

Abstract
Mutant mouse models are indispensable tools for clarifying the functions of genes and elucidating the underlying pathogenic mechanisms of human diseases. We carried out large‐scale mutagenesis using the chemical mutagen N‐ethyl‐N‐nitrosourea. One specific aim of our mutagenesis project was to generate novel cancer models. We screened 7012 animals for dominant traits using a necropsy test and thereby established 17 mutant lines predisposed to cancer. Here, we report on a novel cancer model line that developed osteoma, trichogenic tumor, and breast cancer. Using fine mapping and genomic sequencing, we identified a point mutation in the adenomatous polyposis coli (Apc) gene. The Apc1576 mutants bear a nonsense mutation at codon 1576 in the Apc gene. Although most Apc mutant mice established thus far have multifocal intestinal tumors, mice that are heterozygous for the Apc1576 mutation do not develop intestinal tumors; instead, they develop multifocal breast cancers and trichogenic tumors. Notably, the osteomas that develop in the Apc1576 mutant mice recapitulate the lesion observed in Gardner syndrome, a clinical variant of familial adenomatous polyposis. Our Apc1576 mutant mice will be valuable not only for understanding the function of the Apc gene in detail but also as models of human Gardner syndrome.
N‐Ethyl‐N‐nitrosourea is an effective chemical mutagen that primarily introduces single base‐pair changes.1, 2 The point mutations induced by ENU treatment can result in a large variety of genetic aberrations that range from complete or partial loss‐of‐function to gain‐of‐function. Several large‐scale saturation mutagenesis projects using ENU have been established with the aim of generating large numbers of mutants that will allow gene functions to be systematically investigated in vivo.3, 4, 5 These projects take a phenotype‐driven rather than a gene‐driven approach in that they consist of screening mice that harbor mutations located throughout the genome for altered phenotypes. This approach has the advantage that the screens are not biased by preconceived ideas about gene function. In our Riken mutagenesis project, we screened mutant mice that were generated by ENU mutagenesis for various visible, clinical chemical, hematological, and pathological abnormalities to generate mouse models for common human diseases, including diabetes,6 hypertension, and cancer.
Genetic alterations are some of the major causes of human cancer. Among them, point mutations are often observed in tumor suppressor genes, leading to gain‐of‐function as well as complete or partial loss‐of‐function. The Apc gene was originally isolated as a tumor suppressor gene responsible for FAP, which is characterized by the development of hundreds of colorectal adenomas.7, 8 Several mice lines in which the Apc gene was genetically modified were established by targeted mutation as well as ENU mutagenesis; these mice develop intestinal adenomas and are considered FAP model mice.9, 10 In addition to FAP, mutations in APC are also associated with Gardner syndrome, a clinical variant of FAP. Patients with Gardner syndrome develop desmoid tumors, osteomas, and other types of tumors, including intestinal tumors.11 However, no Apc mutant mice established so far have yet been considered models for Gardner syndrome because they lack osteomas.
Here, we report on the Riken mutagenesis project for the generation of mutant mice predisposed to cancer. We have undertaken a phenotype‐driven screen for dominant mutations and generated 17 dominant hereditary cancer‐predisposed mutant mice. Fine mapping and genome sequencing of one mutant line revealed that a novel nonsense mutation in Apc at codon 1576 was associated with osteomas as well as breast cancer and trichogenic tumors. This unique spectrum of tumors makes Apc1576 mutant mouse a suitable model animal for Gardner syndrome.
Materials and Methods
Animals and ENU mutagenesis
All animal experiments were approved by the Institutional Animal Experiment Committee of Riken BioResource Center (No. 11‐015). We obtained stock mice from Clea Japan (Tokyo, Japan). We injected male B6 mice i.p. at 8–10 weeks of age with 85 or 100 mg/kg body weight of ENU (Sigma‐Aldrich, St. Louis, MO, USA). The injections were carried out twice at weekly intervals. The injected males were mated with D2 females after a sterile period (approximately 10–11 weeks). Phenotypic screens were routinely carried out on the F1 progeny. Early‐onset phenotypic screening tests were carried out at 8–15 weeks, and late‐onset phenotypic screening tests were carried out at 54–77 weeks. Necropsy examinations to detect gross pathological abnormalities were carried out at 78 weeks as the final screening test. In addition to the planned necropsy at 78 weeks, moribund necropsies were carried out where appropriate. The outliers with respect to the manifestation of tumors were assigned M‐numbers. For inheritance testing and mapping, we produced backcrossed progeny by in vitro fertilization using the F1‐outliers' frozen sperm and D2 oocytes. To examine a genetic background effect on the phenotypes, backcrossing to B6 was also carried out.
Mapping and mutated‐gene identification
For mapping, we analyzed the genomic DNA from the N2–N7 progeny backcrossed to D2 using SNP and SSCP markers. We carried out genotype analysis of the Apc mutations by sequencing the genomic DNAs from unaffected and affected backcrossed animals using ABI 3700 and ABI 3100 sequencers (Applied Biosystems, Foster City, CA, USA).
