

Abstract
Cellular senescence is the state of irreversible cell cycle arrest that can be induced by a variety of potentially oncogenic stimuli and has therefore long been considered to suppress tumorigenesis, acting as a guardian of homeostasis. However, surprisingly, emerging evidence reveals that senescent cells also promote secretion of a series of inflammatory cytokines, chemokines, growth factors and matrix remodeling factors, which alter the local tissue environment and contribute to chronic inflammation and cancer. This newly identified senescence phenotype, termed the senescence‐associated secretory phenotype (SASP) or the senescence‐messaging secretome (SMS), is induced by DNA damage that promotes the induction of cellular senescence. All of these senescence‐associated secreting factors are involved in homeostatic disorders such as cancer. Therefore, it is quite possible that accumulation of senescent cells during the aging process in vivo might contribute to age‐related increases in homeostatic disorders. In this review, current knowledge of the molecular and cellular biology of cellular senescence is introduced, focusing on its positive and negative roles in controlling tissue homeostasis in vivo.
Higher eukaryotes, including humans, fulfill their life cycles with embryogenesis, birth, growth, maturity and aging. Human life expectancy is very long, since each life stage is expanded, compared with those of other higher eukaryotes. The major cause for longer human life is thought to be the precise mechanisms of cellular and organismal homeostatic control.1 However, the extended human lifespan could result in a startling rise in the incidence of cancer in later life.2 To meaningfully impact the healthcare and the well‐being of this aging population, there is an urgent need for enhanced understanding of the molecular mechanisms maintaining cellular homeostasis and the consequences of its disruption, which might increase aging‐associated cancer. One of the emerging mechanisms for maintaining cellular homeostasis is “cellular senescence”. Cellular senescence is the state of essentially irreversible cell cycle arrest induced by a variety of potentially oncogenic stimuli, such as telomere erosion, oxidative stress or activation of certain oncogenes, and is considered to act as an important tumor suppression mechanism.3, 4, 5, 6, 7, 8 Thus, cellular senescence has long been considered a fail‐safe mechanism against carcinogenesis by maintaining cellular and organismal homeostasis. However, recently it has been suggested that senescent cells have the potential to secrete various inflammatory cytokines, chemokines and matrix remodelling factors that deleteriously alter tissue homeostasis, leading to chronic inflammation and/or cancer promotion.8, 9 Therefore, it is quite possible that the accumulation of senescent cells in vivo might contribute to age‐related increases in cancer and other aging‐associated illnesses as well. In this review, the molecular events associated with these two faces of cellular senescence are discussed.
What is cellular senescence?
Most normal human somatic cells can divide only a limited number of times in culture. They permanently stop dividing after a finite number of cell divisions and enter a state of irreversible cell proliferation arrest.10, 11 This phenomenon is called “cellular senescence” or “replicative cellular senescence”. Unlike quiescent (G0 phase) cells, which are induced by low serum or contact inhibition conditions, senescent cells are irreversibly arrested predominantly in the G1 phase and are no longer able to divide even with proliferative stimuli, although they remain viable and metabolically active for long periods of time (Fig. 1).3, 4, 5, 6, 7, 8 In contrast, most cancer cells appear to have bypassed this proliferative limit and evaded cellular senescence3, 4, 5, 6, 7, 8. Thus, cellular senescence has long been considered as a barrier to cancer by playing an important role in preventing the extensive cell divisions required for malignant transformation.3, 4, 5, 6, 7, 8
Figure 1.
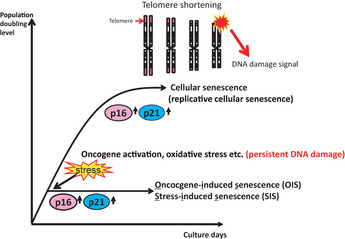
What is cellular senescence? Most normal human somatic cells stop dividing after a finite number of cell divisions and enter a state of irreversible cell proliferation arrest. This phenomenon is called “cellular senescence” or “replicative cellular senescence”. In human cells, the mechanism underlying replicative cellular senescence is thought to be telomere shortening. Recent studies in human cells revealed that a similar irreversible proliferation block can be induced quite rapidly when normal cells are exposed to a variety of potentially oncogenic stimuli, such as excessive levels of reactive oxygen species, treatment with DNA damaging agents or activation of certain oncogenes. Because all of these stimuli induce irreparable DNA damage in both human and murine cells, it is plausible that persistent DNA damage signals induce cellular senescence. These types of oncogenic stress‐induced senescence are now referred to as “oncogene‐induced senescence” or “stress‐induced senescence”. Both p16 and p21 upregulation contributes to the induction of both types of cellular senescence.
