

Abstract
The present study aimed to identify genes associated with patient survival to improve our understanding of the underlying biology of gliomas. We investigated whether the expression of genes selected using random survival forests models could be used to define glioma subgroups more objectively than standard pathology. The RNA from 32 non‐treated grade 4 gliomas were analyzed using the GeneChip Human Genome U133 Plus 2.0 Expression array (which contains approximately 47 000 genes). Twenty‐five genes whose expressions were strongly and consistently related to patient survival were identified. The prognosis prediction score of these genes was most significant among several variables and survival analyses. The prognosis prediction score of three genes and age classifiers also revealed a strong prognostic value among grade 4 gliomas. These results were validated in an independent samples set (n = 488). Our method was effective for objectively classifying grade 4 gliomas and was a more accurate prognosis predictor than histological grading.
Glioblastomas are pathologically the most aggressive form of glioma, with a median survival range of only 9–15 months.1, 2 Even advances in cancer biology, surgical techniques, chemotherapy and radiotherapy have led to little improvement in survival rates of glioblastoma patients.1 Poor prognosis is attributable to difficulties in early detection and to a high recurrence rate after initial treatment. Therefore, more effective therapeutic approaches, a clearer understanding of the biological features of glioblastoma and the identification of novel target molecules are needed for improved diagnosis and therapy of this disease.
Several histological grading schemes exist. The World Health Organization (WHO) system is currently the most widely used; a high WHO grade correlates with clinical progression and decreased survival rate.3 However, individual fates vary within diagnostic categories, even in grade 4 glioma,1, 2 indicating the need for additional prognostic markers. The inadequacy of histopathological grading is evidenced, in part, by the inability to recognize patients prospectively.
Microarray technology has permitted the development of multiorgan cancer classification including gliomas, the identification of glioma subclasses, the discovery of molecular markers and predictions of disease outcomes.4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 Unlike clinicopathological staging, molecular staging can predict long‐term outcomes of individuals based on gene expression profiles of tumors at diagnosis, enabling clinicians to make optimal clinical decisions. The analysis of gene expression profiles in clinical materials is an essential step towards clarifying the detailed mechanisms of oncogenesis and the discovery of target molecules for the development of novel therapeutic drugs.
In the present study, we describe an expression profiling study of a panel of 32 patients with grade 4 gliomas for the identification of genes that predict overall survival (OS) using random survival forests models, with validation in independent data sets.
Materials and Methods
Samples
Tissues were snap‐frozen in liquid nitrogen within 5 min of harvesting and stored thereafter at −80°C. The clinical stage was estimated from accompanying surgical pathology and clinical reports. Samples were specifically re‐reviewed by a board‐certified pathologist at Niigata University, Niigata, Japan according to WHO criteria, by observing sections of paraffin‐embedded tissues that were adjacent or in close proximity to frozen samples from which the RNA was subsequently extracted. The histopathology of each collected specimen was reviewed to confirm the adequacy of the sample (i.e. minimal contamination with non‐neoplastic elements) and to assess the extent of tumor necrosis and cellularity. Informed consent was obtained from all patients for the use of the samples, in accordance with the guidelines of the Ethical Committee on Human Research, Niigata University Medical School (Protocol #70). Overall survival was measured from the date of diagnosis. Survival end‐points corresponded to the dates of death or last follow up.
RNA extraction and array hybridization
Approximately 100 mg of tissue from each tumor was used to extract total RNA using the Isogen method (Nippongene, Toyama, Japan) following the manufacturer's instructions. The quality of RNA obtained was verified with the Bioanalyzer System (Agilent Technologies, Tokyo, Japan) using RNA Pico Chips. Only samples with 28S/18S ratios >0.7 and with no evidence of ribosomal peak degradation were included in the present study. One microgram of each RNA was processed for hybridization using GeneChip Human Genome U133 Plus 2.0 Expression arrays (Affymetrix, Inc., Tokyo, Japan), which comprised approximately 47 000 genes. After hybridization, the chips were processed using a Fluidics Station 450, a High‐Resolution Microarray Scanner 3000 and a GCOS Workstation Version 1.3 (Affymetrix, Inc).
