

Abstract
Topoisomerase II alpha (TOP2A) has a crucial role in proper chromosome condensation and segregation. Here we report the interaction of TOP2A with ataxia telangiectasia mutated (ATM) and its phosphorylation in an ATM‐dependent manner after DNA damage. In vitro kinase assay and site‐directed mutagenesis studies revealed that serine 1512 is the target of phosphorylation through ATM. Serine 1512 to Alanine mutation of TOP2A showed increased stability of the protein, retaining TOP2A activity at least with regard to cell survival activity. Ataxia telangiectasia‐derived cell lines showed high levels of TOP2A that were associated with hypersensitivity to the TOP2 inhibitor etoposide. These findings suggest that ATM‐dependent TOP2A modification is required for proper regulation of TOP2 stability and subsequently of the sensitivity to TOP2 inhibitor. In a lymphoblastoid cell line derived from a patient who developed MLL rearrangement, positive infant leukemia, defective ATM expression, and increased TOP2A expression were shown. It was intriguing that hypersensitivity to TOP2 inhibitor and susceptibility to MLL gene rearrangement were shown by low‐dose etoposide exposure in this cell line. Thus, our findings have clinically important implications for the pathogenesis of infantile acute leukemia as well as treatment‐associated secondary leukemia following exposure to TOP2 inhibitors.
Ataxia telangiectasia (AT) is an autosomal recessive disorder characterized by a pleiotropic phenotype that includes progressive cerebellar degeneration, immunodeficiency, premature aging, genetic instability, and a high incidence of cancer. Heterozygous carriers also appear to be at increased risk of cancer.1, 2 Cells from AT homozygotes lack multiple cell cycle check points, and this leads to hypersensitivity to double‐strand breaks in the DNA.3, 4
DNA topology is controlled and altered by DNA topoisomerases. Topoisomerases are ubiquitous enzymes that resolve topological problems, which arise during the various processes of DNA metabolism including transcription, recombination, replication, and chromosome partitioning during cell division. Topoisomerase I introduces a transient single‐strand break into the DNA, passes an intact single strand of DNA through the broken strand, and re‐ligates the break. Topoisomerase II (TOP2) makes transient double‐strand breaks in one segment of DNA and passes an intact duplex through the broken DNA before resealing the breaks. Human cells express two isoforms of TOP2, topoisomerase II alpha (TOP2A) and topoisomerase II beta (TOP2B). TOP2A is expressed mainly during the S to G2/M phase of the cell cycle and is likely to play a major role in DNA catenation during mitosis. In contrast, TOP2B is expressed constantly throughout the cell cycle. The function of TOP2B is unknown, but the enzyme is speculated to be involved in the metabolism of DNA and/or RNA. Topoisomerase II alpha is highly phosphorylated, and its enzymatic activity has been postulated to be regulated by phosphorylation, by its interaction with other factors, or by its subcellular localization. However, no clearly defined role of phosphorylation has been identified.5
Exposure in utero to TOP2 inhibitor or a similar substance6 is thought to lead to infantile leukemia, which is characterized by frequent chromosomal translocations involving the mixed lineage leukemia (MLL) gene.7, 8, 9 Rearrangement of the MLL gene also occurs in treatment‐related leukemia that arises after treatment with TOP2 inhibitor.10 We reported that dysfunction of ataxia telangiectasia mutated (ATM), responsible for AT, plays an important role in the development of some infantile acute leukemia.11 Our findings led us to speculate that the pathogenesis of infantile acute leukemia involves ATM dysfunction and hypersensitivity to TOP2A inhibitor, and prompted us to investigate the physiologic relation between ATM and TOP2.
Here we report an association between TOP2A and ATM and the phosphorylation of TOP2A in an ATM‐dependent manner. Cell biological data also indicate a functional relation between ATM and TOP2A. Our study provides new evidence for ATM‐dependent regulation of TOP2, a factor that may be involved in the pathogenesis of infantile acute leukemia.
Materials and Methods
Western blot analysis
Western blotting was carried out using the standard method.
Clonogenic assay
Clonogenic assays were carried out as described previously.12 Briefly, cells were transiently treated with etoposide (VP16) (Sigma, St. Louis, MO, USA) or ICRF‐193 (Sigma), then provided with fresh DMEM containing 10% FCS without etoposide or ICRF‐193. After 2 weeks in culture, cells were stained with Giemsa and colony numbers counted.
Complementation assay
Complementation assays were carried out as described previously.13 HTETOP cells, in which >99.5% TOP2A expression can be silenced by the addition of tetracycline, were transfected with pEGFP‐C2, pEGFP‐TOP2A WT, or pEGFP‐TOP2A S1512A by electroporation for complementation. Four hours after transfection, cells were treated with tetracycline. Colonies were counted after 2 weeks selection period.
Long distance inverted PCR
Long distance inverted PCR (LDI‐PCR) was described previously.14, 15 Briefly, cells were treated with 100 μM etoposide for 8 h. DNA was then extracted and subjected to LDI‐PCR. Details of the experimental procedure are also provided in Data S1.
