

Abstract
The novel aroylthiourea analogue of podophyllotoxin HY‐1 (4β‐[benzoyl‐thioureido]‐4‐deoxypodophyllotoxin) was synthesized in our laboratory with the aim of developing multitargeted DNA topoisomerase II inhibitors. The compound showed significant antiproliferative effects on seven cancer cell lines and induced G2/M phase arrest in HCT116 cells. Moreover, HY‐1 showed a potent inhibitory effect on topoisomerase II‐mediated kinetoplast DNA decatenation in a dose‐dependent manner. Our results showed that cdc2 phosphorylation and decreased cdc2 kinase acitivity through the ATR‐Chk1‐Cdc25C and Weel pathways were the central mechanisms for G2/M phase arrest in human colon cancer cells.
Podophyllotoxin (Fig. 1) is a well‐known, naturally occuring antitumor lignan lactone isolated from the genus Podophyllum.1 Several podophyllotoxin derivatives such as etoposide (VP‐16) and teniposide are in clinical use as antineoplastic agents due to their ability to inhibit the enzyme DNA topoisomerase II (TopoII).2 In spite of their impressive clinical efficacy, their therapeutic use is often hindered by poor water solubility, acquired drug resistance, and metabolic inactivation. Multitarget drugs against selective multiple targets improve therapeutic efficacy, safety, and resistance profiles by collective regulation of a primary therapeutic target together with compensatory elements and resistance activities. To develop novel multitargeted DNA TopoII inhibitors, we have designed and synthesized two series of aroylthiourea analogues by coupling a kinase inhibitory group, aroylthiourea, with podophyllotoxin. Among these obtained analogues, 4β‐(benzoyl‐thioureido)‐4‐deoxypodophyllotoxin (HY‐1) possessed potent inhibitory effects on cancer cell proliferation and ability to cell cycle arrest.3
Figure 1.
Podophyllotoxin and 4β‐(benzoyl‐thioureido)‐4‐deoxypodophyllotoxin (HY‐1).
The cell cycle is an intricate sequence of events which enables cells to grow and replicate. The G2/M transition is largely dependent on cyclin B1/cdc2 (Cdk1) activity.4 The activity of cyclin B1/cdc2 is regulated by one positive regulator, cdc25c, and two negative regulators, Weel and Myt1. Cdc25c can dephosphorylate the protein kinase cdc2 at both the Thr14 and Tyr15 residues and activate it. Cdc25c was phosphorylated by Chk1 and Chk2 at Ser‐216. The ataxia telangiectasia mutated (ATM)/ATM Rad3‐related (ATR) signaling pathway involved in checkpoint kinases downstream of ATM/ATR is Chk2 and Chk1.5, 6
The present study is designed to explore in vitro antitumoral acitivities and the underlying mechanism of HY‐1 on G2/M cell cycle arrest. Our results show that ATR‐Chk1‐cdc25 and Weel signal pathways are the central mechanisms in HY‐1‐induced G2/M cell cycle arrest in human colon cancer cell.
Materials and Methods
Reagents and antibodies
HY‐1 was synthesized in our laboratory and dissolved in DMSO (vehicle control) (Beyotime, Shanghai, China) as a stock solution at 10 μM concentration; the final DMSO concentration did not exceed 0.1% for in vitro experiments. Camptothecin and etoposide (VP‐16) were purchased from Sigma‐Aldrich (St. Louis, MO, USA) and dissolved in DMSO. DNA topoisomerase I and plasmid pBR322 were purchased from TaKaRa Biotechnology (Dalian, China). Purified human DNA TopoII and kinetoplast DNA (kDNA) were purchased from Topogen (Port Orange, FL, USA). Horseradish peroxidase‐conjugated secondary antibodies and antibodies to Cdc2, P‐cdc2, Chk1, and Chk2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Weel and Cdc25c were purchased from Cell Signaling Technology (Beverly, MA, USA). The BrdU ELISA kit and annexin V–FLUOS/propidium iodide (PI) staining kit were purchased from Roche (Roche Applied Science, Penzberg, Germany). Hoechst 33342, MTT, and ethidium bromide were purchased from Sigma‐Aldrich. All of the chemical reagents were analytically pure.