Quantitative real‐time RT‐PCR
Left cerebral hemispheres from the mutants (n = 5) and WT littermates (n = 5) were flash‐frozen in liquid nitrogen, and finely pulverized with a Multi‐beads Shocker (Yasui Kikai, Osaka, Japan). Total RNA was extracted using TRIzol reagent (Gibco BRL, Palo Alto, CA, USA). Reverse transcription was carried out using SuperScript II (Invitrogen, Carlsbad, CA, USA) and oligo‐dT primer. Real‐time PCR was carried out using the QuantiTect SYBR Green RT‐PCR system according to the manufacturer's instructions (Qiagen, Hilden, Germany). Real‐time monitoring of the PCR products was carried out using an ABI 7700 real‐time PCR machine (Applied Biosystems). To ensure that the correct product was amplified in the reaction, all samples were separated by 2% agarose gel electrophoresis. To correct for differences in both the quality and quantity of RNA between samples, the data were normalized using the ratio of the target cDNA concentration to that of β‐actin. All data shown are the average of at least two independent experiments. The Apc gene‐specific primer sets were as follows: for the region covering exon 1–2, forward CACCCGCGAGCACAGC and reverse TTTGCACATGCCGGAGC; for the region covering exon 2–3, forward TCCAAGGGTAGCCAAGGATG and reverse TCAATACTTCCCTGTAGCTGCTTAAG; and for the region in exon 15, forward CGACAATGGGAATGAAACTGAA and reverse TTTGGCTTTGCGTGATGACTT. The primer set for mouse β‐actin was as follows: forward primer CACCCGCGAGCACAGC and reverse primer TTTGCACATGCCGGAGC.
Immunoblot analysis of WT and truncated APC products
Left cerebral hemispheres from the mutants (n = 4) and WT littermates (n = 4) were flash‐frozen in liquid nitrogen and finely pulverized with a Multi‐beads Shocker (Yasui Kikai). Lysis buffer (20 mM Tris‐HCl, pH 8.0, 137 mM NaCl, 1% NP‐40, 10% glycerol, 1 mM EDTA) including a proteinase inhibitor tablet (cOmplete Mini in EASY Pack; Roche, Mannheim, Germany) was mixed with the sample, and the mixture was then centrifuged for 10 min at 9000 g and 4°C to remove insoluble debris. Immunoblot analyses were carried out by first combining protein extract (25 μg/well) with sample buffer (EzApply; ATTO, Tokyo, Japan) and heating the samples at 95°C for 5 min. The samples were resolved using 5% SDS‐PAGE. Protein bands were transferred to a PVDF membrane by electroblotting at 100 V for 90 min at 4°C using a wet transfer apparatus (Mini‐Trans‐Blot electrophoresis Transfer Cell, 170‐3930JA; Bio‐Rad, Hercules, CA, USA) in blotting buffer (92 mM glycine, 25 mM Tris, 15% methanol, 0.04% SDS). The membrane was blocked with 5% non‐fat milk in TBST (10 mM Tris‐HCl, pH 7.6, 150 mM NaCl, 0.1% Tween 20) for 1 h. Membranes were then probed with the anti‐APC antibody (clone FE9, Cat No. OP44; Calbiochem, Merck, Darmstadt, Germany), washed, and incubated with the secondary antibody, which was conjugated to HRP. The blots were then developed using an enhanced chemiluminescent protein development system (Millipore, Billerica, MA, USA).
Karyotype analysis
To prepare metaphase spreads from tumors, breast cancer samples were collected and minced between two microscope slides. The released cells were suspended and washed in PBS. The collected cells were then treated with a hypotonic solution (0.075 M KCl) for 10 min, fixed with fixative (3:1 methanol:acetic acid), and dropped onto a humidified microscope slide. The slides were immediately air dried for at least 10 min then stained with Giemsa solution (Kanto Kagaku, Tokyo, Japan).
Histological analyses
Tissues were fixed in 10% buffered formalin, processed routinely, and embedded in paraffin. Sections were then prepared and stained with H&E. Osteomas were decalcified using 10% EDTA solution (pH 7.0) for 24 h at room temperature. Histopathological diagnosis was confirmed by pathologists. For immunohistochemical analysis, the specimens were incubated with a primary antibody raised against β‐catenin (sc‐7199; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a dilution of 1:100 then incubated for 30 min with an Envision labeled polymer reagent (K1491; Dako, Glostrup, Denmark). Antigen retrieval was carried out by microwave treatment for 10 min using 550 W of power in 10 mM citrate buffer (pH 6.0). Final visualization was carried out by treatment with diaminobenzidine tetrachloride (#040‐27001, DAB tablet; Wako Pure Chemical Industries, Osaka, Japan) and 0.05% H2O2 for 8 min. The specimens were counterstained with Mayer's hematoxylin.
Loss of heterozygosity analysis
The tumor samples from the breast cancer and trichogenic tumor were fixed in 10% buffered formalin and embedded in paraffin. Several sections of each were cut and placed onto slides, and one of the sections was stained with H&E to identify tumor tissue. The histologically relevant regions from the unstained sections were isolated using a blade and transferred to an Eppendorf tube. The cellular material was lysed in a proteinase K buffer solution. DNA was isolated and purified using a DNeasy tissue kit (Qiagen). The osteoma and osteosarcoma tissues were flash‐frozen and finely pulverized with a Multi‐bead Shocker, and the DNA was then isolated and purified using the DNeasy tissue kit (Qiagen). Loss of heterozygosity of the Apc locus was estimated by PCR using two microsatellite markers, D18Mit158 and D18Mit68. These two markers are located close to the Apc gene, and their PCR products differed in size between the mutant (B6‐derived) and WT (D2‐derived) alleles. Both markers resulted in the same pattern in all three types of tumors tested, and the results from D18Mit158 are shown in the LOH analysis.