Validation of differential expression using real‐time quantitative PCR
The quantitative PCR (QPCR) was performed using a StepOne Real‐Time PCR System (Applied Biosystems, Tokyo, Japan) and TaqMan Universal PCR Master Mix (Applied Biosystems) according to the manufacturer's protocol. The Assays‐on‐Demand probe/primer sets (Applied Biosystems) used were as follows: ANGPTL1, Hs00559786_m1; ARHGAP39, Hs00286798_m1; ASF1A, Hs00204044_m1; CASP8, Hs01018151_m1; C11orf71, Hs00535489_s1; EFNB2, Hs00187950_m1; GAPDH, Hs99999905_m1; GPNMB, Hs01095679_m1; ITGA7, Hs00174397_m1; LDHA, Hs00855332_g1; LMAN2L, Hs01091681_m1; LOXL3, Hs01046945_m1; MED29, Hs00378316_m1; and MGMT, Hs01037698_m1.
Total RNA (1 μg) was reverse transcribed into cDNA using SuperScript II (Invitrogen, Tokyo, Japan) and 1 μL of the resulting cDNA was used for QPCR. Validation was performed on a subset of tumors that were part of the original tumor data set assessed. Assays were carried out in duplicate. The raw data produced using the QPCR referred to the number of cycles required for reactions to reach the exponential phase. Expression of GAPDH was used to normalize the QPCR data. Mean expression fold change differences between tumor groups were calculated using the 2−ΔΔCT method.15
Immunohistochemistry
Five‐micron sections from formalin‐fixed, paraffin‐embedded tissue specimens were used for immunohistochemistry (IHC). Endogenous peroxidase was blocked with 0.3% H2O2 in methanol. Antigen retrieval was performed by autoclaving at 120°C for 10 min in 50 mM citrate buffer (pH 6.0). The IHC for anti‐O6‐methylguanine‐methyltransferase (MGMT; antibody dilution 1:50; clone MT3.1; Millipore, Billerica, MA, USA) was performed as described previously.16 Immunoreactivity (MGMT staining index [SI]) was quantified by counting stained tumor nuclei in >1000 cells and was expressed as a percentage of positive cells. A MGMT SI >30% was considered positive for MGMT. Averages of three independent measurements were calculated to the first decimal place. Observers were not aware of case numbers.
Analysis of the isocitrate dehydrogenase 1 (IDH1) codon 132 mutation
A 129‐bp fragment of IDH1 that included codon 132 was amplified using IDH1f, 5′‐CGGTCTTCAGAGAAGCCATT‐3′ as the sense primer and IDH1r, 5′‐GCAAAATCACATTATTGCCAAC‐3′ as the antisense primer. A PCR was performed on 20 ng of DNA with Taq DNA Polymerase (Takara, Tokyo, Japan) and standard conditions of 35 cycles were used. The PCR amplification product was sequenced using a BigDyeTerminator v3.1 Sequencing Kit (Applied Biosystems) using the sense primer IDH1f and antisense primer IDH1rc, 5′‐TTCATACCTTGCTTAATGGGTGT‐3′. Sequences were determined using the semiautomated sequencer (ABI 3100 Genetic Analyzer; Applied Biosystems) and Sequence Pilot version 3.1 software (JSI‐Medisys, Kippenheim, Germany) as described previously.17
Bioinformatics analysis
All statistical analyses were performed using R software18 and Bioconductor.19 The Affymetrix GeneChip probe‐level data were preprocessed using MAS 5.0 (Affymetrix Inc.) for background adjustment and log‐transformation (base 2). Each array was normalized using a quantile normalization to impose the same empirical distribution of intensities to each array. Genes that passed the filter criteria below were considered for further analysis. To select predictors (genes) for OS, we first set filtered gene expressions and applied the random survival forests–variable hunting (RSF‐VH) algorithm.20 Among the algorithm parameters, the number of Monte Carlo iterations (nrep) and value to control step size used in the forward process (nstep) were set as nrep = 100 and nstep = 5, respectively, following the method of Ishwaran et al.20 For other parameters such as number of trees and number of variables selected randomly at each node, we used the default settings for varSelfunction within the RandomSurvivalForest package before selection. We classified samples into two survival groups using Ward's minimum variance cluster analysis, inputting ensemble cumulative hazard functions for each individual for all unique death time‐points estimated from the fitted random survival forests model to selected genes.