Cell lines and cell cultures
Human oral epidermoid carcinoma (KB), human hepatocellular carcinoma (HepG2), human non‐small‐cell lung tumor (A549), human colon carcinoma (HCT116), murine prostate carcinoma (RM), human neuroblastoma (SH‐SY5Y), and human breast carcinoma (4T) cell lines were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured on DMEM medium containing 10% FBS plus penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37°C in a humidified atmosphere with 5% CO2.
Topoisomerase activity measurement
Topoisomerase I (TopoI) activity was assessed using TopoI‐mediated supercoiled pBR322 relaxation assay and TopoII activity was measured by the decatenation of kDNA as described.7 The TopoI assay kit (TaKaRa Biotechnology) and TopoII assay kit (Topogen) were used according to the operating manual. The reaction system contained DNA, topoisomerases, reaction buffer, and different concentrations of compounds. Briefly, reactions occurred for 30 min at 37°C, and were terminated with stop buffer. Reaction products were analyzed on a 1% agarose gel containing 0.5 μg/mL ethidium bromide. The results were analyzed by a gel image analysis system.
Cell proliferation and viability assay
Cell proliferation and viability studies were carried out using the MTT assay. Briefly, 1 × 103/well cells were cultured in 96‐well plates. After cell attachment, different concentrations of HY‐1 and VP‐16 in medium were added in 96‐well plates for 72 h at 37°C. The MTT solution (5 mg/mL in PBS) was added (20 μL/well) for an additional 4 h at 37°C. After the purple formazan crystals were dissolved in 100 μL SDS/DMF (dimethylformamide), the plates were read at 570 nm with a microplate reader (Synergy 2; Biotek, Winooski, VT, USA). Assays were carried out in triplicate in three independent experiments. The IC50 values were calculated using SigmaPlot analysis software (Systat Software Inc., Chicago, IL, USA).
Cell proliferation assay by BrdU incorporation
Cell proliferation was measured using a BrdU ELISA kit. HCT116 cells (1 × 103/well) were seeded in 96‐well plates. After attachment, cells were placed in medium with different concentrations of HY‐1 and VP‐16 for 68 h. Then BrdU labeling solution was added at 10 μL per well; after 4 h, cells were dried and fixed. Anti‐BrdU‐POD working solution and substrate solution were added according to the manufacturer's instructions. Finally, the absorbance of the samples was measured by microplate reader at 370 and 492 nm. The inhibition rate of cell growth was calculated by the following formula: Inhibition % = (1−ODt/ODc) × 100%, where ODt and ODc indicate the mean value of the treated group and control group, respectively. Assays were carried out in triplicate in three independent experiments. Quintuplicate wells were analyzed at each concentration and the experiment was repeated in triplicate.
Cell cycle analysis
The cell cycle was analyzed using PI staining for flow cytometry. After treatment with HY‐1 and VP‐16 for 24 h, cells were collected and washed with PBS three times, then fixed with 70% ethanol overnight at 4°C, washed three more times with PBS, and resuspended. RNase was added and incubated for 30 min to eliminate the interference of RNA. Cells were treated with PI for a further 30 min and washed three times with PBS. The DNA contents were detected by FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA) flow cytometer at 488–630 nm. The relative DNA content was analyzed by CellQuest software (Becton Dickinson) based on the presence of a red fluorescence.
Nuclear staining assay
Cells (1 × 105/well) were cultured in 6‐well plates and incubated for 24 h before treatment with different concentrations of HY‐1 for 72 h. After removing the medium, cells were washed with PBS three times. Hoechst solution (diluted in PBS) was added for 10 min in the dark. Cell nuclei were observed under fluorescence microscope with 200 × magnification.
Apoptosis assay
Apoptosis of HCT116 cells was detected by flow cytometry with an annexin V–FLUOS/PI staining kit. After treatment with HY‐1 and VP‐16 for 72 h, cells were collected, washed with PBS three times, then resuspended in 100 μL binding buffer plus annexin V–FITC and PI. After 15 min of incubation in the dark, fluorescence was analyzed by a FACSCalibur flow cytometer. The DNA content of the nuclei was registered on a logarithmic scale.