Results
Generation and identification of Apc mutant mouse lines
To generate various medically relevant mutants in mice and to identify their mutations, we carried out a genome‐wide mutagenesis screen. Male B6 mice were treated with ENU then mated with D2 females. To identify dominant mutations, 7012 F1 progeny derived from the ENU‐treated males were screened: 6478 progenies with planned necropsy tests at 78 weeks of age; and 534 progenies with moribund necropsy tests at 13–77 weeks of age (Table 1). Among the 7012 F1 progeny, 72 phenodeviants showed tumor predisposition. We then carried out inheritance testing in 43 lines and obtained 17 dominant hereditary cancer‐predisposed mutants. Histological analysis of the tumors in the 17 mutant lines revealed a range of tumor types, such as squamous carcinoma, osteoma, breast cancer, trichogenic tumor, pituitary tumor, thyroid tumor, renal carcinoma, lymphoma, pheochromocytoma, duodenal adenoma, and colorectal adenoma.
Table 1.
Phenotypic screening for cancer models in N‐ethyl‐N‐nitrosourea mutagenesis project
| Necropsy tests | Screened age (weeks) | Screened mice | Tumor‐predisposed mice | Inheritance tested | Inheritance confirmed |
|---|---|---|---|---|---|
| Planned | 78 | 6478 | 38 | 23 | 8 |
| Moribund | 13–77 | 534 | 34 | 20 | 9 |
| Total | 7012 | 72 | 43 | 17 |
Phenotype of novel cancer mouse model
In this study, we focused on one mutant that developed osteoma, which has rarely been reported in currently established mouse models. The analysis of other lines will be reported elsewhere. In the inheritance test using N2 progeny with the D2 strain, 76% of the male progeny survived up to 60 weeks of age (Fig. 1). The planned necropsy at 79 weeks of age of these males revealed the onset of multiple osteomas, similar to those detected in the founder mutant. In contrast to the male mice, only half of the females survived up to 50 weeks of age (Fig. 1). Moribund necropsies of these females indicated the onset of multiple breast cancers. In addition to these types of tumor, we noticed that some animals of both genders had multiple trichogenic tumors.
Figure 1.
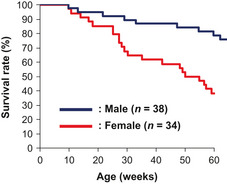
Survival curves of novel cancer model mouse generated by N‐ethyl‐N‐nitrosourea mutagenesis. Survival curves for female (red line) and male (blue line) progeny backcrossed for two generations to the DBA/2J strain.
To correctly estimate the incidence and characterize the histopathology of the phenotype, we backcrossed the mutant mice to the B6 strain and carried out moribund necropsies in the progeny from each generation (Table 2). Osteomas were detected in 66.7% of the females and 100% of males examined in the B6N3 progeny (Fig. 2a, upper panel). These osteomas were mainly found in the skulls and mandibular bones, and some were observed in the ribs and scapulas; histopathological analysis revealed the typical expansile growth pattern of osteomas (Fig. 2a, middle panel). Some animals developed bone tumors containing proliferating spindle cells, and such tumors were diagnosed as osteosarcomas (Fig. 2a, lower panel). Similar to the D2N2 progeny, the first B6N3 female animal became moribund as at 9 weeks of age, and in this generation, breast cancers were found in 100% of the female progeny examined but in none of the male progeny (Table 2). These breast cancers were diagnosed as adenosquamous carcinoma with ductal structures, invasive foci, and poorly differentiated features (Fig. 2b, middle and lower panels). Trichogenic tumors were detected in 80.0% of the males but were not detected in any females of B6N3 progeny (Table 2). These trichogenic tumors were diagnosed as trichoepithelioma and pilomatricoma (Fig. 2c, middle and lower panels, respectively). As shown in Table 2, no significant difference in the incidence of these tumors was observed among generations (B6N2 to B6N18), indicating that there is no genetic background effect on the phenotypes. We note that the lower incidences of osteomas and trichogenic tumor in females than in males are most likely affected by the age of morbidity; breast cancers in female mice arose from 2 months of age, whereas osteomas and trichogenic tumors in males arose from 9 months of age. No other types of tumor or cancer were detected in any of the generations of either gender.
Table 2.