The two classified survival groups were used to compute the prognosis prediction score (PPS) from a simple form (linear combination of gene expressions). To do this, we used principal component analysis and receiver operating characteristic analysis. Briefly, we computed the first principal component of gene expressions selected by the RSF‐VH algorithm as a risk score and then searched for the optimal value to predict survival groups with maximum accuracy using the Youden index.21 Validation for this method is used in the validation set (n = 488; Table 1), which is derived from glioblastoma patients in four external data sets.8, 10, 12, 22
Table 1.
Patient characteristics of grade 4 glioma
| Variable | Test set | Validation set | P |
|---|---|---|---|
| (n = 32) | (n = 488) | ||
| Age (years) | |||
| Average | 54.5 | 55.0 | 0.84 |
| Range | 18–80 | 10–86 | |
| Gender | |||
| Male | 20 | 305 | 1.00 |
| Female | 12 | 183 | |
| Survival time (days) | 411 | 364 | 0.41 |
The survival tree method23 constructs prognostic groups based on PPS and age among those with grade 4 glioma. This method is based on a recursive partition of the PPS and age values while splitting patients into the subset. Final output results in groups of patients with similar prognoses, which are represented as combinations of binarized PPS or age. This was executed using the rpart package of the R software.
The Kaplan–Meier method was used to estimate the survival distribution for each group. A log‐rank test was used to test differences between survival groups. The association of the PPS with OS was evaluated using multivariate analyses with clinical characteristics and with other predictors using the Cox proportional hazards regression model. P < 0.05 was considered statistically significant.
Results
Patient characteristics
Thirty‐two non‐treated primary glioblastomas (WHO grade IV) came from patients who underwent surgical resections between 2000 and 2005 (Table 1). The median age of patients was 54.5 years (range,18–80 years). Twenty patients were male and 12 were female. The preoperative Karnofsky performance status (KPS) was at least 70 in 25 (78%) patients. The IDH1 mutation was negative in 31 cases, but was detected in one patient who remains alive 2365 days after the onset of disease. The MGMT IHC was positive in 21 cases and negative in 11 cases. After maximum surgical tumor resections, patients received external beam radiation therapy (standard dose of 60 Gy to the tumor with a 2‐cm margin) and first‐line chemotherapy with nimustine and temozolomide at recurrence. Patients were monitored for tumor recurrence during initial and maintenance therapy using MRI or computed tomography. Treatments were carried out at the Department of Neurosurgery, Niigata University Hospital. The median survival time was 13.7 months.
Selection of predictive genes
Microarray data were deposited in the Gene Expression Omnibus (accession number GSE 43378) and 25 genes were selected as predictors. Table 2 shows a list of the genes with their variable importance values. The scatter plot in Supporting Information Figure S1 shows the relationships between the estimated ensemble mortalities and expression for six selected genes (AFTPH, ARHGAP39, CASP8, ITGA7, LDHA and LOXL3). Validation of the microarray results was accomplished using QPCR. These 10 genes were also found to be differentially expressed between short‐term (survival time, ≤1.5 years) and long‐term (survival time, ≥2.5 years) survivors (Table S1). The heat map (Fig. S2) shows patients clustered by estimated ensemble mortalities (columns) and genes clustered by their expression levels (rows). For patients with low survival (blue bar), the lower genes are overexpressed while the upper genes are underexpressed. For patients with improved survival (red bar), these patterns were reversed; thus, the indicated genes might be effective in distinguishing between patients with different survival rates.
Table 2.