Results
Topoisomerase I alpha interacts with ATM
To look for ATM‐interacting proteins, we immunoprecipitated ATM from non‐irradiated and 10‐Gy irradiated wild‐type lymphoblastoid cell line (WT‐LCL) and AT lymphoblastoid cell line (AT‐LCL) using anti‐ATM antibody. Several proteins that co‐immunoprecipitated with ATM were detected by Coomassie Brilliant Blue staining. We noted the presence of a protein with an apparent molecular size of almost 170 kDa only in lysates from irradiated WT‐LCL (Fig. 1a). This protein was in gel digested with trypsin, eluted from the gel, purified by HPLC, and subjected to peptide microsequencing, leading to identification of TOP2A. To determine whether ATM and TOP2A form a complex in the same molecular weight fraction, we subjected cell extracts to sucrose density gradient analysis. This assay revealed that ATM and TOP2A exist in density fractions of similar molecular weight (Fig. 1b). The ATM–TOP2A interaction was confirmed by immunoprecipitation with anti‐ATM antibody followed by Western blotting with anti‐TOP2A antibody (Fig. 1c, left panel). Topoisomerase II alpha was not co‐immunoprecipitated by anti‐ATM antibody from AT‐LCL cell lysate. Reciprocally, ATM was co‐immunoprecipitated by anti‐TOP2A antibody only from WT cell lysate. The ATM–TOP2A interaction was also confirmed by anti‐TOP2A and anti‐ATM antibody immunoprecipitation but not control IgG from WT‐LCL lysate (Fig. 1c, right panel). Although ATM–TOP2A interaction was initially identified in irradiated samples, the interaction between ATM and TOP2A illustrated by co‐immunoprecipitation was not affected by irradiation (Fig. S1). These findings indicate that ATM associates with TOP2A in cells.
Figure 1.
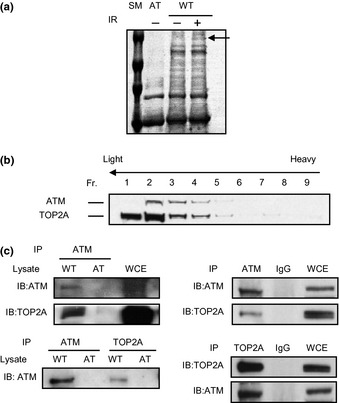
(a) Ataxia telangiectasia mutated (ATM) immunoprecipitant contains a 170‐kDa protein. ATM was immunoprecipitated from unirradiated (−) and irradiated (IR) (+) wild‐type lymphoblastoid cell lines (WT) and ATR65RM cells (AT). Immunoprecipitants were visualized by Coomassie Brilliant Blue staining. The band at approximately 170 kDa (arrow) was subjected to peptide sequencing. SM, size marker. (b) Separation of ATM and topoisomerase II alpha (TOP2A) by sucrose density gradient (1, lightest fraction [Fr.]; 9, heaviest fraction). (c) Left panel, WT lymphoblastoid cell line (WT) and AT65RM (AT) cell lysates were immunoprecipitated (IP) with anti‐ATM or anti‐TOP2A antibody, and immunoprecipitants were immunoblotted (IB) with anti‐ATM or anti‐TOP2A antibody, respectively. Whole‐cell extract (WCE) was included as a size control. Right panel, cell lysates from 293T cells were immunoprecipitated with anti‐ATM or anti‐TOP2A antibody, with control IgG.
Topoisomerase II alpha is phosphorylated in an ATM‐dependent manner after irradiation
Ataxia telangiectasia mutated is a protein kinase activated by DNA damage. An association between ATM and TOP2A suggests that TOP2A is a substrate of ATM. To evaluate this hypothesis, we used [32P] orthophosphate metabolic labeling to determine whether DNA damage induces TOP2A phosphorylation, and we detected increased incorporation of [32P] orthophosphate into TOP2A after irradiation (Fig. 2a). Phosphorylation of TOP2A is dependent on the cell cycle; therefore, to exclude the possibility that cell cycle distributions affected the phosphorylation status of TOP2A, we analyzed the cell cycle distributions of WT‐LCL and AT‐LCL at the time TOP2A was analyzed, 20 min after irradiation. There was no difference in cell cycle distribution between WT and AT cells (data not shown). These results suggest that TOP2A is phosphorylated as a result of DNA damage in an ATM‐dependent manner in vivo, and this phosphorylation is not due to differences in cell cycle distribution.
Figure 2.
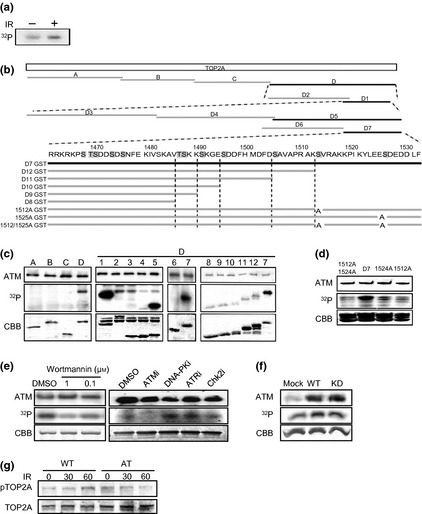
(a) Incorporation of [32 P] orthophosphate (32 P) into topoisomerase II alpha (TOP2A). Autoradiography of TOP2A immunoprecipitant from irradiated (IR) (+) or unirradiated (−) cells. (b) Schematic diagram of GST‐TOP2A fragment. The phosphorylated fragments are shown in black. (c) In vitro phosphorylation of TOP2A fragments by ataxia telangiectasia mutated (ATM) immunoprecipitants. Fragment numbers correspond to (b). Immunoprecipitated ATM (upper panel), 32 P incorporation into GST‐TOP2A (middle panel), and GST‐TOP2A fragment loading by Coomassie Brilliant Blue (CBB) staining (bottom panel) is shown. (d) Ser1512 or Ser1524 substituted GST‐TOP2A were subjected to in vitro kinase assay. (e) In vitro kinase assay of GST‐TOP2A with or without wortmannin, and with 10 μM ATM inhibitor (ATMi; KU55933), 10 μM DNA‐PK inhibitor (DNA‐Pki; NU7026), 10 μM ATM and Rad3‐related inhibitor (ATRi; VE‐821), and 10 μM Chk2 inhibitor (Chk2i; BML‐277). Dimethylsulfoxide was used as control. (f) In vitro phosphorylation of GST‐TOP2A by FLAG‐mock (mock), FLAG‐ATM wild‐type (WT), and FLAG‐ATM kinase‐dead (KD) immunoprecipitants. (g) In cellulo TOP2A Ser1512 phosphorylation after 10‐Gy irradiation was analyzed using phospho‐specific antibody.