In human cells, the mechanism underlying replicative cellular senescence is thought to be telomere shortening. Telomeres are located at the ends of eukaryotic chromosomes and they have special DNA structures with repetitive DNA elements to protect the DNA ends from degradation and/or chromosomal end‐to‐end fusion.12 The telomere length is maintained by a specific enzyme called telomerase, which is expressed in human germ‐line cells or stem cells, but not in most normal somatic cells. Due to the nature of the DNA replication process and the lack of telomerase, telomeres become shorter with each round of cell division.13 Eventually, progressive telomere erosion results in telomere dysfunction and this is thought to initiate DNA damage response signals to activate p53‐dependent checkpoints that contribute to cellular senescence.12, 14
In contrast to human cells, there is no strong evidence that rodent cells undergo replicative cellular senescence by telomere erosion,15, 16 because the telomeres in rodent cells prepared from most laboratory animals are quite long and many somatic rodent cells have telomerase activity. However, the senescence‐like proliferative block in rodent cells occurs without detectable telomere shortening. This phenomenon suggests that a mechanism other than telomere shortening can cause cellular senescence in rodent cells. In this regard, it is interesting to note that primary mouse embryonic fibroblasts reportedly proliferate indefinitely if the cells are maintained under appropriate culture conditions, such as low oxygen conditions.17 In addition, rat oligodendrocyte precursor cells and rat Schwann cells do not senesce in serum‐free medium, but the presence of serum induces senescence.18 These findings clearly demonstrate that cellular senescence can be induced without apparent telomere shortening when cells are exposed to non‐physiological conditions in vitro. Notably, the fact that anti‐oxidant treatment delays cellular senescence induction strongly suggests that reactive oxygen species (ROS) can trigger cellular senescence.1, 2, 8 Recent studies in human cells revealed that a similar irreversible proliferation block can be induced quite rapidly when normal cells are exposed to a variety of potentially oncogenic stimuli, such as excessive levels of ROS, treatment with DNA damaging agents or activation of certain oncogenes.3, 4, 5, 6, 7, 8, 19, 20, 21 Because all of these stimuli induce irreparable DNA damage in both human and murine cells, it is plausible that persistent DNA damage signals induce cellular senescence. These types of oncogenic stress‐induced senescence are now referred to as “oncogene‐induced senescence” (OIS) or “stress‐induced senescence” (SIS) (Fig. 1).
Molecular machinery of cellular senescence for tumor suppression
The basic mechanisms for the induction of senescence cell cycle arrest have been well documented recently.22,23 In mammalian cells, the RB and p53 tumor suppressor proteins play critical roles in the induction of cellular senescence.22,23 In particular, RB and its family members, p107 and p130, are essential for the onset of senescence cell cycle arrest.24, 25, 26, 27 The activities of RB‐family proteins are precisely regulated by phosphorylation, protein–protein interactions and other protein modifications.28,29 A series of cyclin‐dependent kinases (CDK) including CDK2, CDK4 and CDK6, play key roles in regulating the activities of RB‐family proteins.30 When RB is phosphorylated by these CDK, it loses its ability to bind to and repress the functions of the E2F family of transcription factors, thereby resulting in gene transcription required for the initiation of DNA replication and cell cycle progression (Fig. 2).28, 29, 30
Figure 2.
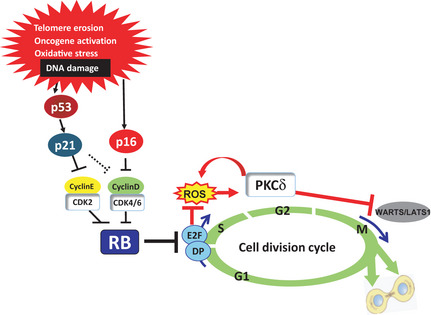
Molecular mechanisms of cellular senescence. A series of cyclin‐dependent kinases (CDK) including CDK2, CDK4 and CDK6 play key roles in regulating the activities of RB‐family proteins. When RB is phosphorylated by these CDK, it loses its ability to bind to and repress the functions of the E2F family of transcription factors, thereby resulting in gene transcription required for the initiation of DNA replication and cell cycle progression. In response to irreparable DNA damage caused by a variety of oncogenic stimuli, expression levels of the p21 Cip1 and p16 Ink4a CDK inhibitors are significantly upregulated by p53‐dependent and p53‐independent mechanisms, respectively. Activation of the p16Ink4a–Rb pathway cooperates with mitogenic signals to induce elevated intracellular levels of reactive oxygen species (ROS), thereby causing activation of PKCδ, a critical downstream mediator of the ROS signaling pathway, and leading to a cytokinetic block that might provide an additional safeguard against the proliferation of senescent cells, especially in the case of RB and p53 subsequently become inactivated.
Senescent cells arrest themselves in the G1 phase of the cell cycle and are no longer able to replicate by synthesizing DNA. In many cases, this is because the CDK are inactivated in senescent cells by CDK inhibitors (CDKI).30 There are two different classes of CDKI, the KIP/CIP family CDKI (p21Cip1, p27Kip1 and p57KipII) and the INK4 family CDKI (p16Ink4a, p15Ink4b, p18Ink4c and p19Ink4d). The KIP/CIP family members are known to inhibit a broad range of CDK, whereas the INK4 family proteins specifically bind and inactivate CDK4 and CDK6.30 In normal proliferating cells, the expression levels of CDKI are very low.31, 32, 33 However, in response to irreparable DNA damage caused by a variety of oncogenic stimuli, expression levels of the p21 Cip1 and p16 Ink4a genes are significantly upregulated by p53‐dependent and p53‐independent mechanisms, respectively.31, 32, 33, 34, 35 In addition, simultaneous induction of p21Cip1and p16Ink4a cooperatively and efficiently inactivates all CDK that phosphorylate RB‐family proteins,36, 37, 38 thereby causing senescence cell cycle arrest (Fig. 2). It is noteworthy that the senescence pathway including p16Ink4a, CDK and RB are highly deregulated in nearly all human cancers,39 illustrating the importance of the p16/RB pathway in tumor suppression through senescence induction.
Once RB is fully activated, particularly by p16Ink4a expression, senescence cell cycle arrest becomes irreversible and is no longer revoked by the subsequent inactivation of RB and p53 in human somatic cells.40 Interestingly, inactivation of RB and p53 enables human senescent cells to reinitiate DNA synthesis, but they subsequently fail to complete the cell cycle, suggesting that there might be additional block(s) in the G2 and/or M phase.40, 41, 42 We have previously shown that activation of the p16Ink4a–Rb pathway cooperates with mitogenic signals to induce elevated intracellular levels of reactive oxygen species (ROS), thereby causing activation of PKCδ, a critical downstream mediator of the ROS signaling pathway and leading to a cytokinetic block.43 Moreover, once it is activated by ROS, PKCδ in turn promotes further production of ROS, thus establishing a positive feedback loop to maintain ROS–PKCδ activation in senescent cells.43 Sustained activation of ROS–PKCδ signaling irreversibly blocks cytokinesis, at least partly by reducing the level of LATS1 (also known as WARTS), a mitotic exit network kinase essential for cytokinesis.43 Moreover, p21Cip1 is also required for autonomous ROS production in senescent cells,44 indicating that both p21Cip1and p16Ink4a play critical roles in inhibiting not only DNA replication but also cytokinesis in senescent cells. These lines of evidences suggest that the cytokinetic block might provide an additional safeguard against the proliferation of senescent cells, especially if RB and p53 subsequently become inactivated (Fig. 2).
Risk of chromosomal instability in senescent cells
As mentioned above, we have shown that an increased level of ROS and activation of ROS–PKCδ signaling in senescent cells causes the degradation of LATS1. The reduction of LATS1 in senescent cells could contribute to blocking cytokinesis of cells. However, importantly, the excess level of ROS is a well‐known DNA damaging agent45 and LATS1 is reported to have a tumor suppressive role.46 Moreover, the cytokinetic block is known to cause aneuploidy, which is associated with chromosomal instability, a hallmark of tumorigenesis.47
It is also worthwhile emphasizing that another mechanism linking cellular senescence and aneuploidy has recently emerged, which is associated with the accumulation of telomere damage in stress‐induced senescence.48, 49 Intriguingly, since DNA damage in the telomeric sequence resists repair, it has been reported that a large fraction of exogenously induced DNA damage tends to be accumulated in telomeres.48 Moreover, this DNA damage in telomeres is shown to be irrespective of telomere length or telomerase activity both in vitro and in vivo and is correlated with age.48, 49 In addition, Davoli et al.50 have recently shown that telomere damage has the potential to induce the bypass of mitosis through APC/CCdh1‐dependent degradation of Geminin, thereby resulting in whole genome reduplication and tetraploidy. Furthermore, they showed that the tetraploidization driven by telomere damage, together with inactivation of RB and/or p53 tumor suppressors, enhanced the tumorigenic transformation in mouse cells that resembled human aneuploid cancer cells.51 These findings strongly suggest that the “persistent” DNA damage of telomeres observed in senescent cells could cause cancerous aneuploidization.
It is well known that aneuploidy is a major cause of the transformation and progression of most human cancers. It is therefore possible that many of the aneuploid senescent cells could transform into malignant cancer cells. Indeed, it has been reported that tumorigenic cells spontaneously emerged from senescent cells.52 These notions imply that although the onset of cellular senescence initially acts as a barrier to cancer, accumulation of senescent cells might eventually promote tumorigenesis in later life, contributing to an age‐related increase in cancer.
Senescence‐associated secretome
There is no doubt that cellular senescence functions as an important tumor suppression mechanism. However, unlike apoptotic cells, senescent cells remain viable for long periods of time and accumulate with age in various organs and tissues. Indeed, we and others confirmed that senescent cells with high p16 expression accumulate in various aged organs (Fig. 3),53, 54 with accumulation of the DNA damage markers. Of particular biological importance, it has recently become apparent that senescent cells exhibit increased expression of genes encoding a series of secreted proteins, such as inflammatory cytokines, chemokines and matrix remodeling factors, which alter the local tissue environment and/or contribute to chronic inflammation and tumorigenesis.55, 56, 57 This newly identified senescent phenotype, termed the senescence‐associated secretory phenotype (SASP) or the senescence‐messaging secretome (SMS)57, 58 (hereafter referred to as senescence‐associated secretome), is observed in both replicative and oncogene‐and/or stress‐induced cellular senescence.57 The senescence‐associated secretome is known to be induced by DNA damage59 and can be beneficial or deleterious, depending on the biological context (Fig. 4).8, 9 Although the mechanisms of senescence‐associated secretomes have been investigated mainly using normal fibroblasts,55, 56, 57 a series of papers reported that the secretome by cellular senescence is observed in other types of cells such as human melanocytes,56 human endothelial cells60 and human breast epithelial cells,57 as well as cancerous cells such as human melanoma cells61 and mouse lymphoma cells62.
Figure 3.
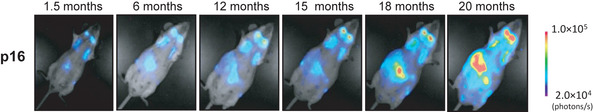
Dynamics of p16 Ink4a gene expression during the aging process in vivo. The p16‐luc mice, an imaging mice for p16 gene expression in vivo, were subjected to non‐invasive bioluminescence imaging throughout their entire life span. Accumulation of p16 was confirmed in the aged mouse.
Figure 4.
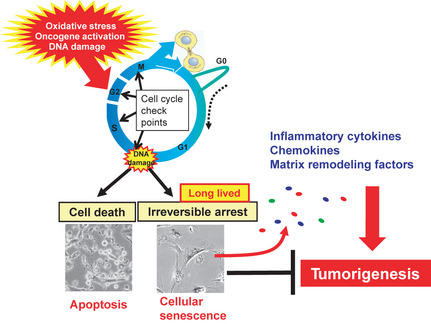
Senescence‐associated secretome. Irreparable DNA damage provokes either apoptosis or cellular senescence depending on the strength of stress and/or cellular context. Recently it has become apparent that long‐lived senescent cells exhibit increased expression of genes encoding a series of secreted proteins, such as inflammatory cytokines, chemokines and matrix remodeling factors, which alter the local tissue environment and/or contribute to chronic inflammation and tumorigenesis. Cellular senescence initially prevents proliferation of damaged cells, thereby acting as a fail‐safe mechanism. However, in the long term, senescent cells might eventually promote tumorigenesis by accelerating chromosomal instability and/or production of senescence‐associated secretome factors.
Factors such as IL‐6, IL‐8 and plasminogen activator inhibitor‐1 (PAI‐1), which are secreted from senescent fibroblasts, can reportedly promote tumor suppression by reinforcing the senescence proliferation block induced by an activated oncogene or oxidative stress.55, 56, 63 However, IL‐6 and IL‐8 are also known to promote malignant transformation in cooperation with certain oncogenes.64, 65, 66 Moreover, secreted factors from senescent fibroblasts have been shown to induce an epithelial–mesenchymal transition, an important step in cancer progression and metastasis.57 These findings, together with observations that the proteins secreted by senescent cells can promote degenerative hyper‐proliferative or metastatic changes in neighboring cells,57, 61 indicate that the release of the senescence‐associated secretome factors results in both beneficial and harmful consequences. Thus, these secreted factors can act in an autocrine manner to reinforce the senescence cell cycle arrest,55, 56, 62, 63 while in contrast they can have deleterious cell non‐autonomous side‐effects that promote tumorigenesis.9, 57, 61, 64, 65, 66 The senescence‐associated secretome might also be involved in the effect of the microenvironment of cancer tissue. Indeed, the stromal fibroblasts of human ovarian cancer tissue were reportedly senescent cells and secreted a tumor‐promoting chemokine,67 implying that the senescence‐associated secretome might promote human ovarian cancer. Therefore, considering the accumulation of senescent cells with age, it is possible that the deleterious side‐effects of the senescence‐associated secretome might also contribute to the age‐related increase in cancer (Fig. 4).
Epigenetic regulation of the senescence‐associated secretome
Both the DNA damage response (DDR) and the activation of NF‐κB and C/EBPβ transcription factors are reported to play key roles in the onset of the senescence secretome phenotype.55, 56, 57, 58, 61, 62, 68, 69, 70 However, it remains unclear how these two events cooperate to establish senescence‐associated secretome. Recently, we noted that the levels of histone 3 lysine 9 di‐methylation (H3K9me2), an epigenetic mark for euchromatic gene silencing,53, 71, 72, 73 were strikingly reduced around the IL‐6 and IL‐8 gene promoters in senescent human diploid fibroblasts (HDF).71 Moreover, we found that this phenomenon was due to the proteasomal degradation of G9a and GLP, the major H3K9 mono‐ and di‐methyltransferases, through DDR‐dependent activation of the APC/CCdh1 ubiquitin ligase.71 It is also noteworthy that GLP has been shown to bind to the NF‐κB subunit RelA and represses chromatin at RelA‐occupied genes, such as the IL‐6 gene, by promoting H3K9 methylation.74 Intriguingly, G9a has been shown to block the transcriptional activation potential of C/EBPβ through the methylation of lysine‐39 of C/EBPβ.75 All of these observations suggest that G9a/GLP might regulate senescence‐associated secretome factor gene expression through multiple mechanisms in response to persistent DNA damage in senescent cells.
Conclusions
It is clear that cellular senescence plays an important role in tumor suppression,3, 4, 5, 6, 7, 8, 9 as illustrated by the increase in tumor incidence in mice with an engineered block in senescence induction.38 Moreover, several lines of evidence suggest that senescent cells can facilitate tumor clearance in certain settings by promoting immunological responses in vivo.76, 77, 78 Thus, with no doubt, cellular senescence has a positive contribution to organismal defences against cancer. However, sustained cellular senescence is also associated with the secretion of pro‐inflammatory and pro‐proliferative factors, as well as a cytokinesis block that might lead to chromosomal instability and DNA damage, which are phenomena associated with cancer. Although there is the idea that the facilitation of cellular senescence is an attractive method for cancer therapy, the harmful side‐effects need to be seriously considered. Greater understanding of the molecular mechanisms linking cellular senescence and cellular homeostatic disorders will provide valuable new insights into their roles in the generation of age‐associated cancers, the avoidance of undesirable side‐effects and the development of novel therapeutic strategies for aging‐associated diseases.
Disclosure statement
The authors have no conflict of interest.
Acknowledgments
The authors thank members of the Hara lab for helpful discussion during the preparation of this manuscript. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Ministry of Health, Labor, and Welfare of Japan, Mitsubishi Foundation, Uehara Memorial Foundation, Princess Takamatsu Cancer Research Fund, Kao Research Council for the Study of Healthcare Science, the Vehicle Racing Commemorative Foundation and Japan Science Technology Agency.
(Cancer Sci 2013; 104: 525–530)
References
- 1. Martin GM. The biology of aging: 1985–2010 and beyond. FASEB J 2011; 25: 3756–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature 2007; 448: 767–74. [DOI] [PubMed] [Google Scholar]
- 3. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120: 513–22. [DOI] [PubMed] [Google Scholar]
- 4. Herbig U, Sedivy JM. Regulation of growth arrest in senescence: telomere damage is not the end of the story. Mech Ageing Dev 2006; 127: 16–24. [DOI] [PubMed] [Google Scholar]
- 5. Sharpless NE, DePinho RA. Cancer: crime and punishment. Nature 2005; 436: 636–7. [DOI] [PubMed] [Google Scholar]
- 6. Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell 2009; 36: 2–14. [DOI] [PubMed] [Google Scholar]
- 7. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10: 51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev 2010; 24: 2463–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192: 547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haff RF, Swim HE. Serial propagation of 3 strains of rabbit fibroblasts; their susceptibility to infection with vaccinia virus. Proc Soc Exp Biol Med 1956; 93: 200–4. [DOI] [PubMed] [Google Scholar]
- 11. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965; 37: 614–36. [DOI] [PubMed] [Google Scholar]
- 12. Deng Y, Chan SS, Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer 2008; 8: 450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature 1990; 345: 458–60. [DOI] [PubMed] [Google Scholar]
- 14. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19: 2100–10. [DOI] [PubMed] [Google Scholar]
- 15. Wright WE, Shay JW. Telomere dynamics in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nat Med 2000; 6: 849–51. [DOI] [PubMed] [Google Scholar]
- 16. Sherr CJ, Depinho RA. Cellular senescence: mitotic clock or culture shock? Cell 2000; 102: 407–10. [DOI] [PubMed] [Google Scholar]
- 17. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003; 5: 741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lloyd AC. Limits to lifespan. Nat Cell Biol 2002; 4: E25–7. [DOI] [PubMed] [Google Scholar]
- 19. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a . Cell 1997; 88: 593–602. [DOI] [PubMed] [Google Scholar]
- 20. Serrano M, Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol 2001; 13: 748–53. [DOI] [PubMed] [Google Scholar]
- 21. d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA‐damage response. Nat Rev Cancer 2008; 8: 512–22. [DOI] [PubMed] [Google Scholar]
- 22. Shay JW, Pereira‐Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res 1991; 196: 33–9. [DOI] [PubMed] [Google Scholar]
- 23. Hara E, Tsurui H, Shinozaki A, Nakada S, Oda K. Cooperative effect of antisense‐Rb and antisense‐p53 oligomers on the extension of life span in human diploid fibroblasts, TIG‐1. Biochem Biophys Res Commun 1991; 179: 528–34. [DOI] [PubMed] [Google Scholar]
- 24. Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth‐restricting conditions. Genes Dev 2000; 14: 3051–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sage J, Mulligan GJ, Attardi LD et al Targeted disruption of the three Rb‐related genes leads to loss of G(1) control and immortalization. Genes Dev 2000; 14: 3037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sage J, Miller AL, Perez‐Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re‐entry. Nature 2003; 424: 223–8. [DOI] [PubMed] [Google Scholar]
- 27. Dirac AM, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem 2003; 278: 11731–4. [DOI] [PubMed] [Google Scholar]
- 28. Cobrinik D, Dowdy SF, Hinds PW, Mittnacht S, Weinberg RA. The retinoblastoma protein and the regulation of cell cycling. Trends Biochem Sci 1992; 17: 312–5. [DOI] [PubMed] [Google Scholar]
- 29. Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2002; 2: 910–7. [DOI] [PubMed] [Google Scholar]
- 30. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev 1999; 13: 1501–12. [DOI] [PubMed] [Google Scholar]
- 31. Noda A, Ning Y, Venable SF, Pereira‐Smith OM, Smith JR. Cloning of senescent cell‐derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res 1994; 211: 90–8. [DOI] [PubMed] [Google Scholar]
- 32. Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol 1996; 16: 859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin‐dependent kinase inhibitor p16 INK4a in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 1996; 93: 13742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohtani N, Zebedee Z, Huot TJ et al Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001; 409: 1067–70. [DOI] [PubMed] [Google Scholar]
- 35. El‐Deiry WS, Tokino T, Velculescu VE et al WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–25. [DOI] [PubMed] [Google Scholar]
- 36. McConnell BB, Gregory FJ, Stott FJ, Hara E, Peters G. Induced expression of p16INK4a inhibits both CDK4‐ and CDK2‐associated kinase activity by reassortment of cyclin‐CDK‐inhibitor complexes. Mol Cell Biol 1999; 19: 1981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mitra J, Dai CY, Somasundaram K et al Induction of p21WAF1/CIP1 and inhibition of Cdk2 mediated by the tumor suppressor p16INK4a . Mol Cell Biol 1999; 19: 3916–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takeuchi S, Takahashi A, Motoi N et al Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo . Cancer Res 2010; 70: 9381–90. [DOI] [PubMed] [Google Scholar]
- 39. Gil J, Peters G. Regulation of the INK4b‐ARF‐INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7: 667–77. [DOI] [PubMed] [Google Scholar]
- 40. Beausejour CM, Krtolica A, Galimi F et al Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22: 4212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gorman SD, Cristofalo VJ. Reinitiation of cellular DNA synthesis in BrdU‐selected nondividing senescent WI‐38 cells by simian virus 40 infection. J Cell Physiol 1985; 125: 122–6. [DOI] [PubMed] [Google Scholar]
- 42. Dai CY, Enders GH. p16INK4a can initiate an autonomous senescence program. Oncogene 2000; 19: 1613–22. [DOI] [PubMed] [Google Scholar]
- 43. Takahashi A, Ohtani N, Yamakoshi K et al Mitogenic signalling and the p16INK4a‐Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 2006; 8: 1291–7. [DOI] [PubMed] [Google Scholar]
- 44. Passos JF, Nelson G, Wang C et al Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 2010; 6: 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol 2003; 15: 247–54. [DOI] [PubMed] [Google Scholar]
- 46. Visser S, Yang X. LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle 2010; 9: 3892–903. [DOI] [PubMed] [Google Scholar]
- 47. Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012; 13: 189–203. [DOI] [PubMed] [Google Scholar]
- 48. Fumagalli M, Rossiello F, Clerici M et al Telomeric DNA damage is irreparable and causes persistent DNA‐damage‐response activation. Nat Cell Biol 2012; 14: 355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hewitt G, Jurk D, Marques FD et al Telomeres are favoured targets of a persistent DNA damage response in ageing and stress‐induced senescence. Nat Commun 2012; 3: 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010; 141: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Davoli T, de Lange T. Telomere‐driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 2012; 21: 765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gosselin K, Martien S, Pourtier A et al Senescence‐associated oxidative DNA damage promotes the generation of neoplastic cells. Cancer Res 2009; 69: 7917–25. [DOI] [PubMed] [Google Scholar]
- 53. Yamakoshi K, Takahashi A, Hirota F et al Real‐time in vivo imaging of p16Ink4a reveals cross talk with p53. J Cell Biol 2009; 186: 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krishnamurthy J, Torrice C, Ramsey MR et al Ink4a/Arf expression is a biomarker of aging. J Clin Invest 2004; 114: 1299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Acosta JC, O'Loghlen A, Banito A et al Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133: 1006–18. [DOI] [PubMed] [Google Scholar]
- 56. Kuilman T, Michaloglou C, Vredeveld LC et al Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell 2008; 133: 1019–31. [DOI] [PubMed] [Google Scholar]
- 57. Coppe JP, Patil CK, Rodier F et al Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6: 2853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kuilman T, Peeper DS. Senescence‐messaging secretome: SMS‐ing cellular stress. Nat Rev Cancer 2009; 9: 81–94. [DOI] [PubMed] [Google Scholar]
- 59. Rodier F, Coppe JP, Patil CK et al Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol 2009; 11: 973–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maier JA, Voulalas P, Roeder D, Maciag T. Extension of the life‐span of human endothelial cells by an interleukin‐1 a antisense oligomer. Science 1990; 249: 1570–4. [DOI] [PubMed] [Google Scholar]
- 61. Ohanna M, Giuliano S, Bonet C et al Senescent cells develop a PARP‐1 and nuclear factor‐kB‐associated secretome (PNAS). Genes Dev 2011; 25: 1245–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chien Y, Scuoppo C, Wang X et al Control of the senescence‐associated secretory phenotype by NF‐κB promotes senescence and enhances chemosensitivity. Genes Dev 2011; 25: 2125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor‐1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 2006; 8: 877–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sparmann A, Bar‐Sagi D. Ras‐induced interleukin‐8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004; 6: 447–58. [DOI] [PubMed] [Google Scholar]
- 65. Ancrile B, Lim KH, Counter CM. Oncogenic Ras‐induced secretion of IL6 is required for tumorigenesis. Genes Dev 2007; 21: 1714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Park EJ, Lee JH, Yu GY et al Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL‐6 and TNF expression. Cell 2010; 140: 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang G, Rosen DG, Zhang Z et al The chemokine growth‐regulated oncogene 1 (Gro‐1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A 2006; 103: 16472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jing H, Kase J, Dorr JR et al Opposing roles of NF‐kB in anti‐cancer treatment outcome unveiled by cross‐species investigations. Genes Dev 2011; 25: 2137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface‐bound IL‐1alpha is an upstream regulator of the senescence‐associated IL‐6/IL‐8 cytokine network. Proc Natl Acad Sci U S A 2009; 106: 17031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J 2011; 30: 1536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Takahashi A, Imai Y, Yamakoshi K et al DNA damage signaling triggers degradation of histone methyltransferases through APC/CCdh1 in senescent cells. Mol Cell 2012; 45: 123–31. [DOI] [PubMed] [Google Scholar]
- 72. Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 2005; 6: 838–49. [DOI] [PubMed] [Google Scholar]
- 73. Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev 2011; 25: 781–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Levy D, Kuo AJ, Chang Y et al Lysine methylation of the NF‐kappaB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF‐kappaB signaling. Nat Immunol 2011; 12: 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pless O, Kowenz‐Leutz E, Knoblich M et al G9a‐mediated lysine methylation alters the function of CCAAT/enhancer‐binding protein‐beta. J Biol Chem 2008; 283: 26357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ventura A, Kirsch DG, McLaughlin ME et al Restoration of p53 function leads to tumour regression in vivo . Nature 2007; 445: 661–5. [DOI] [PubMed] [Google Scholar]
- 77. Xue W, Zender L, Miething C et al Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445: 656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Krizhanovsky V, Yon M, Dickins RA et al Senescence of activated stellate cells limits liver fibrosis. Cell 2008; 134: 657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]