Identification of survival related 25 genes
| Probe | Symbol | Description | VI |
|---|---|---|---|
| 225708_at | MED29 | Mediator complex subunit 29 | 0.0202 |
| 217939_s_at | AFTPH | Aftiphilin | 0.0101 |
| 227876_at | ARHGAP39 | Rho GTPase activating protein 39 | 0.0101 |
| 228821_at | ST6GAL2 | ST6 beta‐galactosamide alpha‐2,6‐sialyltranferase 2 | 0.0101 |
| 200650_s_at | LDHA | Lactate dehydrogenase A | 0.0081 |
| 220260_at | TBC1D19 | TBC1 domain family, member 19 | 0.0060 |
| 218981_at | ACN9 | ACN9 homolog (S. cerevisiae) | 0.0060 |
| 231773_at | ANGPTL1 | Angiopoietin‐like 1 | 0.0060 |
| 201141_at | GPNMB | Glycoprotein (transmembrane) nmb | 0.0040 |
| 228255_at | ALS2CR4 | Amyotrophic lateral sclerosis 2 (juvenile) chromosome region, candidate 4 | 0.0040 |
| 203427_at | ASF1A | ASF1 anti‐silencing function 1 homolog A (S. cerevisiae) | 0.0020 |
| 222108_at | AMIGO2 | Adhesion molecule with Ig‐like domain 2 | 0.0020 |
| 1562527_at | LOC283027 | Hypothetical protein LOC283027 | 0.0000 |
| 218789_s_at | C11orf71 | Chromosome 11 open reading frame 71 | 0.0000 |
| 219240_s_at | C10orf88 | Chromosome 10 open reading frame 88 | −0.0020 |
| 213373_s_at | CASP8 | Caspase 8, apoptosis‐related cysteine peptidase | −0.0020 |
| 225126_at | MRRF | Mitochondrial ribosome recycling factor | −0.0020 |
| 209663_s_at | ITGA7 | Integrin, alpha 7 | −0.0040 |
| 223222_at | SLC25A19 | Solute carrier family 25 (mitochondrial thiamine pyrophosphate carrier), member 19 | −0.0040 |
| 214271_x_at | RPL12 | Ribosomal protein L12 | −0.0040 |
| 229648_at | ARHGAP32 | Rho GTPase activating protein 32 | −0.0040 |
| 228253_at | LOXL3 | Lysyl oxidase‐like 3 | −0.0060 |
| 202669_s_at | EFNB2 | Ephrin‐B2 | −0.0081 |
| 206172_at | IL13RA2 | Interleukin 13 receptor, alpha 2 | −0.0101 |
| 221274_s_at | LMAN2L | Lectin, mannose‐binding 2 like | −0.0141 |
VI, variable importance.
Identification of a PPS associated with survival
The gene expression predictor PPS was computed from a linear combination of the 25 genes and was calculated for each tumor as follows:
The Z 1 score of the expression value for each individual gene was adapted in this formula. The Z 1 scores ranged from −4.91 to 4.28, with high scores associated with poor outcomes. The optimal cut‐off was a Z score of −1.17. As expected, the predictor performed well in terms of patient prognosis; the improved prognosis group (Z ≤ −1.17) had a median survival time of 721 days, while the poor prognosis group (Z > −1.17) had a significantly lower median survival time of 335 days (P < 0.0001; Fig. 1a).
Figure 1.
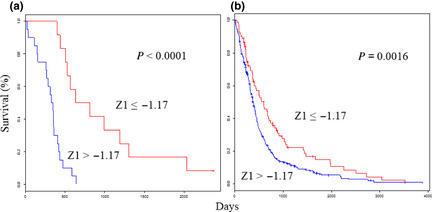
Survival analyses using the selected 25‐gene classifiers show the prognostic value for glioblastoma. Kaplan–Meier curves that compare groups classified using the Z 1 prognosis prediction score with the 25‐gene model in the test (a) and validation (b) sets.
Identification of a PPS with a three‐gene set associated with survival
For more practical purposes, the gene expression predictor PPS was computed from a linear combination of three genes and was calculated for each tumor as follows:
The Z 2 score of the expression value for each individual gene was adapted in this formula. The Z 2 scores ranged from −2.53 to 2.27, with high scores associated with poor outcomes. The optimal cut‐off was a Z score of −0.76. As expected, the predictor performed well in terms of patient prognosis; the improved prognosis group (Z ≤ −0.76) had a median survival time of 721 days, while the poor prognosis group (Z > −0.76) had a significantly lower median survival time of 335 days (P < 0.0001; Fig. 2a). Classification using cell‐of‐origin is associated with survival. We classified our cases into proneural, neural, classical and mesenchymal subtypes using a gene expression‐based method according to Verhaak et al.13 (Fig. S3A). These four groups differed significantly in survival rates (P = 0.0093; Fig. S3B) and classification by cell‐of‐origin was found to be significantly associated with patient survival.
Figure 2.
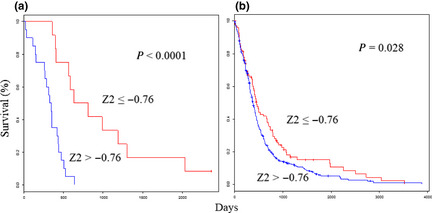
Survival analyses using the selected three‐gene classifiers show the prognostic value for glioblastoma. Kaplan–Meier curves that compare groups classified using the Z 2 prognosis prediction score with the three‐gene model in the test (a) and validation (b) sets.
The gene expression predictor is the most significant feature
The Z PPS results were compared with traditional individual indicators. As shown in Table 3, Z 1, Z 2, age, KPS and subtype were significantly associated with OS in univariate analyses. Table 4 shows the results of the multivariate analyses, which found that the gene expression predictor Z 1 was significantly associated with OS. The PPS was the most significant feature of these clinical parameters.
Table 3.
Prognostic value of clinical factors stratified by overall survival (OS) in patients with grade 4 glioma
| Variable | n | Median OS (days) | P |
|---|---|---|---|
| Age (years) | |||
| ≥60 | 17 | 352 | <0.05 |
| <60 | 15 | 525 | |
| Gender | |||
| Male | 20 | 434 | 0.48 |
| Female | 12 | 337 | |
| KPS | |||
| ≥70 | 25 | 474 | <0.01 |
| <70 | 7 | 268 | |
| IDH1 | |||
| Wild | 31 | 405 | 0.05 |
| Mutated | 1 | N.D. | |
| MGMT IHC | |||
| Positive | 21 | 352 | 0.09 |
| Negative | 11 | 630 | |
| MGMT mRNA | |||
| >0.017 | 16 | 407 | 0.42 |
| ≤0.017 | 16 | 419 | |
| Subtype | |||
| CL | 7 | 432 | 0.009 |
| MES | 11 | 308 | |
| NL | 7 | 988 | |
| PN | 7 | 417 | |
| Z 1 Score | |||
| >−1.17 | 20 | 335 | <0.0001 |
| ≤−1.17 | 12 | 721 | |
| Z 2 Score | |||
| >−0.76 | 20 | 335 | <0.0001 |
| ≤−0.76 | 12 | 721 | |
CL, classical; IDH1, isocitrate dehydrogenase 1; IHC, immunohistochemistry; KPS, Karnofsky performance status; MES, mesenchymal; MGMT, O6‐methylguanine‐methyltransferase; ND, not determined; NL, neural; PN, proneural.
Table 4.
Multivariate analysis: prognosis prediction score and clinical and therapeutic variables associated with overall survival in patients with grade 4 glioma
| Variable | Subgroup | Entire series (n = 32) | ||
|---|---|---|---|---|
| Hazard ratio | 95% CI | P | ||
| Z 1 | Continuous variable | 1.34 | 1.03–1.77 | 0.026 |
| Z 2 | Continuous variable | 1.48 | 0.95–2.38 | 0.081 |
| MGMT IHC | Positive/Negative | 1.72 | 0.70–4.30 | 0.228 |
| Age (years) | ≥60, <60 | 2.22 | 0.95–5.37 | 0.065 |
| KPS | ≥70, <70 | 2.76 | 0.88–8.50 | 0.078 |
CI, confidence interval; IHC, immunohistochemistry; MGMT, anti‐O6‐methylguanine‐methyltransferase.
The PPS formula was validated in the independent sample set
The PPS formula was validated in the validation set (n = 488; Table 1), which is derived from glioblastoma patients in four external data sets.8, 10, 12, 22 The Z 1 scores ranged from −5.43 to 5.33. As expected, the OS was significantly higher in the improved prognosis group (Z ≤ −1.17) than in the poor prognosis group (Z > −1.17; P = 0.0016; Fig. 1b). The Z 2 scores ranged from −3.98 to 2.66. As expected, the OS was significantly higher in the improved prognosis group (Z ≤ −0.76) than in the poor prognosis group (Z > −0.76; P = 0.028; Fig. 2b).