Western blot analysis
Cytosolic extracts were prepared by suspension in ice‐cold lysis buffer (50 mM Tris, pH 7.4, 0.8 M NaCl, 5 mM MgCl2, 0.5% Nonidet P‐40, 1 mM PMSF, and 20 μg/mL each proteinase inhibitor aprotinin, leupeptin, and pepstatin), and incubated on ice for 30 min. After centrifugation at 10 000 g for 20 min at 4°C, the protein in the supernatant was quantitated using a protein assay kit from Bio‐Rad Laboratories (Hercules, CA, USA). Sixty micrograms of protein was electrophoresed on 10% SDS‐polyacrylamide gels, then transferred to PVDF membranes. The membranes were blocked for 2 h at room temperature in TBS plus 0.5% Tween 20 containing 5% fat‐free milk, then incubated with primary antibodies overnight at 4°C. After washing, the membranes were incubated with anti‐mouse or anti‐rabbit secondary antibodies for 2 h. Then the membranes were washed, detected by the ECL method and visualized on KodakBio‐MAX MR film (Rochester, NY, USA).
Results
Effect of HY‐1 on TopoI and TopoII activity
Topoisomerase I‐mediated supercoiled pBR322 relaxation assay and TopoII‐mediated kDNA decatenation were used to determine the inhibitory effects of HY‐1 on the catalysis acitivity of TopoI and TopoII. As shown in Figure 2(a), HY‐1 had no inhibitory effect on the relaxation activity of TopoI towards supercoiled PBR322 at 100 μM. The TopoI inhibitor, camptothecin, effectively inhibited the catalytic activity of TopoI at 10 μM. However, HY‐1 showed a potent inhibitory effect on TopoII‐mediated kDNA decatenation in a dose‐dependent manner (Fig. 2b). HY‐1 completely inhibited the catalytic activity of TopoII catalytic activity at 100 μM, equating to the effect of VP‐16 at 500 μM. These results indicated HY‐1 might have the same mechanism of action as VP‐16.
Figure 2.
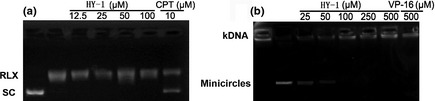
(a) Effects of 4β‐(benzoyl‐thioureido)‐4‐deoxypodophyllotoxin (HY‐1) and camptothecin (CPT) on human topoisomerase I‐mediated decatenation activity of pBR322. RLX, relaxed pBR322; SC, supercoiled pBR322. (b) Effects of HY‐1 and etoposide (VP‐16) on human topoisomerase II‐mediated decatenation activity of kinetoplast DNA (kDNA). Minicircles indicate no drug. DNA samples were separated by electrophoresis on agarose gels containing ethidium bromide (1 mg/mL). Gels were photographed under UV light.
Effects of HY‐1 on cell proliferation and viability
Seven cancer cell lines from various solid tumors were used to determine the effects of HY‐1 on cell proliferation and viability, using an MTT assay. The average inhibitory concentrations of 50% are shown in Figure 3(a). The results indicated that HY‐1 displayed a potent inhibitory effect on cell proliferation in a dose‐dependent manner and showed distinct specificity. HY‐1 showed a strong inhibitory effect on HCT116, A549, KB, and HepG2 cell lines with IC50 values from 0.1 to 0.3 μM. The average IC50 values for HY‐1 and VP‐16 against these cell lines were 1.2 and 15.6 μM, respectively.
Figure 3.
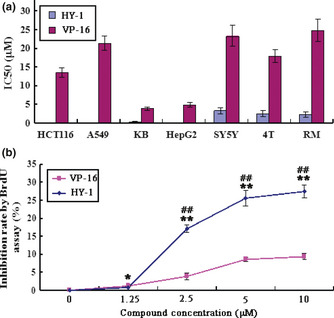
(a) Antiproliferation effects of 4β‐(benzoyl‐thioureido)‐4‐deoxypodophyllotoxin (HY‐1) and etoposide (VP‐16) on different human cancer cells by MTT assay. Cells were treated with different concentrations of compounds for 72 h. The IC 50 values were calculated using the mean results of three separate experiments, expressed as the mean ± SD (μM). (b) