM chimerism after transplantation | Quantitative; reflects function; clinically relevant | Dependent on transplantation method (i.e., lethal vs sub-lethal irradiation, purified HSCs vs whole BM, competitive vs noncompetitive setting, etc.); similar effects observed on regenerative and inflammatory stresses; readout takes months |
| Lineage potential | Lymphoid deficiency with age [1] | Flow cytometry-based analyses of lymphoid and myeloid blood and BM populations | Quantitative; reflects function; clinically relevant | Dependent on engraftment efficiency; HSC exhaustion can also result in improved lymphomyeloid ratio since lymphoid cells have longer turnover time than myeloid cells; similar effects observed on regenerative and inflammatory stress; readout takes months |
| Replication stress | Increased with age [7,15,145] | Tracking division kinetics in culture; immunofluorescence-based visualization of γH2A.x and fibrillarin colocalization | Purified HSCs; scalable; quantitative, molecular feature | Dependent on cell cycle status |
| DNA damage | Increased with age [1] | Immunofluorescence-based γH2A.x staining; alkaline COMET assays | Purified HSCs; scalable; quantitative, molecular feature | Unclear how it relates to functionality |
| Cell polarity | Loss of polarity with age [15,17,70,189]. | Immunofluorescence-based visualization of Cdc42 and β-actin | Purified HSCs; scalable; quantitative, molecular feature | Unclear how it relates to functionality |
| Surface marker | Increased expression with age [54,102,190] | Flow cytometry-based analyses of CD150 and CD41 levels in BM populations | Quantitative | Unclear how it relates to functionality; CD41 is also upregulated in inflammatory conditions and CD150 on HSC activation |
| Cell cycle status | Loss of quiescence with age [7,147] | Flow cytometry-based cell cycle analyses; BrdU/EdU incorporation assays | Quantitative | Unclear how it relates to functionality; influenced by inflammation |
Moreover, the role of dysregulated pathways identified in aging is usually assessed in genetic models in young mice and needs to be directly validated in aged organisms, as the same triggering event can result in different phenotypes in young and old organisms. For instance, transformation of young and old BM cells with the BCR-ABL oncogene leads to the development of different types of MPN, with recipients of transformed hematopoietic cells from young mice developing both B cell leukemia and MPN and recipients of old transformed BM developing MPN without lymphoid involvement [154]. Additionally, recent reports show that the transcriptional, epigenetic, and mutational profile of adult AML is significantly different from that of pediatric AML, regarding their metabolic and inflammatory gene signatures and stromal remodeling capacities [155,156]. It is also plausible that our ability to effectively intervene against age-associated dysfunction becomes limited after a certain stage of the lifespan, and therefore time-course studies are required to establish the optimal timing for interventions. Additionally, while attrition of stem cell function has been suggested as one of the drivers of tissue degeneration [60], it remains to be proved that targeting the HSC compartment will be sufficient to meaningfully delay the onset or reverse blood aging phenotypes.
Concluding Remarks and Future Perspectives
The identification of epigenetic drift and metabolic activation as drivers of cell-intrinsic HSC aging and leukemic transformation opens exciting new therapeutic routes to restore hematopoietic function. The role of the deteriorating BM microenvironment in promoting HSC dysfunction in aging, and the dependence of LSCs on a perturbed BM niche, open complementary avenues. Future research (see Outstanding Questions) should focus on developing an integrated perspective on the complex interplay of the altered niche in the mediation of cell-intrinsic changes to HSCs. Given that these changes are interconnected and occur concurrently, the key questions are the extent to which they are causal or compensatory, whether any of them are rate limiting in the promotion of cellular and organismal dysfunction, and, as a corollary, whether targeting a specific age-related perturbation will be sufficient to promote health span or organismal longevity. Building an integrated perspective and the development of rejuvenation standards will benefit the hematopoietic field greatly and will help in developing a coherent model and actionable therapeutic strategies for this universal, yet poorly understood, process.
Outstanding Questions.
How are different features of cell-intrinsic HSC aging interrelated? Which features are rate limiting in HSC functional decline?
What comes first: the aging of the BM microenvironment/inflammation or intrinsic changes in the hematopoietic system, in particular HSCs?
Is aging of the hematopoietic system reversible or preventable? If yes, at which age should interventions be targeted?
How representative are mouse models of hematopoietic aging or leukemia development? Should further modeling be conducted in aged mice instead of young animals to understand the context dependency of genetic and pharmacological perturbations?
How does blood aging and HSC aging fit into our general understanding of organismal aging?
Highlights.
Metabolic activation and epigenetic alterations emerge as major contributors to the decline of hematopoietic stem cell (HSC) function with age. It has also become evident that epigenetic and metabolic regulation of HSCs are tightly linked.
Recent data provide new insights into the role of the bone marrow niche microenvironment in regulating HSC function. In particular, the role of the inflammatory microenvironment in enforcing the preferential transformation and survival of mutation-bearing clones has emerged during aging and leukemogenesis.
Mutations in epigenetic modifier and metabolic genes are associated with age-related clonal hematopoiesis (ARCH). Specific mutational signatures of ARCH can predict leukemia onset years before diagnosis and ARCH has been found associated with a range of nonhematopoietic morbidities and high mortality.
Restoration of metabolic and epigenetic profiles and blocking of inflammatory signaling have been shown to be prime targets for the rejuvenation of aged HSCs and prevention of leukemic transformation.
Clinician’s Corner.
Aging interventions need to hit clinically tangible endpoints to be pragmatically translated.
Mutations in epigenetic readers in age-related clonal hematopoiesis are frequent initiating events that support clonal competition and preneoplastic stem cells [128,158], suggesting that epigenetic deregulation is one of the central promoters of leukemic transformation.
Recent evidence suggests that targeting metabolic cofactors such as ascorbate can influence the epigenetic poising of HSCs and allow them to compensate for mutations in Tet2, potentially preventing or slowing leukemogenesis [38].
Blocking inflammatory signaling pathways with therapeutic agents may have the capacity to delay or restore age-related niche deterioration, which may be sufficient to prevent age-related leukemic transformation.
Analysis of peripheral blood mutation profiles in the elderly can be used to predict and monitor leukemia development.
Acknowledgments
E.V.V. is supported by a Rubicon Grant from The Netherlands Organisation for Scientific Research, a Stem Cell Grant from BD Biosciences, and the Empire State Stem Cell Fund through New York State Department of Health Contract #DOH01-C30291GG-3450000 to Columbia University. This work was supported by an NIH 1R35HL135763 grant and LLS Scholar Award to E.P.
Glossary
- Additional sex combs-like transcriptional regulator 1 (ASXL1)
a protein associated with PRCs that maintains the methylation of histone H3K27, an important repressive histone mark.
- Age-related clonal hematopoiesis (ARCH)
expansion of clones that acquired fitness advantage as a result of mutation that is observed in older individuals; 90% of ARCH mutations occur in enzymes involved in the regulation of epigenetics and metabolism, including TET2, DNMT3A, and ASXL1. ARCH-associated mutations are also frequently found in blood cancers.
- Clonal expansion
while in the context of lymphopoiesis, clonal expansion describes the specific amplification of antigen-specific cells, in the context of myelopoiesis the term usually reflects the outgrowth of a mutation-bearing clone that acquires competitive advantage over other hematopoietic clones.
- DNA methyltransferase 3 alpha (DNMT3A)
catalyst of methylation of CpG islands.
- Epigenetic drift
accumulation of epigenetic changes through successive cell divisions that accompanies aging. It can be caused by defects in the transmission of epigenetic information [157].
- Genomic instability
increased levels of acquisition of mutations particularly during cell division.
- Glycolysis
oxygen-independent metabolic pathway located in the cytosol.
- Hematopoietic stem cell (HSC) niche
component of the BM microenvironment that provides cell-cell interactions and soluble factors that are essential for the regulation of HSC fates, including quiescence, cycling, migration, and differentiation.
- Histone marks
covalent post-translational modifications of histone proteins (including methylation, acetylation, etc.) that can affect DNA confirmation and accessibility and therefore lead to changes in gene expression.
- Inflammaging
chronic low-level sterile inflammation observed in aged individuals that is associated with increased levels of morbidity and mortality.
- Isocitrate dehydrogenase 1 and 2 (IDH1/2)
convert isocitrate into αKG and produce NADPH. Mutant forms of IDH1/2 produce oncogenic R-2-hydroxyglutarate, an inhibitor of αKG-dependent dioxygenases such as TET2.
- Methylcytosine dioxygenase 2 (TET2)
an important player in DNA demethylation (especially in CpG motifs) that can result in transcriptional regulation. Catalyzes the conversion of 5-methylcytosine into 5-hydroxymethylcytosine. TET2 function is dependent on αKG as its cofactor.
- Oxidative phosphorylation (OXPHOS)
oxygen-dependent metabolic pathway located in mitochondria.
- Quiescence
The G0 stage of the cell cycle; a low activity state of reversible growth arrest.
- Replicative history
number of divisions that HSCs encounter over a certain time. Generally, such divisional history is negatively associated with HSC functionality.
- Tumor protein p53 (TP53)
tumor suppressor gene; a transcriptional activator protein that is mutated in the majority of human cancers.
References
- 1.de Haan G and Lazare SS (2018) Aging of hematopoietic stem cells. Blood 131, 479–487 [DOI] [PubMed] [Google Scholar]
- 2.Geiger H et al. (2013) The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol 13, 376–389 [DOI] [PubMed] [Google Scholar]
- 3.McKerrell T and Vassiliou GS (2015) Aging as a driver of leukemogenesis. Sci. Transl. Med 7, 306fs38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busch K et al. (2015) Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518, 542–546 [DOI] [PubMed] [Google Scholar]
- 5.Sawai CM et al. (2016) Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity 45, 597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schepers K et al. (2015) Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell 16, 254–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flach J et al. (2014) Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho TT et al. (2017) Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohrin M et al. (2015) A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown K et al. (2013) SIRT3 reverses aging-associated degeneration. Cell Rep. 3, 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen C et al. (2009) mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal 2, ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun D et al. (2014) Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14, 673–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chambers SM et al. (2007) Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 5, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ergen AV et al. (2012) Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 119, 2500–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maryanovich M et al. (2018) Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med 24, 782–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poulos MG et al. (2017) Endothelial transplantation rejuvenates aged hematopoietic stem cell function. J. Clin. Invest 127, 4163–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guidi N et al. (2017) Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. EMBO J. 36, 840–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kusumbe AP et al. (2016) Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 532, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandel NS et al. (2016) Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol 18, 823–832 [DOI] [PubMed] [Google Scholar]
- 20.López-Otín C et al. (2016) Metabolic control of longevity. Cell 166, 802–821 [DOI] [PubMed] [Google Scholar]
- 21.Shyh-Chang N et al. (2013) Stem cell metabolism in tissue development and aging. Development 140, 2535–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito K and Suda T (2014) Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol 15, 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snoeck H-W (2017) Mitochondrial regulation of hematopoietic stem cells. Curr. Opin. Cell Biol 49, 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takubo K et al. (2010) Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell 7, 391–402 [DOI] [PubMed] [Google Scholar]
- 25.Suda T et al. (2011) Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9, 298–310 [DOI] [PubMed] [Google Scholar]
- 26.Simsek T et al. (2010) The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7, 380–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takubo K et al. (2013) Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halvarsson C et al. (2017) Pyruvate dehydrogenase kinase 1 is essential for transplantable mouse bone marrow hematopoietic stem cell and progenitor function. PLoS One 12, e0171714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maryanovich M et al. (2015) An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat. Commun 6, 7901. [DOI] [PubMed] [Google Scholar]
- 30.Yu W-M et al. (2013) Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 12, 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ansó E et al. (2017) The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol 19, 614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guitart AV et al. (2017) Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. J. Exp. Med 214, 719–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cabezas-Wallscheid N et al. (2014) Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522 [DOI] [PubMed] [Google Scholar]
- 34.Martínez-Reyes I et al. (2016) TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 61, 199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shyh-Chang N and Ng H-H (2017) The metabolic programming of stem cells. Genes Dev. 31, 336–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko M et al. (2011) Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. U. S. A 108, 14566–14571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cimmino L et al. (2017) Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agathocleous M et al. (2017) Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Challen GA et al. (2012) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet 44, 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong M et al. (2018) Loss of Dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep. 23, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beerman I and Rossi DJ (2015) Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell 16, 613–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan W-I et al. (2011) The transcriptional coactivator CBP regulates self-renewal and differentiation in adult hematopoietic stem cells. Mol. Cell. Biol 31, 5046–5060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katsumoto T et al. (2008) Roles of the histone acetyltransferase monocytic leukemia zinc finger protein in normal and malignant hematopoiesis. Cancer Sci. 99, 1523–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loizou JI et al. (2009) Histone acetyltransferase cofactor Trrap is essential for maintaining the hematopoietic stem/progenitor cell pool. J. Immunol 183, 6422–6431 [DOI] [PubMed] [Google Scholar]
- 45.Li X et al. (2018) Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol 19, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schvartzman JM et al. (2018) Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol 217, 2247–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beerman I et al. (2013) Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan T et al. (2015) An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet. 11, e1004996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warr MR et al. (2013) FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehta A et al. (2015) The microRNA-132 and microRNA-212 cluster regulates hematopoietic stem cell maintenance and survival with age by buffering FOXO3 expression. Immunity 42, 1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rimmelé P et al. (2014) Aging-like phenotype and defective lineage specification in SIRT1-deleted hematopoietic stem and progenitor cells. Stem Cell Rep. 3, 44–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H et al. (2016) SIRT6 controls hematopoietic stem cell homeostasis through epigenetic regulation of Wnt signaling. Cell Stem Cell 18, 495–507 [DOI] [PubMed] [Google Scholar]
- 53.Luo H et al. (2019) Mitochondrial stress-initiated aberrant activation of the NLRP3 inflammasome regulates the functional deterioration of hematopoietic stem cell aging. Cell Rep. 26, 945–954.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernitz JM et al. (2016) Hematopoietic stem cells count and remember self-renewal divisions. Cell 167, 1296–1309.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu VWC et al. (2016) Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell 167, 1310–1322.e17 [DOI] [PubMed] [Google Scholar]
- 56.Qiu J et al. (2014) Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Rep. 2, 473–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Säwén P et al. (2016) Mitotic history reveals distinct stem cell populations and their contributions to hematopoiesis. Cell Rep. 14, 2809–2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beerman I et al. (2014) Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 15, 37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teschendorff AE et al. (2010) Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20, 440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.López-Otín C et al. (2013) The hallmarks of aging. Cell 153, 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodgers JT et al. (2014) mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert. Nature 510, 393–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qian P et al. (2016) The Dlk1-Gtl2 locus preserves LT-HSC function by inhibiting the PI3K–mTOR pathway to restrict mitochondrial metabolism. Cell Stem Cell 18, 214–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kharas MG et al. (2010) Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 115, 1406–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh S et al. (2018) Id1 ablation protects hematopoietic stem cells from stress-induced exhaustion and aging. Cell Stem Cell 23, 252–265.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saito Y et al. (2015) AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell 17, 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pei S et al. (2018) AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell 23, 86–100.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ito K et al. (2016) Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354, 1156–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ito K et al. (2012) A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med 18, 1350–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McDonnell E et al. (2016) Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 17, 1463–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Florian MC et al. (2012) Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 10, 520–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Florian MC et al. (2018) Aging alters the epigenetic asymmetry of HSC division. PLoS Biol. 16, e2003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y-H et al. (2014) Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158, 1309–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lagadinou ED et al. (2013) BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12, 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Medeiros BC et al. (2017) Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 31, 272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DiNardo CD et al. (2018) Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, Phase 1b study. Lancet Oncol. 19, 216–228 [DOI] [PubMed] [Google Scholar]
- 76.Pollyea DA et al. (2018) Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med 24, 1859–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jones CL et al. (2018) Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 34, 724–740.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taya Y et al. (2016) Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science 354, 1152–1155 [DOI] [PubMed] [Google Scholar]
- 79.Hattori A et al. (2017) Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 545, 500–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raffel S et al. (2017) BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388 [DOI] [PubMed] [Google Scholar]
- 81.Sykes DB et al. (2016) Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 167, 171–186.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ricciardi MR et al. (2015) Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood 126, 1925–1929 [DOI] [PubMed] [Google Scholar]
- 83.Karigane D and Takubo K (2017) Metabolic regulation of hematopoietic and leukemic stem/progenitor cells under homeostatic and stress conditions. Int. J. Hematol 106, 18–26 [DOI] [PubMed] [Google Scholar]
- 84.Crane GM et al. (2017) Adult haematopoietic stem cell niches. Nat. Rev. Immunol 17, 573–590 [DOI] [PubMed] [Google Scholar]
- 85.Kunisaki Y et al. (2013) Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bruns I et al. (2014) Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med 20, 1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao M et al. (2014) Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med 20, 1321–1326 [DOI] [PubMed] [Google Scholar]
- 88.Greenbaum A et al. (2013) CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ding L et al. (2012) Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamazaki S et al. (2011) Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147, 1146–1158 [DOI] [PubMed] [Google Scholar]
- 91.Ding L and Morrison SJ (2013) Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cordeiro Gomes A et al. (2016) Hematopoietic stem cell niches produce lineage-instructive signals to control multipotent progenitor differentiation. Immunity 45, 1219–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rankin EB et al. (2012) The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell 149, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Omatsu Y et al. (2010) The essential functions of adipoosteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33, 387–399 [DOI] [PubMed] [Google Scholar]
- 95.Hérault A et al. (2017) Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 544, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Almeida M et al. (2007) Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem 282, 27285–27297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.El Assar M et al. (2013) Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med 65, 380–401 [DOI] [PubMed] [Google Scholar]
- 98.Almeida M and O’Brien CA (2013) Basic biology of skeletal aging: role of stress response pathways. J. Gerontol. A Biol. Sci. Med. Sci 68, 1197–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gomariz A et al. (2018) Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat. Commun 9, 2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tanaka T et al. (2018) Plasma proteomic signature of age in healthy humans. Aging Cell 17, e12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Franceschi C and Campisi J (2014) Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci 69, S4–S9 [DOI] [PubMed] [Google Scholar]
- 102.Kovtonyuk LV et al. (2016) Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front. Immunol 7, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Loi F et al. (2016) Inflammation, fracture and bone repair. Bone 86, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vandoorne K et al. (2018) Imaging the vascular bone marrow niche during inflammatory stress. Circ. Res 123, 415–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prendergast ÁM et al. (2017) IFNα-mediated remodeling of endothelial cells in the bone marrow niche. Haematologica 102, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mirantes C et al. (2014) Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp. Cell Res 329, 248–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mann M et al. (2018) Heterogeneous responses of hematopoietic stem cells to inflammatory stimuli are altered with age. Cell Rep. 25, 2992–3005.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Duarte D et al. (2018) The interplay of leukemia cells and the bone marrow microenvironment. Blood 131, 1507–1511 [DOI] [PubMed] [Google Scholar]
- 109.Walkley CR et al. (2007) A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell 129, 1097–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Raaijmakers MHGP et al. (2010) Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464, 852–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zambetti NA et al. (2016) Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell 19, 613–627 [DOI] [PubMed] [Google Scholar]
- 112.Kode A et al. (2014) Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 506, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang L et al. (2014) Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-κB dependent manner. Cell Stem Cell 15, 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dong L et al. (2016) Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 539, 304–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duan C-W et al. (2014) Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 25, 778–793 [DOI] [PubMed] [Google Scholar]
- 116.Hameed A et al. (2014) Bone disease in multiple myeloma: pathophysiology and management. Cancer Growth Metastasis 7, 33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hawkins ED et al. (2016) T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 538, 518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schepers K et al. (2013) Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 13, 285–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hanoun M et al. (2014) Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 15, 365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Duarte D et al. (2018) Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell 22, 64–77.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arranz L et al. (2014) Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 512, 78–81 [DOI] [PubMed] [Google Scholar]
- 122.Wang W et al. (2016) Aberrant Notch signaling in the bone marrow microenvironment of acute lymphoid leukemia suppresses osteoblast-mediated support of hematopoietic niche function. Cancer Res. 76, 1641–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mead AJ et al. (2017) Niche-mediated depletion of the normal hematopoietic stem cell reservoir by Flt3-ITD-induced myeloproliferation. J. Exp. Med 214, 2005–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shastri A et al. (2018) Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J. Clin. Invest 128, 5479–5488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mitchell K et al. (2018) IL1RAP potentiates multiple oncogenic signaling pathways in AML. J. Exp. Med 215, 1709–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Meisel M et al. (2018) Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cai Z et al. (2018) Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell 23, 833–849.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jaiswal S et al. (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xie M et al. (2014) Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med 20, 1472–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Laurie CC et al. (2012) Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet 44, 642–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jacobs KB et al. (2012) Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet 44, 651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Busque L et al. (2012) Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet 44, 1179–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Genovese G et al. (2014) Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fuster JJ et al. (2017) Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Coombs CC et al. (2017) Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 21, 374–382.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jaiswal S et al. (2017) Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med 377, 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Risques RA and Kennedy SR (2018) Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 14, e1007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jan M et al. (2012) Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med 4, 149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Corces-Zimmerman MR et al. (2014) Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. U. S. A 111, 2548–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Desai P et al. (2018) Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med 24, 1015–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Abelson S et al. (2018) Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Marusyk A et al. (2010) Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 8, e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen J et al. (2018) Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med 25, 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vas V et al. (2012) Aging of the microenvironment influences clonality in hematopoiesis. PLoS One 7, e42080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ganuza M et al. (2019) The global clonal complexity of the murine blood system declines throughout life and after serial transplantation. Blood Published online February 19, 2019 10.1182/blood-2018-09-873059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pietras EM et al. (2016) Chronic interleukin-1 drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol 18, 607–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pietras EM et al. (2015) Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 17, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Walter D et al. (2015) Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552 [DOI] [PubMed] [Google Scholar]
- 149.Parekh C and Crooks GM (2013) Critical differences in hematopoiesis and lymphoid development between humans and mice. J. Clin. Immunol 33, 711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rübe CE et al. (2011) Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One 6, e17487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pang WW et al. (2011) Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. U. S. A 108, 20012–20017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nilsson AR et al. (2016) Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS One 11, e0158369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kuranda K et al. (2011) Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell 10, 542–546 [DOI] [PubMed] [Google Scholar]
- 154.Signer RAJ et al. (2007) Age-related defects in B lymphopoiesis underlie the myeloid dominance of adult leukemia. Blood 110, 1831–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bolouri H et al. (2018) The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med 24, 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chaudhury S et al. (2018) Age-specific biological and molecular profiling distinguishes paediatric from adult acute myeloid leukaemias. Nat. Commun 9, 5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fraga MF et al. (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U. S. A 102, 10604–10609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shlush LI (2018) Age-related clonal hematopoiesis. Blood 131, 496–504 [DOI] [PubMed] [Google Scholar]
- 159.Chung SS and Park CY (2017) Aging, hematopoiesis, and the myelodysplastic syndromes. Blood Adv. 1, 2572–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mohrin M et al. (2010) Hematopoietic stem cell quiescence promotes error prone DNA repair and mutagenesis. Cell Stem Cell 7, 174–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Garaycoechea JI et al. (2012) Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489, 571–575 [DOI] [PubMed] [Google Scholar]
- 162.Garaycoechea JI et al. (2018) Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 553, 171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rossi DJ et al. (2007) Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725–729 [DOI] [PubMed] [Google Scholar]
- 164.Moehrle BM and Geiger H (2016) Aging of hematopoietic stem cells: DNA damage and mutations? Exp. Hematol 44, 895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kottemann MC and Smogorzewska A (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493, 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nijnik A et al. (2007) DNA repair is limiting for haematopoietic stem cells during ageing. Nature 447, 686–690 [DOI] [PubMed] [Google Scholar]
- 167.Gutierrez-Martinez P et al. (2018) Diminished apoptotic priming and ATM signalling confer a survival advantage onto aged haematopoietic stem cells in response to DNA damage. Nat. Cell Biol 20, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Moehrle BM et al. (2015) Stem cell-specific mechanisms ensure genomic fidelity within HSCs and upon aging of HSCs. Cell Rep. 13, 2412–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Norddahl GL et al. (2011) Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell 8, 499–510 [DOI] [PubMed] [Google Scholar]
- 170.Mortensen M et al. (2011) The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med 208, 455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.de Almeida MJ et al. (2017) Dye-independent methods reveal elevated mitochondrial mass in hematopoietic stem cells. Cell Stem Cell 21, 725–729.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jin G et al. (2018) Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol 19, 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.TeSlaa T et al. (2016) α-Ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab. 24, 485–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Laukka T et al. (2015) Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem 291, 4256–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gaude E and Frezza C (2014) Defects in mitochondrial metabolism and cancer. Cancer Metab. 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang H et al. (2016) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443 [DOI] [PubMed] [Google Scholar]
- 177.Challen GA et al. (2014) Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 15, 350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rao RC and Dou Y (2015) Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 15, 334–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dent SY and Chandra J (2013) The lasting influence of LSD1 in the blood. eLife 2, e00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sun H et al. (2016) Lysine-specific histone demethylase 1 inhibition promotes reprogramming by facilitating the expression of exogenous transcriptional factors and metabolic switch. Sci. Rep 6, 30903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fistonich C et al. (2018) Cell circuits between B cell progenitors and IL-7+ mesenchymal progenitor cells control B cell development. J. Exp. Med 215, 2586–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kusumbe AP et al. (2014) Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507, 323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Farr JN et al. (2017) Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med 23, 1072–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Naveiras O et al. (2009) Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460, 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhou BO et al. (2017) Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol 19, 891–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ambrosi TH et al. (2017) Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell 20, 771–784.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Quail DF and Dannenberg AJ (2019) The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol 15, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hilpert M et al. (2014) p19INK4d controls hematopoietic stem cells in a cell-autonomous manner during genotoxic stress and through the microenvironment during aging. Stem Cell Rep. 3, 1085–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Grigoryan A et al. (2018) LaminA/C regulates epigenetic and chromatin architecture changes upon aging of hematopoietic stem cells. Genome Biol. 19, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gekas C and Graf T (2013) CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 121, 4463–4472 [DOI] [PubMed] [Google Scholar]