Phenotype of C57BL/6J (B6) backcrossed progeny of a novel cancer model mouse generated by N‐ethyl‐N‐nitrosourea mutagenesis
| B6 backcross generation | Sex | No. of animals tested | Osteoma | Breast cancer | Trichogenic tumor | |||
|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |||
| B6N3 | Female | 9 | 6 | 66.7 | 9 | 100.0 | 0 | 0.0 |
| Male | 10 | 10 | 100.0 | 0 | 0.0 | 8 | 80.0 | |
| B6N4 | Female | 7 | 4 | 57.1 | 7 | 100.0 | 0 | 0.0 |
| Male | 9 | 8 | 88.9 | 1 | 11.1 | 4 | 44.4 | |
| B6N5 | Female | 12 | 3 | 25.0 | 12 | 100.0 | 1 | 8.3 |
| Male | 13 | 13 | 100.0 | 0 | 0.0 | 8 | 61.5 | |
| B6N6 | Female | 9 | 6 | 66.7 | 9 | 100.0 | 1 | 11.1 |
| Male | 7 | 7 | 100.0 | 1 | 14.3 | 5 | 71.4 | |
| B6N7 | Female | 4 | 4 | 100.0 | 4 | 100.0 | 0 | 0.0 |
| Male | 7 | 7 | 100.0 | 0 | 0.0 | 6 | 85.7 | |
| B6N8 | Female | 8 | 5 | 62.5 | 8 | 100.0 | 0 | 0.0 |
| Male | 14 | 11 | 78.6 | 3 | 21.4 | 14 | 100.0 | |
| B6N9 | Female | 13 | 5 | 38.5 | 13 | 100.0 | 2 | 15.4 |
| Male | 18 | 17 | 94.4 | 2 | 11.1 | 13 | 72.2 | |
| B6N10 | Female | 9 | 4 | 44.4 | 9 | 100.0 | 0 | 0.0 |
| Male | 6 | 6 | 100.0 | 2 | 33.3 | 5 | 83.3 | |
| B6N11 | Female | 10 | 5 | 50.0 | 10 | 100.0 | 1 | 10.0 |
| Male | 17 | 17 | 100.0 | 1 | 5.9 | 16 | 94.1 | |
| B6N12 | Female | 12 | 6 | 50.0 | 12 | 100.0 | 3 | 25.0 |
| Male | 9 | 8 | 88.9 | 1 | 11.1 | 9 | 100.0 | |
| B6N13 | Female | 5 | 2 | 40.0 | 5 | 100.0 | 1 | 20.0 |
| Male | 7 | 7 | 100.0 | 0 | 0.0 | 7 | 100.0 | |
| B6N14 | Female | 9 | 2 | 22.2 | 9 | 100.0 | 3 | 33.3 |
| Male | 6 | 6 | 100.0 | 1 | 16.7 | 6 | 100.0 | |
| B6N15 | Female | 6 | 5 | 83.3 | 6 | 100.0 | 2 | 33.3 |
| Male | 9 | 8 | 88.9 | 1 | 11.1 | 9 | 100.0 | |
| B6N16 | Female | 9 | 6 | 66.7 | 9 | 100.0 | 1 | 11.1 |
| Male | 13 | 11 | 84.6 | 1 | 7.7 | 11 | 84.6 | |
| B6N17 | Female | 2 | 1 | 50.0 | 2 | 100.0 | 0 | 0.0 |
| Male | 19 | 17 | 89.5 | 3 | 15.8 | 19 | 100.0 | |
| B6N18 | Female | 7 | 7 | 100.0 | 7 | 100.0 | 1 | 14.3 |
| Male | 14 | 13 | 92.9 | 1 | 7.1 | 14 | 100.0 | |
| B6 backcross total | Female | 131 | 71 | 54.2 | 131 | 100.0 | 16 | 12.2 |
| Male | 178 | 166 | 93.3 | 18 | 10.1 | 154 | 86.5 | |
| Total | 309 | 237 | 76.7 | 149 | 48.2 | 170 | 55.0 | |
Incidence of cancer phenotypes was examined in B6 backcrossed progeny by moribund necropsies.
Figure 2.
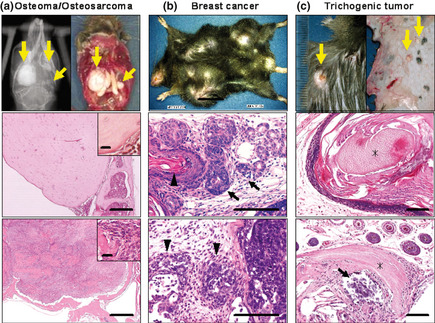
Cancer phenotype of novel cancer model mouse generated by N‐ethyl‐N‐nitrosourea mutagenesis. (a) Osteoma. Upper panel, macroscopic and X‐ray image. Middle panel, osteoma, an expansile growth pattern with a sharp demarcation from the surrounding tissue was observed. Lower panel, osteosarcoma. Bar = 200 μm. Inset, proliferating spindle cells. Bar = 20 μm. (b) Breast cancer. Upper panel, macroscopic image. Middle panel, ductal structure (arrow) and keratinization (arrowhead) revealed in a breast cancer in Apc1576. Lower panel, poorly differentiated adenosquamous carcinoma. Invasion of the subcutaneous area was observed (arrowheads). Bar = 100 μm. (c) Trichogenic tumor. Upper panel, macroscopic image. Middle panel, trichoepithelioma. Lower panel, pilomatricoma. Shadow cells (asterisk) and calcification (arrow) were observed. Bar = 100 μm.
Mapping and identification of the mutation
To identify the mutation responsible for the phenotype, genome‐wide mapping was initially carried out with SSCP markers and the genomic DNA from the N2–N7 progeny backcrossed to D2, and the cancer phenotype was mapped to mouse chromosome 18. We then carried out a fine mapping study using SNP markers. Three phenotypes, osteomas, breast cancer, and trichogenic tumors, were mapped to the same 0.9 Mb region between D18Mit158 and an SNP in the Pkd2 l2 gene (Fig. 3). There are 10 genes in this region (Table 3), according to the Ensembl Mouse Genome assembly (http://may2012.archive.ensembl.org/Mus_musculus/Info/Index): NCBI m37. We carried out genomic DNA sequencing of 71 exons and splice sites of these 10 genes and detected 18 single‐base substitutions that were not listed in the public database as the SNPs lying between B6 and D2 (Table 4). Of these 18 base substitutions, 12 were detected in introns or in a 3′ downstream region but not in splicing regulation sites. A further six substitutions were detected in exons. Of these, two were in non‐coding regions and three were detected in exon 16 of the Ebp4.1l4a gene, resulting in synonymous amino acid changes. One substitution detected in exon 15 of the Apc gene, T4728A, resulted in a nonsense mutation that changed Cys1576 (TGT) to a stop codon (TGA). We carried out genome sequencing on wild B6 and D2 animals that did not receive ENU treatment, and the 17 substitutions (substitutions other than that in the Apc gene) were found in these ENU‐untreated animals. Therefore, these 17 substitutions were not ENU‐induced mutations but were instead SNPs between the mouse strains. We confirmed that the T4728A substitution in the Apc gene was not detected in the normal littermates of the mutants. Thus, we concluded that this mutation, which we hereafter term Apc1576, was responsible for the predisposition to the tumors. In addition, to address the potential role of other cancer driver genes in the Apc1576 mouse tumors, sequence‐based analyses for the coding region of K‐ras and p53 were carried out in tumors developed in Apc1576 mutants, and no mutations of these genes were identified. Immunohistochemical analyses showed no significant elevation of p53 expression in the breast cancers or osteosarcomas of the Apc1576 mutants.
Figure 3.
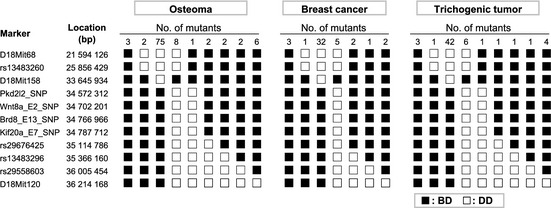
Fine mapping study on chromosome 18 using single nucleotide polymorphism markers. Haplotype analysis of DBA/2J (D2)‐backcrossed progeny (generations N2–N7) of a novel cancer mutant mouse. Results from mutants that developed osteomas are shown on the left, results from mutants that developed breast cancer are shown in the middle, and results from mutants that developed trichogenic tumors are shown on the right. Black boxes (■) represent heterozygosity of the C57BL/6J and D2 allele (BD), and white boxes (□) represent homozygosity of the D2 allele (DD). The numbers of mutant animals that inherited each haplotype are shown at the top of each column.
Table 3.
List of genes located in the narrowed genomic region by fine mapping
| Gene name | Gene start (bp) | Gene end (bp) | No. of exons | Coding (bp) | cDNA (bp) |
|---|---|---|---|---|---|
| 2410004N09Rik | 33 954 575 | 33 955 640 | 3 | 0 | 401 |
| Epb4.1l4a | 33 955 981 | 34 166 860 | 24 | 2050 | 3853 |
| U7 | 34 183 874 | 34 183 934 | 1 | 0 | 61 |
| SNORA17 | 34 253 099 | 34 253 224 | 1 | 0 | 126 |
| U7 | 34 348 532 | 34 348 593 | 1 | 0 | 62 |
| Gm10548 | 34 367 429 | 34 381 426 | 2 | 0 | 2629 |
| Apc | 34 380 578 | 34 481 843 | 15 | 8529 | 8867 |
| Srp19 | 34 490 501 | 34 496 253 | 4 | 435 | 876 |
| Reep5 | 34 504 543 | 34 533 069 | 5 | 570 | 2894 |
| Pkd2 l2 | 34 569 077 | 34 602 445 | 15 | 1866 | 2463 |
| Total | 71 | 13 450 | 22 232 |
There are 10 genes located in a region of approximately 0.9 Mb in mouse chromosome 18.
Table 4.
Eighteen base substitutions detected by genomic sequencing of the fine mapping region
| SNP name | Gene | Location(bp in Chr18) | Base detected in mutants | Base detected in WT | Detected region | Amino acid change | |
|---|---|---|---|---|---|---|---|
| C57BL/6J | DBA/2J | ||||||
| novel_1 | 241004N09Rik | 33 955 722 | G | G | A | 3′ downstream sequence | – |
| novel_2 | Epb4.1l4a | 34 051 055 | T | T | C | Intron 4–5 | – |
| novel_3 | Epb4.1l4a | 34 051 039 | C | C | G | Intron 4–5 | – |
| novel_4 | Epb4.1l4a | 34 046 147 | C | C | A | Intron 4–5 | – |
| novel_5 | Epb4.1l4a | 34 039 809 | A | A | G | Intron 6–7 | – |
| novel_6 | Epb4.1l4a | 34 038 517 | G | G | A | Intron 7–8 | – |
| rs31625359 | Epb4.1l4a | 34 020 741 | A | A | G | Intron 11–12 | – |
| novel_7 | Epb4.1l4a | 33 987 863 | T | T | C | Intron 15–16 | – |
| rs51317212 | Epb4.1l4a | 33 987 871 | C | C | T | Exon 16 | S421S (synonymous) |
| rs31627049 | Epb4.1l4a | 33 987 790 | T | T | C | Exon 16 | S448S (synonymous) |
| rs31627050 | Epb4.1l4a | 33 987 775 | G | G | A | Exon 16 | S453S (synonymous) |
| novel_8 | Epb4.1l4a | 33 969 823 | G | G | A | Intron 19–20 | – |
| novel_9 | Gm10548 | 34 369 526 | C | C | T | Exon 2 (non‐coding) | – |
| novel_10 | Gm10548 | 34 368 236 | A | A | G | Exon 2 (non‐coding) | – |
| – | Apc | 34 474 434 | A | T | T | Exon 15 | C1576X (Stop) |
| novel_11 | Srp19 | 34 491 540 | A | A | G | Intron 2–3 | – |
| novel_12 | Reep5 | 34 509 476 | T | T | G | Intron 4–5 | – |
| novel_13 | Pkd2 l2 | 34 572 312 | G | G | A | Intron 2–3 | – |
Genomic DNA sequencing on all exons and splicing sites of all genes within the mapped region was carried out and 18 base substitutions were detected. The 17 substitutions were not N‐Ethyl‐N‐nitrosourea‐induced mutations but were instead single nucleotide polymorphisms (SNPs) for the C57BL/6J and DBA/2J strains. Chr18, chromosome 18. Bold text indicates the mutation identified in Apc gene. “– ” indicates that no amino acid change was induced by the base substitution because the substitution was in non‐coding sequence region.
Effects of mutation
To elucidate the effects of the Apc1576 mutation on its mRNA stability in vivo, we investigated the level of Apc mRNA from the mutant mouse brains. As shown in Figure 4(a), real‐time PCR revealed no significant change in the levels of Apc mRNA in these mutants, indicating that the nonsense‐mediated decay pathway was not activated by the Apc1576 mutation. Additionally, immunoblot analysis showed that the Apc1576 mutant expressed the predicted truncated protein (Fig. 4b). Furthermore, LOH of the Apc gene locus was confirmed in all of the cancerous specimens tested (Fig. 4c, lower panels). Because it has been shown that Apc suppresses canonical Wnt signaling by stimulating the degradation of β‐catenin, we used immunohistochemistry to probe for β‐catenin levels. In all three types of lesions (breast cancer, trichogenic tumor, and osteoma), strong nuclear staining of β‐catenin was detected, suggesting that the Apc1576 mutation affects β‐catenin degradation (Fig. 4c). Taken together, these observations further support our conclusion that the Apc1576 mutation was responsible for the onset of these tumors.
Figure 4.
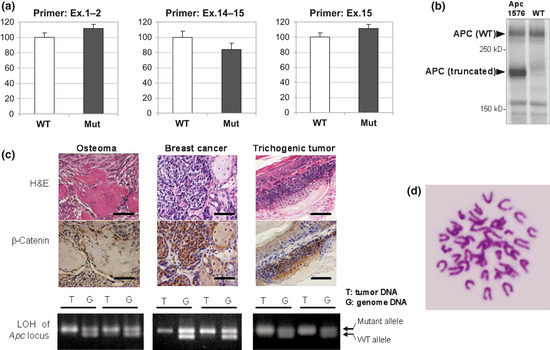
Effects of Apc1576 mutation. (a) Real‐time PCR of Apc mRNA levels in the brains of Apc1576 hetero mutant animals (Mut) or of wild‐type littermates (WT; n = 5 for each genotype). We examined the expression levels using three primer pairs that amplify the region covering exons (Ex.) 1–2 (left), exons 14–15 (center), and exon 15 (right). No significant difference was seen between the hetero mutants and their WT littermates. (b) Immunoblot analysis using an anti‐Apc antibody of protein extracted from the brains of the Apc1576 mutant (left lane) or a WT animal (right lane). In Apc1576 mutants, both truncated product (176 kDa) and WT product (312 kDa) were detected. (c) Nuclear accumulation of β‐catenin and loss of heterozygosity (LOH) of the Apc locus in tumor tissues of Apc1576 mutant animals. As shown in the upper two panels, strong nuclear staining of β‐catenin was detected in all three types of lesions, osteosarcoma (left), breast cancer (center), and trichogenic tumor (right). Bar = 50 μm. In the lower panels, LOH of the Apc gene locus was confirmed in all types of tumors detected in the Apc1576 mutants. Each panel shows the representative results from tumor DNA (T) and genomic DNA (G) of two different mutants in one tumor type. (d) Karyotype analysis of breast cancer cells derived from the Apc1576 mutant. No aneuploidy was observed.
Because previous studies on Apc knockout mice have suggested that adenoma–carcinoma progression proceeds under genomic instability,12, 13, 14 we carried out karyotype analyses on freshly collected breast cancer cells from the 42‐week‐old female mutant. However, all examined cells had the normal number of chromosomes; therefore, no signs of aneuploidy were detected (Fig. 4d).
To assess the viability of the Apc1576 homozygous mutant mice, heterozygous mutant animals were interbred, and the resulting 101 embryos were analyzed (data not shown). Homozygous embryos were detected in E6.5–E9.5 in accordance with Mendelian inheritance; however, no homozygous embryo was found in E10.5–E13.5, indicating that the Apc1576 mutation results in embryonic lethality, thus confirming the previous reports of Apc mutant mice.
Discussion
The human APC gene was originally identified as responsible for FAP, an autosomal dominant disease characterized by a predisposition to cancer, and affected individuals develop thousands of adenomatous polyps in the intestine.7, 8 The APC gene encodes a multidomain protein and its major role in tumor suppression is to downregulate Wnt signaling by stimulating the degradation of β‐catenin. To date, several mouse models for FAP have been established by targeting the mouse Apc gene or by ENU mutagenesis. Most of the resulting mutants lack 20‐amino acid repeats and β‐catenin downregulation domains. These mutants result in the development of intestinal tumors with high multiplicity (Fig. 5).15, 16, 17, 18, 19, 20, 21, 22 However, the Apc1576 mice obtained in this study retained three of the seven domains and did not develop intestinal tumors, supporting a link between the onset of intestinal tumors and β‐catenin degradation. Instead, we observed a high incidence of breast cancer, particularly in female animals. Of note, the absence of intestinal tumors and the development of breast cancer are similar to those reported in Apc1572T mutant mice.23 These observations indicate that the nonsense mutation at codon 1576 of the Apc gene is responsible for the tumorigenesis of the breast cancer developed in these mice.
Figure 5.
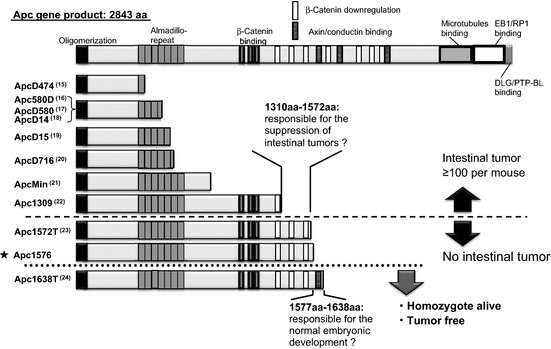
Summary of the relationship between the length of the truncated adenomatous polyposis coli (APC) proteins expressed in mutant mice and their phenotypes. Full‐length APC protein is shown together with the major functional domains at the top. The 10 main Apc mutations reported previously and our novel mutation, Apc1576 (★), are described to the left of the truncated APC proteins that they encode. The top eight mutants developed multiple intestinal tumors,15, 16, 17, 18, 19, 20, 21, 22 whereas the lower three did not. Both Apc1572T23 and Apc1576 similarly developed breast cancer, but only Apc1576 developed osteomas. Of all these mutants, only in Apc1638T are the heterozygous mutants tumor‐free and the homozygote mutants viable.24 a.a., amino acids.
Unlike the reports for breast cancer, osteomas have been found to develop in Apc1576 mice at high incidence but have not been reported in Apc1572T mutant mice. Because the phenotypes of both mutants were analyzed in the B6‐backcrossed animals (Ref. 23 and Table 2), it is less likely that the phenotypic difference is due to the effect of the genetic background. A possible explanation for this difference could be that additional mutations were introduced during the course of ENU mutagenesis within the 0.9‐Mb region as defined by backcrossing with the D2 strain followed by fine mapping using SNP markers (Fig. 3). Such additional and unknown mutations could be responsible for osteoma independently of Apc mutation or, alternatively, they may have additional effects to the mutation in Apc. In fact, synergistic mutations have previously been reported in a mutant mouse line generated by ENU mutagenesis.25 Therefore, we carried out extensive genomic sequencing of all exons and their adjacent introns. In this region, five protein‐coding genes and five non‐coding genes were located, which were encoded by 71 exons. A total of approximately 18 kb of sequencing revealed 18 nucleotide substitutions. However, the genomic sequencing of WT animals from the parental strains indicated that all substitutions except Apc1576 were SNPs (Table 4). Although we cannot completely exclude the possibility of additional mutation, these results support the idea that the nonsense mutation of Apc gene at codon 1576 is responsible for the onset of the osteomas. This hypothesis is further supported by the fact that N18 progeny that were backcrossed to the B6 strain, which retains the Apc1576 mutation, developed osteomas (in 92.9% of male mutants, Table 2). These observations support our conclusion that the difference between Apc1576 and Apc1572T results from the four‐amino acid difference. Alternatively, because we focused on the late‐onset lesion in our mutagenesis project, we were able to identify osteomas, a relatively late‐onset phenotype compared to breast cancer. Further analysis is required to address this issue.
Human FAP is characterized by the development of intestinal adenomas but also has phenotypic variants that differ in the onset of tumor development, tumor number, and the tumor spectrum. Several factors, such as genetic polymorphisms, epigenetic changes, and additional mutations, potentially affect the phenotypic variations. Gardner syndrome is a clinical variant of FAP in which extracolonic features, including osteomas, are prominent. Although the molecular basis for the characteristic manifestations of Gardner syndrome remains to be fully elucidated, several studies indicate a genotype–phenotype correlation. Some mutational analyses on the human APC gene showed that mutations contributing to classical FAP occur in the 5′ region of the APC gene (reviewed in Ref. 26), whereas other mutations associated with Gardner syndrome tend to cluster in the 3′ region of the gene.27, 28, 29 Thus, our findings that mutation at codon 1576 leads to extracolonic manifestations are in good agreement with the genotype–phenotype correlations found in human FAP patients. Because the truncated protein expressed in Apc1576 mutant mouse partially contains 20‐amino acid repeats that are involved in the downregulation of β‐catenin but lack SAMP repeats that contribute to Axin binding, Wnt signaling may be differently modulated in Apc1576 compared to the other classical Apc mutants that do not show the osteoma‐prone phenotype. In addition, because the product of APC is a multifunctional protein that is ubiquitously expressed and interacts with a variety of proteins in addition to Wnt signaling molecules, the unique truncation mutation may affect these interactions, thus contributing to the development of extracolonic tumors. Taken together, the results suggest that the genetically modified Apc1576 mutant mice generated in this study will be a powerful tool to elucidate the molecular mechanism underlying Gardner syndrome and aid in understanding the in vivo function of the Apc tumor suppressor gene.
Abbreviations
- APC
adenomatous polyposis coli
- FAP
familial adenomatous polyposis
- ENU
N‐ethyl‐N‐nitrosourea
- LOH
loss of heterozygosity
- SNP
single nucleotide polymorphism
- SSCP
single‐strand conformation polymorphism
- B6
C57BL/6J
- D2
DBA/2J
Disclosure Statement
The authors have no conflict of interest.
(Cancer Sci, doi: 10.1111/cas.12161, 2013)
References
- 1. Hitotsumachi S, Carpenter DA, Russell WL. Dose‐repetition increases the mutagenic effectiveness of N‐ethyl‐N‐nitrosourea in mouse spermatogonia. Proc Natl Acad Sci U S A 1985; 82: 6619–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Noveroske JK, Weber JS, Justice MJ. The mutagenic action of N‐ethyl‐N‐nitrosourea in the mouse. Mamm Genome 2000; 11: 478–83. [DOI] [PubMed] [Google Scholar]
- 3. Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A. Mouse ENU mutagenesis. Hum Mol Genet 1999; 8: 1955–63. [DOI] [PubMed] [Google Scholar]
- 4. Nolan PM, Peters J, Strivens M et al A systematic, genome‐wide, phenotype‐driven mutagenesis programme for gene function studies in the mouse. Nat Genet 2000; 25: 440–3. [DOI] [PubMed] [Google Scholar]
- 5. Hrabe de Angelis MH, Flaswinkel H, Fuchs H et al Genome‐wide, large‐scale production of mutant mice by ENU mutagenesis. Nat Genet 2000; 25: 444–7. [DOI] [PubMed] [Google Scholar]
- 6. Inoue M, Sakuraba Y, Motegi H et al A series of maturity onset diabetes of the young, type 2 (MODY2) mouse models generated by a large‐scale ENU mutagenesis program. Hum Mol Genet 2004; 13: 1147–57. [DOI] [PubMed] [Google Scholar]
- 7. Groden J, Thliveris A, Samowitz W et al Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991; 66: 589–600. [DOI] [PubMed] [Google Scholar]
- 8. Nishisho I, Nakamura Y, Miyoshi Y et al Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991; 253: 665–9. [DOI] [PubMed] [Google Scholar]
- 9. Taketo MM, Edelmann W. Mouse models of colon cancer. Gastrienterology 2009; 136: 780–98. [DOI] [PubMed] [Google Scholar]
- 10. McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract 2008; 204: 479–90. [DOI] [PubMed] [Google Scholar]
- 11. Gardner EJ. A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum. Am J Hum Genet 1951; 3: 167–76. [PMC free article] [PubMed] [Google Scholar]
- 12. Hinoi T, Akyol A, Theisen BK et al Mouse model of colonic adenoma‐carcinoma progression based on somatic Apc inactivation. Cancer Res 2007; 67: 9721–30. [DOI] [PubMed] [Google Scholar]
- 13. Dikovskaya D, Schiffmann D, Newton IP et al Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. J Cell Biol 2007; 176: 183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aoki K, Taketo M. Adenomatous polyposis coli (APC): a multi‐functional tumor suppressor gene. J Cell Sci 2007; 120: 3327–35. [DOI] [PubMed] [Google Scholar]
- 15. Sasai H, Masaki M, Wakitani K. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. Carcinogenesis 2000; 21: 953–8.10783317 [Google Scholar]
- 16. Shibata H, Toyama K, Shioya H et al Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 1997; 278: 120–3. [DOI] [PubMed] [Google Scholar]
- 17. Kuraguchi M, Wang XP, Bronson RT et al Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet 2006; 2: 1362–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colnot S, Niwa‐Kawakita M, Hamard G et al Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest 2004; 84: 1619–30. [DOI] [PubMed] [Google Scholar]
- 19. Robanus‐Maandag EC, Koelink PJ, Breukel C et al A new conditional Apc‐mutant mouse model for colorectal cancer. Carcinogenesis 2010; 31: 946–52. [DOI] [PubMed] [Google Scholar]
- 20. Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Ape gene. Proc Natl Acad Sci U S A. 1995; 92: 4482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990; 247: 322–4. [DOI] [PubMed] [Google Scholar]
- 22. Ito M, Miura S, Noda T. Mouse model for familial adenomatous polyposis coli and APC gene. Protein, Nucleic acid and Enzyme 1995; 40: 2035–44. (Japanese.) [PubMed] [Google Scholar]
- 23. Gaspar C, Franken P, Molenaar L et al A targeted constitutive mutation in the Apc tumor suppressor gene underlies mammary but not intestinal tumorigenesis. PLoS Genet 2009; 5: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smits R, Kielman MF, Breukel C et al Apc1638T: a mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev 1999; 13: 1309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Douglas DS, Moran JL, Bermingham JR Jr et al Concurrent Lpin1 and Nrcam mouse mutations result in severe peripheral neuropathy with transitory hindlimb paralysis. J Neurosci 2009; 29: 12089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis 2009; 4: 22–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Enomoto M, Konishi M, Iwama T et al The relationship between frequencies of extracolonic manifestations and the position of APC germline mutation in patients with familial adenomatous polyposis. Jpn J Clin Oncol 2000; 30: 82–8. [DOI] [PubMed] [Google Scholar]
- 28. Bisgaard ML, Bülow S. Familial adenomatous polyposis (FAP): genotype correlation to FAP phenotype with osteomas and sebaceous cysts. Am J Med Genet 2006; 140: 200–4. [DOI] [PubMed] [Google Scholar]
- 29. Dobbie Z, Spycher M, Mary JL et al Correlation between the development of extracolonic manifestations in FAP patients and mutations beyond codon 1403 in the APC gene. J Med Genet 1996; 33: 274–80. [DOI] [PMC free article] [PubMed] [Google Scholar]