Survival analyses using the PPS with a three‐gene set and age classifiers shows a prognostic value for patients with grade 4 glioma
Even among Grade 4 gliomas in both test (n = 32) and validation sets (n = 488), the OS ranged between 0 and 3880 days. Fifty‐two patients (10%) survived for longer than 1000 days. As predicted by the survival tree, the OS differed significantly between the improved prognosis group (−0.76 ≥ Z 2 or −0.76 < Z 2 with age <57 years) and the poor prognosis group (−0.76 < Z 2 with age ≥57 years) in the test and validation set (P = 0.0006 and P < 0.0001, respectively; Fig. 3). The median OS using test and validation data sets was 641 and 490 days, respectively, for the improved prognosis group and 347 and 302 days, respectively, for the poor prognosis group. The two‐year survival rates were 36.3% and 30.8% in the improved prognosis group and 4.7% and 11.8% in the poor prognosis group, using the test and validation data sets, respectively.
Figure 3.

Survival analyses using the Z 2 prognosis prediction score (PPS) and age classifiers reveal a prognostic value for glioblastoma. Kaplan–Meier curves compare groups classified using the Z 2 PPS and age in the test (a) and validation (b) sets.
Discussion
We assessed relationships between gene expression and survival time using a random survival forests model. This is classified into a tree‐based method, which aids the detection of interactions. As discussed by Cordell, the functional form should contain gene‐by‐gene interaction terms.24 The model was developed for use with datasets in which several variables (genes, in the present case) greatly outnumber patients; a framework of random forests is needed for such an analysis. Genes were selected using the RSF‐VH algorithm, which eliminates the need to screen the genes.20
Many studies of microarray data use univariate analyses for screening in which potential genes that interact with other genes may be dropped from the analyses. However, the RSF‐VH algorithm is more appropriate for our application and we previously reported its usefulness20 in the identification of a gene‐expression signature that predicts outcomes in patients with malignant glioma and primary central nervous system lymphoma.14, 25
Although our predictor was mainly based on cases from first‐line nitrosourea‐based chemotherapy, results from the combined four external data sets,8, 10, 12, 22 in which first‐line temozolomide‐based chemotherapy was used, support the universal performance of the predictor, irrespective of the chemotherapeutic regimen. Survival benefit by chemotherapy is relatively small in most grade 4 gliomas, so it is important to elucidate the differences in the intrinsic biological characteristics of the tumors. Genetic differences within malignant gliomas also underscore the heterogeneity of these tumor types. Compared with our previous report of oligodendrocytic tumor patients,14 three (GPNMB, LOXL3 and IL13RA2) out of 25 genes are identical to the present study.
The value of gene expression‐based predictors in estimating the prognosis of malignant glioma patients will not be fully realized until more efficacious therapies are available for those in whom current treatment is less successful. In this regard, although the biological investigation of these genes is important, expression profiles might predict long‐term survival as well as yielding clues about individual genes involved in tumor development, progression and response to therapy. Moreover, the ability to distinguish between histologically ambiguous gliomas will enable appropriate therapies to be tailored to specific tumor subtypes. Class prediction models based on defined molecular profiles allow the classification of malignant gliomas in a manner that will better correlate with clinical outcomes than with standard pathology. Glioblastomas have wide‐ranging survival times, which require a more precise prognostic scoring system to study novel therapeutic approaches. Therefore, the identification of molecular subclasses could greatly facilitate prognosis and our ability to develop effective treatment protocols. As our PPS involves a small number of genes, quantitative reverse transcriptase PCR assays or customized DNA microarrays could be developed for clinical applications. Molecular targeted therapies that specifically target disabled pathways might then be tailored for those patients with poor prognoses.
In summary, we identified gene signatures associated with outcome in patients with glioblastoma. Adaptation of subsets of these genes for use in clinical assays could result in improved outcome prediction. We have extended our observations to validate these signatures using independent data sets from other institutions. Our profiling results should help construct a new classification scheme that better assesses clinical malignancies compared with the conventional histological classification system.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Marginal plots for the six genes.
Fig. S2. Heat map for selected genes.
Fig. S3. Classification by cell‐of‐origin associated with survival.
Table S1. Quantitative PCR validation on survival‐related genes.
Acknowledgments
This work was supported in part by JSPS KAKENHI grant number 21700312 to A.K. and 17390394 to R.Y.
(Cancer Sci, doi: 10.1111/cas.12214, 2013)
References
- 1. Stewart LA. Chemotherapy in adult high‐grade glioma: a systematic review and meta‐analysis of individual patient data from 12 randomised trials. Lancet 2002; 359: 1011–8. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Hegi ME, Mason WP et al Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol 2009; 10: 459–66. [DOI] [PubMed] [Google Scholar]
- 3. Kleihues P, Louis DN, Wiestler OD et al WHO grading of tumours of the central nervous system In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. World Health Organization Classification of Tumours of the Nervous System. Lyon, France: IARC Press, 2007; 10–11. [Google Scholar]
- 4. van de Vijver MJ, He YD, van't Veer LJ et al A gene‐expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347: 1999–2009. [DOI] [PubMed] [Google Scholar]
- 5. Godard S, Getz G, Delorenzi M et al Classification of human astrocyticgliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res 2003; 63: 6613–25. [PubMed] [Google Scholar]
- 6. Nutt CL, Mani DR, Betensky RA et al Gene expression‐based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 2003; 63: 1602–7. [PubMed] [Google Scholar]
- 7. Sorlie T, Tibshirani R, Parker J et al Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 2003; 100: 8418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Freije WA, Castro‐Vargas FE, Fang Z et al Gene expression profiling of gliomas strongly predicts survival. Cancer Res 2004; 64: 6503–10. [DOI] [PubMed] [Google Scholar]
- 9. Rich JN, Hans C, Jones B et al Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res 2005; 65: 4051–8. [DOI] [PubMed] [Google Scholar]
- 10. Phillips HS, Kharbanda S, Chen R et al Molecular subclasses of high‐grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006; 9: 157–73. [DOI] [PubMed] [Google Scholar]
- 11. Yamanaka R, Arao T, Yajima N et al Identification of expressed genes characterizing long‐term survival in malignant glioma patients. Oncogene 2006; 25: 5994–6002. [DOI] [PubMed] [Google Scholar]
- 12. Petalidis LP, Oulas A, Backlund M et al Improved grading and survival prediction of human astrocytic brain tumors by artificial neural network analysis of gene expression microarray data. Mol Cancer Ther 2008; 7: 1013–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verhaak RGW, Hoadley KA, Purdom E et al Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kawaguchi A, Yajima N, Komohara Y et al Identification and validation of a gene expression signature that predicts outcome in malignant glioma patients. Int J Oncol 2012; 40: 721–30. [DOI] [PubMed] [Google Scholar]
- 15. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- 16. Nakasu S, Fukami T, Baba K, Matsuda M. Immunohistochemical study for O6‐methylguanine‐DNA methyltransferase in the non‐neoplastic and neoplastic components of gliomas. J Neurooncol 2004; 70: 333–40. [DOI] [PubMed] [Google Scholar]
- 17. Balss J, Meyer J, Mueller W et al Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 2008; 116: 597–602. [DOI] [PubMed] [Google Scholar]
- 18. R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2011. [Cited 10 Dec 2012.] Available from URL: http://www.R-project.org. [Google Scholar]
- 19. Gentleman R, Carey V, Bates D et al Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishwaran H, Kogalur UB, Gorodeski EZ et al High‐dimensional variable selection for survival data. J Amer Stat Assoc 2010; 105: 205–17. [Google Scholar]
- 21. Youden WJ. Index for rating diagnostic tests. Cancer 1950; 3: 32–5. [DOI] [PubMed] [Google Scholar]
- 22. Cancer Genome Atlas Research Network . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. LeBlanc M, Crowley J. Relative risk trees for censored survival data. Biometrics 1992; 48: 411–25. [PubMed] [Google Scholar]
- 24. Cordell HJ. Detecting gene–gene interactions that underlie human diseases. Nat Rev Genet 2009; 10: 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kawaguchi A, Iwadate Y, Komohara Y et al Gene expression signature‐based prognostic risk score in patients with primary central nervous system lymphoma. Clin Cancer Res 2012; 18: 5672–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Marginal plots for the six genes.
Fig. S2. Heat map for selected genes.
Fig. S3. Classification by cell‐of‐origin associated with survival.
Table S1. Quantitative PCR validation on survival‐related genes.