Topoisomerase II alpha is an in vitro substrate of ATM immunoprecipitants
To extend the above findings, we explored the possibility that ATM phosphorylates TOP2A in vitro. We generated several GST‐fused TOP2A fragments and determined whether these fragments are phosphorylated by ATM in vitro. We immunoprecipitated ATM from 293 cells using anti‐ATM antibody and carried out an immune complex kinase assay using these GST‐fused TOP2A fragments as the substrates. Results showed that only fragment D (C‐terminal fragment) was phosphorylated by the ATM immunoprecipitants (Fig. 2b,c). We then generated GST‐fused deletion mutants of fragment D to determine the site of phosphorylation. The results of this experiment indicated that Serine (Ser)1512 or Ser1524 were possible sites of phosphorylation by ATM immunoprecipitants (Fig. 2b,c). To determine which site is phosphorylated, we generated Ser1512Alanine (Ser1512Ala), Ser1524Ala, and Ser1512/1524Ala mutants of fragment D7. Phosphorylation of the C‐terminal fragment of GST‐TOP2A was reduced to the background level when Ser1512 was replaced with Ala and also moderately reduced when Ser1524 was replaced with Ala. Mutation at both sites abolished 32P incorporation into this fragment (Fig. 2d). These findings suggest that Ser1512 rather than Ser1524 is the preferential target site of phosphorylation by ATM immunoprecipitants. Serine 1512 is highly conserved from yeast to mammalian cells (Fig. 2e). Ataxia telangiectasia mutated prefers to phosphorylate Ser/threonine (Thr) followed by a glutamine (Gln) residue, but this position is occupied by valine (Val) in TOP2A. Therefore, we carried out experiments to determine whether Ser1512 is directly phosphorylated by ATM. First, we treated the in vitro kinase complex with wortmannin, which inhibits phosphatidyl inositol (PI)‐3 kinase families, including ATM. This treatment abolished GST‐TOP2A phosphorylation in a dose‐dependent manner (Fig. 2f). Next, the involvement of other PI‐3 kinase‐related kinases was investigated. DNA‐PK, ATM and Rad3‐related (ATR), or ATM downstream kinase Chk2 were inhibited with their specific inhibitors, and in vitro kinase assay was carried out. Genuinely, ATM inhibitor (KU55933) suppressed its kinase activity to the TOP2A fragment. However, DNA‐PK inhibitor (NU7027), ATR inhibitor (VE‐821), and Chk2 inhibitor (BML‐277) did not suppress its kinase activity (Fig. 2f). Casein kinase II inhibitor (5,6‐dichloro‐1‐β‐D‐ribofuranosylbenzimidazole), MEK inhibitor (PD98059), p38 MAPK inhibitor (SB203580), and a wide‐range protein kinase inhibitor (staurosporine) also did not alter kinase activity (data not shown). Next, we transfected 293 cells with FLAG‐tagged WT or kinase‐dead constructs of ATM with dominant negative activity. The FLAG‐tagged WT ATM immunoprecipitant phosphorylated GST‐TOP2A, and the ATM kinase‐dead construct also phosphorylated GST‐TOP2A at almost a similar level to WT ATM immunoprecipitant (Fig. 2g). These experiments suggest that the ATM immunoprecipitants include some other PI‐3 kinase like protein(s) that phosphorylate TOP2A. But phosphorylation of TOP2A by these unknown protein kinases is still dependent on the ATM molecule, irrespective of the presence or absence of its kinase activity. To detect in cellulo TOP2A phosphorylation, phosphor‐Ser1512‐specific antibody was generated. After irradiation, Ser1512 phosphorylation was augmented in WT cells but not in AT cells (Fig. 2h).
Functional significance of Ser1512 residue in TOP2A
For further evaluation of the functional significance of the Ser1512 residue, complementation assays for cell survival activity were carried out using HTETOP cells. The expression of TOP2A can be silenced in HTETOP cells by the addition of tetracycline. The TOP2A‐depleted HTETOP cells enter mitosis and undergo chromosome condensation, albeit with delayed kinetics, but normal anaphases and cytokinesis are completely prevented, and all cells die. Cells can be rescued by expression of GFP‐fused TOP2A. The function of mutant TOP2A can be evaluated by clonogenic survival.13 We evaluated whether this Ser1512Ala mutant could complement TOP2A function by transfection of this mutant construct, and silenced endogenous TOP2A by addition of tetracycline. This experiment revealed that Ser1512Ala mutant could complement depletion of TOP2A (Table 1). This result suggests that the Ser1512Ala mutant retains TOP2A activity at least with regard to cell survival activity.
Table 1.
HTETOP complementation with topoisomerase II alpha (TOP2A) mutants
| Plasmid | Dox + puro |
|---|---|
| GFP‐TOP2A WT | 16, 14 |
| GFP‐TOP2A S1512A | 18, 15 |
| GFP‐mock | 1, 2, 0 |
Topoisomerase II alpha‐complemented 0.5 million HTETOP cells were seeded on six‐well plates and cells were selected by the addition of doxycycline and puromycine (Dox + puro). Each number represents the number of colonies from independent experiments.
Expression level of TOP2A in AT cells
The sensitivity of TOP2 inhibitor is correlated with the TOP2 expression level.16, 17, 18 Therefore, we examined TOP2A expression levels in WT‐LCL and AT‐LCL by Western blotting. More TOP2A was expressed in AT‐LCL than in WT‐LCL (Fig. 3a). We saw similar cell cycle distributions in asynchronous cell cultures of both AT‐LCL and WT‐LCL. This observation should be noted because TOP2A is expressed during S to G2/M phase. To show that the difference in TOP2A expression is not simply dependent on the difference in cell cycle distribution, we used 2D flow cytometry analysis to compare the amount of TOP2A expressed during S and G2/M phases. During G2/M phase, larger amounts of TOP2A were expressed in AT65RM cells than in WT‐LCL (Fig. 3b). The TOP2A mRNA levels were indistinguishable between WT‐LCL and AT65RM (data not shown). Protein stability of TOP2A was also compared using asynchronous GM0637 (WT) and GM05849C (AT) cells after cycloheximide (protein synthesis inhibitor) treatment. The half‐life of TOP2A was estimated to be approximately 6 h in GM0637 cells, and approximately 12 h in GM05849C cells (Fig. 3c). Therefore, increased expression of TOP2A in AT cells appears to be due to post‐transcriptional regulation. Next, the half‐life of TOP2A in WT and Ser15112Ala mutant was investigated. After transfection of GFP‐TOP2A WT and GFP‐TOP2A S1512A plasmid in 293T cells, expression of TOP2A was chronologically monitored using anti‐GFP antibody after cycloheximide treatment. Interestingly Ser1512Ala mutated TOP2A showed an increase in the half‐life of protein (Fig. 3d).
Figure 3.
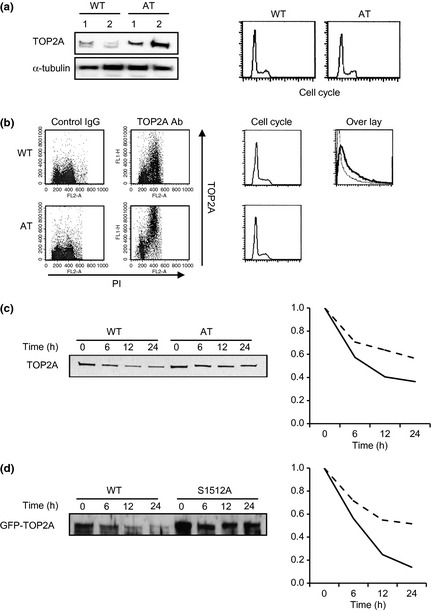
(a) Western blot analysis of wild‐type (WT) cells and ataxia telangiectasia (AT) cells. Right panel, cell cycle distribution analyzed by flow cytometry. (b) Flow cytometric analysis of nuclear topoisomerase II content. Ten thousand cells of the G 2/M population were subjected to analysis. Topoisomerase II alpha (TOP2A) expression (FITC labeling) and cell cycle distribution (propidium iodide [PI] staining) are shown. Cell cycle histograms corresponding to each dot blot are shown (middle panel). Far right, overlaid histograms of TOP2A expression in WT (fine line) and AT (bold line) cells. (c) Half‐life of TOP2A was evaluated in GM0637 and GM05849C cells. Cells were treated for cycloheximine (1 μg/mL) for the indicated time period, and harvested. The TOP2A expression was determined using Western blotting. Right panel, quantitative analysis of TOP2A expression. (d) Half‐life of TOP2A WT and Ser1512Ala mutant transfected in 293T cell, evaluated in the same way as (c).
Topoisomerase II alpha inhibitor sensitivity of AT cells
We next determined whether AT cells carry an abnormality that depends on TOP2A function. We treated the ATM‐deficient cell line GM05849C, GM05849C cells transfected with FLAG‐tagged WT ATM (GM05849C/ATM), and GM0637 cells (WT for ATM) with the TOP2 inhibitor etoposide (VP16). These experiments showed that sensitivity to etoposide was enhanced in GM05849C cells. The GM05849C/ATM cells had the same level of resistance to etoposide as GM0637 cells (Fig. 4). ICRF‐193 is a catalytic non‐cleavable complex‐forming inhibitor of DNA TOP2 that does not produce protein‐linked DNA strand breaks. We have tested whether AT cells show ICRF‐193 sensitivity. Similar to etoposide treatment, AT cells showed hypersensitivity to ICRF‐193. After treatment with ICRF‐193, AT fibroblasts show a significantly attenuated G2 decatenation checkpoint response.19 Cells lacking the G2 decatenation checkpoint override G2 arrest by DNA TOP2 catalytic inhibitor or poison, then DNA breaks are produced. DNA breaks created by defective G2 decatenation checkpoints may sensitize AT cells to ICRF‐193.
Figure 4.
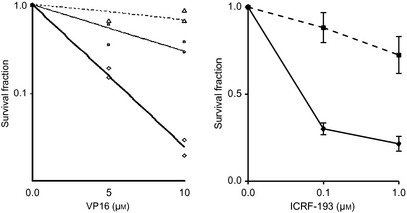
Left panel, survival responses of GM05849C/mock (diamonds), GM05849C/ATM (triangles), and GM0637 cells (squares) to etoposide (VP16). Right panel, survival responses of GM05849C (diamonds) and GM0637 cells (squares) to ICRF‐193. Data from two independent experiments are shown.
Functional abnormality of TOP2A in an EBV‐transformed cell line from a patient who developed infant leukemia
We previously described an EBV‐transformed B‐lymphoblastic cell line derived from a patient who developed infant leukemia.11 This cell line carries ATM heterogenous single nucleotide polymorphism with reduced ATM expression (LCL‐L2) compared with LCL‐WT. This observation prompted us to investigate the functional relationship between ATM and TOP2A in this cell line. The expression levels of TOP2A in these cell lines were investigated using Western blotting. The LCL‐L2 cells showed increased levels of TOP2A expression compared with WT‐LCL (Fig. 5a). To evaluate whether increased levels of TOP2A may correlate to etoposide sensitivity, and whether MLL fusion can be generated by etoposide treatment, we treated these cell lines with etoposide and investigated cell death and MLL rearrangement by LDI‐PCR. As expected, the AT cell line showed increased frequencies of cell death (35%) as compared with WT (18%). The LCL‐L2 cells showed intermediated sensitivity (23%) between AT and WT cell lines (Fig. 5b). Interestingly, the LCL‐L2 cell line showed increased susceptibility to MLL rearrangement by etoposide treatment compared with WT cells (Fig. 5c).
Figure 5.
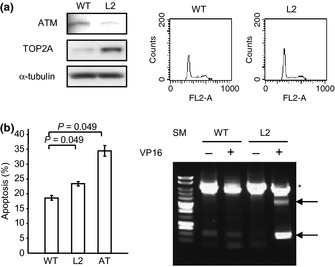
(a) Western blot analysis of wild‐type lymphoblastoid cells (WT) and cells carrying ataxia telangiectasia mutated (ATM) heterogenous single nucleotide polymorphism with reduced ATM expression (L2). Extracts from LCLs probed with antibodies to topoisomerase II alpha (TOP2A), and α‐tubulin. Right panel, histograms of each cell cycle distribution. (b) Apoptosis percentages after etoposide treatment. The WT, L2, and ataxia telangiectasia (AT) cells were treated with 10 μM etoposide for 16 h and subjected flow cytometric analysis. Apoptosis percentages were measured by counting subdiploid fractions after staining with propidium iodide. The results were obtained after three independent experiments. (c) MLL rearrangements were analyzed by long distance inverted PCR. Arrow, rearranged bands; asterisk, germline bands; SM, size marker.
Discussion
We found that ATM and TOP2A bind to each other, and this finding suggested the possibility of ATM dependent TOP2A phosphorylation, nevertheless its kinase activity. We also found increased TOP2A expression in AT cells that are defective in ATM expression. This increased expression of TOP2 was associated with hypersensitivity to TOP2 inhibitor.
Ataxia telangiectasia cells are hypersensitive not only to irradiation, but also to TOP2 inhibitor,20, 21, 22 and defective activation of the replication checkpoint machinery occurs after exposure to TOP2 inhibitor.23 Inhibitors of TOP2 induce DNA double‐strand breaks, but TOP2 catalytic inhibitor also induces chromatin structural changes without DNA breaks. For example, ATM is activated by chromatin structural changes, which is induced by the TOP2 catalytic inhibitor chloroquine.24 The function of TOP2 in AT cells has been much investigated, but contradictory findings, such as decreased25, 26, 27 or increased TOP2 activity,22, 28 have been reported. Nevertheless, these findings strongly suggest that ATM is involved in the checkpoint of DNA damage induced by TOP2 inhibition; they also suggest a functional relationship between ATM and TOP2.
Recent research suggested that TOP2A and BRCA1 interact with each other, and TOP2A decatenation activity is regulated by BRCA1 interaction and ubiquitination.29 BRCA1 is one of the best characterized ATM substrates, and forms BRCA1‐associated genome surveillance complex that contains ATM protein.30 These two reports support the potential connection of molecular interaction between ATM and TOP2A shown by our experiment. Several proteins have also been reported to interact with topoisomerase. Topoisomerase II‐binding protein 1 (TOPBP1), for example,31 contains a breast cancer suppressor protein, C‐terminal domain and is phosphorylated in response to DNA damage in an ATM‐dependent manner.32 It has been shown that TOPBP1, TOP2A, and TOP2B interact with the C‐terminus of p53,33 and this finding allows us to speculate that ATM, TOP2, TOPBP1, and p53 function as a protein complex.
Topoisomerase II alpha is phosphorylated on multiple Ser and Thr residues, most of which are located in the C‐terminal domain.34, 35, 36, 37, 38, 39 Several potential ATM‐dependent or ‐regulated phosphorylation sites (Ser1424, 1466, 1522, and 1524) were reported.40 Our findings showed phosphorylation of a new target site, Ser1512, induced by DNA damage in an ATM‐dependent manner. The ATM family proteins such as ATM, ATR, and DNA‐PK prefer to phosphorylate the Ser/Thr‐Gln motif.41 Topoisomerase II alpha contains three Ser/Thr‐Gln motifs, but ATM immunoprecipitants did not phosphorylate these sites. The site phosphorylated by the ATM immunoprecipitants was a Ser residue followed by Val at the C‐terminal of TOP2A. DNA‐PK phosphorylates proline–serine in vitro.42 Ataxia telangiectasia mutated also phosphorylates the Ser residue with the adjacent glycine residue of BRCA1 in vitro and in cellulo.43 A recent study identified numerous ATM‐dependent and ‐regulated phosphorylation sites.40 Target proteins are complexly regulated in an ATM‐dependent manner, involving either direct or indirect phosphorylation by ATM. Our finding that TOP2A is phosphorylated by ATM kinase‐dead construct immunoprecipitants to some extent suggests the possibility that the ATM immunoprecipitants include some other ATM‐like unknown protein kinases, which phosphorylate TOP2A. The analysis using phospho‐specific Ser1512 TOP2A antibody supported the finding that phosphorylation of Ser1512 depends on the presence of ATM molecules in cellulo. A previous report suggested that mutations at phosphorylation sites in the C‐terminal of TOP2A (Ser1106, 1247, 1354, and 1393) do not impair its function.13 The TOP2A Ser1512Ala mutant could also complement the loss of TOP2A function for cell survival. It remains unknown whether phosphorylation at this site contributes to TOP2 function. Several TOP2A mutations associated with TOP2 inhibitor resistance have been reported. For example, mutations in the C‐terminal region of TOP2A confer defects in its nuclear localization and lead to a decreased sensitivity to TOP2 inhibitor.44, 45, 46, 47 Several missense mutations of TOP2A have also been shown to render TOP2A resistant to inhibitors.48, 49, 50, 51 Interestingly, substitution of Ser1512 residue to Ala strikingly stabilized TOP2A expression. Increased expression of TOP2A observed in AT cells may result from defective phosphorylation of Ser1512. It is still unclear whether ATM activation directly conducts TOP2A to degradation. However, it is clear that AT cells show elevated TOP2A expression by increased protein stability, and disruption of the ATM‐dependent phosphorylation site of TOP2A stabilizes the protein. We can hypothesize that cell cycle progression from G2 to M, mediated by TOP2A, needs to be halted to prevent carry‐over DNA breaks after DNA damage. For that purpose, TOP2A expression may be suppressed after DNA damage. Further investigation is required into the consequences of phosphorylation at the C‐terminal region of TOP2A, including Ser1512.
Tumor cell lines acquire resistance to topoisomerase inhibitors through various mechanisms, including expression of multidrug‐resistance genes,52 mutation of the TOP2 gene,44, 48, 51 distinct extranuclear localization,46, 47, 53 and reduction of TOP2 expression.52 Decreased TOP2A expression by heterozygous gene targeting confers increased resistance to TOP2A inhibitor,16 and increased expression of TOP2A correlates with increased sensitivity to TOP2 inhibitor.17, 18 Ataxia telangiectasia cells expressed more TOP2A than WT cells and showed hypersensitivity to TOP2A inhibitor. We also observed that the sensitivity of AT cells to etoposide was normalized by introduction of ATM. Therefore, these results indicate that ATM‐associated phosphorylation and reduction of TOP2A expression play a crucial role in the regulation of sensitivity of AT cells to TOP2A inhibitor.
Epidemiological data have suggested that the development of infantile leukemia is tightly associated with in utero exposure to TOP2 inhibitor, which causes rearrangement of the MLL gene.6, 7, 8, 9 We can hypothesize that increased sensitivity to TOP2 inhibitor due to genetic reasons also induces the development of infantile leukemia. Indeed, we identified one infant leukemia patient with defective ATM expression by germline ATM single nucleotide polymorphism.11 As expected, the EBV‐LCL of this patient showed increased TOP2A sensitivity. These observations are in accord with our hypothesis that defective expression of ATM correlates with increased levels of TOP2A, which sensitizes to TOP inhibitor and leads to the susceptibility to MLL gene breaks and rearrangement. Previously, we reported our observation that MLL rearrangement is induced by treatment with etoposide in AT cells but not WT cells.54
On the basis of the data presented here and our previous report, ATM‐dependent TOP2A regulation appears to have important clinical implications for the pathogenesis of infantile leukemia as well as treatment‐associated secondary leukemia, both of which are associated with TOP 2 inhibitor related MLL rearrangement.
Disclosure Statement
The authors have no conflicts of interest.
Supporting information
Data S1. Supplemental experimental procedure.
Fig. S1. Ataxia telangiectasia mutated (ATM) and topoisomerase II alpha (TOP2A) interaction after DNA damage.
Acknowledgments
We thank Yoshiko Kamiya, Kaoru Oguchi (Tokyo Medical and Dental University), Hikari Taka, and Kimie Murayama (Juntendo University) for technical assistance, and Minoru Asada (Nihon Medical University) for helpful comments. The GFP‐TOP2A expression vector pT104‐1 was a kind gift from Dr T. Beck (University of Illinois at Chicago). pCDNA3 YZ5 was a kind gift from Dr M. B. Kastan (Duke University). This work was supported by grants from the Ministry of Education, Science, and Culture (Japan).
(Cancer Sci, doi: 10.1111/cas.12067, 2013)
References
- 1. Athma P, Rappaport R, Swift M. Molecular genotyping shows that ataxia‐telangiectasia heterozygotes are predisposed to breast cancer. Cancer Genet Cytogenet 1996; 92: 130–4. [DOI] [PubMed] [Google Scholar]
- 2. Broeks A, Urbanus JH, Floore AN et al ATM‐heterozygous germline mutations contribute to breast cancer‐susceptibility. Am J Hum Genet 2000; 66: 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 2001; 11: 71–7. [DOI] [PubMed] [Google Scholar]
- 4. Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol 2000; 1: 179–86. [DOI] [PubMed] [Google Scholar]
- 5. Isaacs RJ, Davies SL, Sandri MI, Redwood C, Wells NJ, Hickson ID. Physiological regulation of eukaryotic topoisomerase II. Biochim Biophys Acta 1998; 1400: 121–37. [DOI] [PubMed] [Google Scholar]
- 6. Alexander FE, Patheal SL, Biondi A et al Transplacental chemical exposure and risk of infant leukemia with MLL gene fusion. Cancer Res 2001; 61: 2542–6. [PubMed] [Google Scholar]
- 7. Ross JA, Davies SM, Potter JD, Robison LL. Epidemiology of childhood leukemia, with a focus on infants. Epidemiol Rev 1994; 16: 243–72. [DOI] [PubMed] [Google Scholar]
- 8. Ross JA, Potter JD, Robison LL. Infant leukemia, topoisomerase II inhibitors, and the MLL gene. J Natl Cancer Inst 1994; 86: 1678–80. [DOI] [PubMed] [Google Scholar]
- 9. Ross JA, Potter JD, Reaman GH, Pendergrass TW, Robison LL. Maternal exposure to potential inhibitors of DNA topoisomerase II and infant leukemia (United States): a report from the Children's Cancer Group. Cancer Causes Control 1996; 7: 581–90. [DOI] [PubMed] [Google Scholar]
- 10. Broeker PL, Super HG, Thirman MJ et al Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment‐related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood 1996; 87: 1912–22. [PubMed] [Google Scholar]
- 11. Oguchi K, Takagi M, Tsuchida R et al Missense mutation and defective function of ATM in a childhood acute leukemia patient with MLL gene rearrangement. Blood 2003; 101: 3622–7. [DOI] [PubMed] [Google Scholar]
- 12. Ziv Y, Bar‐Shira A, Pecker I et al Recombinant ATM protein complements the cellular A‐T phenotype. Oncogene 1997; 15: 159–67. [DOI] [PubMed] [Google Scholar]
- 13. Carpenter AJ, Porter AC. Construction, characterization, and complementation of a conditional‐lethal DNA topoisomerase IIalpha mutant human cell line. Mol Biol Cell 2004; 15: 5700–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meyer C, Schneider B, Reichel M et al Diagnostic tool for the identification of MLL rearrangements including unknown partner genes. Proc Natl Acad Sci USA 2005; 102: 449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blanco JG, Edick MJ, Relling MV. Etoposide induces chimeric Mll gene fusions. FASEB J 2004; 18: 173–5. [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi M, Adachi N, Aratani Y, Kikuchi A, Koyama H. Decreased topoisomerase IIalpha expression confers increased resistance to ICRF‐193 as well as VP‐16 in mouse embryonic stem cells. Cancer Lett 2001; 166: 71–7. [DOI] [PubMed] [Google Scholar]
- 17. Asano T, An T, Zwelling LA, Takano H, Fojo AT, Kleinerman ES. Transfection of a human topoisomerase II alpha gene into etoposide‐resistant human breast tumor cells sensitizes the cells to etoposide. Oncol Res 1996; 8: 101–10. [PubMed] [Google Scholar]
- 18. Zhou Z, Zwelling LA, Kawakami Y et al Adenovirus‐mediated human topoisomerase IIalpha gene transfer increases the sensitivity of etoposide‐resistant human breast cancer cells. Cancer Res 1999; 59: 4618–24. [PubMed] [Google Scholar]
- 19. Bower JJ, Zhou Y, Zhou T et al Revised genetic requirements for the decatenation G2 checkpoint: the role of ATM. Cell Cycle 2010; 9: 1617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caporossi D, Porfirio B, Nicoletti B et al Hypersensitivity of lymphoblastoid lines derived from ataxia telangiectasia patients to the induction of chromosomal aberrations by etoposide (VP‐16). Mutat Res 1993; 290: 265–72. [DOI] [PubMed] [Google Scholar]
- 21. Petrinelli P, Elli R, Marcucci L, Barbieri C, Ambra R, Antonelli A. VP16 hypersensitivity and increased faulty recombination in ataxia telangiectasia lymphocytes characterized by the tandem translocation t(14;14)(q11;q32). Carcinogenesis 1996; 17: 203–7. [DOI] [PubMed] [Google Scholar]
- 22. Smith PJ, Makinson TA. Cellular consequences of overproduction of DNA topoisomerase II in an ataxia‐telangiectasia cell line. Cancer Res 1989; 49: 1118–24. [PubMed] [Google Scholar]
- 23. Kaufmann WK, Boyer JC, Estabrooks LL, Wilson SJ. Inhibition of replicon initiation in human cells following stabilization of topoisomerase‐DNA cleavable complexes. Mol Cell Biol 1991; 11: 3711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421: 499–506. [DOI] [PubMed] [Google Scholar]
- 25. Mohamed R, Pal Singh S, Kumar S, Lavin MF. A defect in DNA topoisomerase II activity in ataxia‐telangiectasia cells. Biochem Biophys Res Commun 1987; 149: 233–8. [DOI] [PubMed] [Google Scholar]
- 26. Singh SP, Lavin MF. Study of DNA topoisomerase II in ataxia‐telangiectasia cells. Carcinogenesis 1989; 10: 1215–8. [DOI] [PubMed] [Google Scholar]
- 27. Lavin MF, Singh SP. Defective DNA topoisomerase II in ataxia‐telangiectasia cells. Acta Biol Hung 1990; 41: 125–35. [PubMed] [Google Scholar]
- 28. Davies SM, Harris AL, Hickson ID. Overproduction of topoisomerase II in an ataxia telangiectasia fibroblast cell line: comparison with a topoisomerase II‐overproducing hamster cell mutant. Nucleic Acids Res 1989; 17: 1337–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lou Z, Minter‐Dykhouse K, Chen J. BRCA1 participates in DNA decatenation. Nat Struct Mol Biol 2005; 12: 589–93. [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1‐associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 2000; 14: 927–39. [PMC free article] [PubMed] [Google Scholar]
- 31. Yamane K, Tsuruo T. Conserved BRCT regions of TopBP1 and of the tumor suppressor BRCA1 bind strand breaks and termini of DNA. Oncogene 1999; 18: 5194–203. [DOI] [PubMed] [Google Scholar]
- 32. Yamane K, Wu X, Chen J. A DNA damage‐regulated BRCT‐containing protein, TopBP1, is required for cell survival. Mol Cell Biol 2002; 22: 555–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cowell IG, Okorokov AL, Cutts SA et al Human topoisomerase IIalpha and IIbeta interact with the C‐terminal region of p53. Exp Cell Res 2000; 255: 86–94. [DOI] [PubMed] [Google Scholar]
- 34. Wells NJ, Addison CM, Fry AM, Ganapathi R, Hickson ID. Serine 1524 is a major site of phosphorylation on human topoisomerase II alpha protein in vivo and is a substrate for casein kinase II in vitro. J Biol Chem 1994; 269: 29746–51. [PubMed] [Google Scholar]
- 35. Wells NJ, Fry AM, Guano F, Norbury C, Hickson ID. Cell cycle phase‐specific phosphorylation of human topoisomerase II alpha. Evidence of a role for protein kinase C. J Biol Chem 1995; 270: 28357–63. [DOI] [PubMed] [Google Scholar]
- 36. Ishida R, Iwai M, Marsh KL et al Threonine 1342 in human topoisomerase IIalpha is phosphorylated throughout the cell cycle. J Biol Chem 1996; 271: 30077–82. [DOI] [PubMed] [Google Scholar]
- 37. Escargueil AE, Plisov SY, Filhol O, Cochet C, Larsen AK. Mitotic phosphorylation of DNA topoisomerase II alpha by protein kinase CK2 creates the MPM‐2 phosphoepitope on Ser‐1469. J Biol Chem 2000; 275: 34710–8. [DOI] [PubMed] [Google Scholar]
- 38. Wells NJ, Hickson ID. Human topoisomerase II alpha is phosphorylated in a cell‐cycle phase‐dependent manner by a proline‐directed kinase. Eur J Biochem 1995; 231: 491–7. [DOI] [PubMed] [Google Scholar]
- 39. Shapiro PS, Whalen AM, Tolwinski NS et al Extracellular signal‐regulated kinase activates topoisomerase IIalpha through a mechanism independent of phosphorylation. Mol Cell Biol 1999; 19: 3551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bensimon A, Schmidt A, Ziv Y et al ATM‐dependent and ‐independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal 2010; 3: rs3. [DOI] [PubMed] [Google Scholar]
- 41. Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 1999; 274: 37538–43. [DOI] [PubMed] [Google Scholar]
- 42. Watanabe F, Teraoka H, Iijima S, Mimori T, Tsukada K. Molecular properties, substrate specificity and regulation of DNA‐dependent protein kinase from Raji Burkitt's lymphoma cells. Biochim Biophys Acta 1994; 1223: 255–60. [DOI] [PubMed] [Google Scholar]
- 43. Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM‐dependent phosphorylation of brca1 in the DNA damage response to double‐strand breaks. Science 1999; 286: 1162–6. [DOI] [PubMed] [Google Scholar]
- 44. Matsumoto Y, Takano H, Kunishio K, Nagao S, Fojo T. Incidence of mutation and deletion in topoisomerase II alpha mRNA of etoposide and mAMSA‐resistant cell lines. Jpn J Cancer Res 2001; 92: 1133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Feldhoff PW, Mirski SE, Cole SP, Sullivan DM. Altered subcellular distribution of topoisomerase II alpha in a drug‐resistant human small cell lung cancer cell line. Cancer Res 1994; 54: 756–62. [PubMed] [Google Scholar]
- 46. Wessel I, Jensen PB, Falck J, Mirski SE, Cole SP, Sehested M. Loss of amino acids 1490Lys‐Ser‐Lys1492 in the COOH‐terminal region of topoisomerase IIalpha in human small cell lung cancer cells selected for resistance to etoposide results in an extranuclear enzyme localization. Cancer Res 1997; 57: 4451–4. [PubMed] [Google Scholar]
- 47. Oloumi A, MacPhail SH, Johnston PJ, Banath JP, Olive PL. Changes in subcellular distribution of topoisomerase IIalpha correlate with etoposide resistance in multicell spheroids and xenograft tumors. Cancer Res 2000; 60: 5747–53. [PubMed] [Google Scholar]
- 48. Chan VT, Ng SW, Eder JP, Schnipper LE. Molecular cloning and identification of a point mutation in the topoisomerase II cDNA from an etoposide‐resistant Chinese hamster ovary cell line. J Biol Chem 1993; 268: 2160–5. [PubMed] [Google Scholar]
- 49. Patel S, Keller BA, Fisher LM. Mutations at arg486 and glu571 in human topoisomerase IIalpha confer resistance to amsacrine: relevance for antitumor drug resistance in human cells. Mol Pharmacol 2000; 57: 784–91. [DOI] [PubMed] [Google Scholar]
- 50. Lee MS, Wang JC, Beran M. Two independent amsacrine‐resistant human myeloid leukemia cell lines share an identical point mutation in the 170 kDa form of human topoisomerase II. J Mol Biol 1992; 223: 837–43. [DOI] [PubMed] [Google Scholar]
- 51. Mao Y, Yu C, Hsieh TS et al Mutations of human topoisomerase II alpha affecting multidrug resistance and sensitivity. Biochemistry 1999; 38: 10793–800. [DOI] [PubMed] [Google Scholar]
- 52. Long BH, Wang L, Lorico A, Wang RC, Brattain MG, Casazza AM. Mechanisms of resistance to etoposide and teniposide in acquired resistant human colon and lung carcinoma cell lines. Cancer Res 1991; 51: 5275–83. [PubMed] [Google Scholar]
- 53. Mirski SE, Cole SP. Cytoplasmic localization of a mutant M(r) 160,000 topoisomerase II alpha is associated with the loss of putative bipartite nuclear localization signals in a drug‐resistant human lung cancer cell line. Cancer Res 1995; 55: 2129–34. [PubMed] [Google Scholar]
- 54. Nakada S, Katsuki Y, Imoto I et al Early G2/M checkpoint failure as a molecular mechanism underlying etoposide‐induced chromosomal aberrations. J Clin Invest 2006; 116: 80–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental experimental procedure.
Fig. S1. Ataxia telangiectasia mutated (ATM) and topoisomerase II alpha (TOP2A) interaction after DNA damage.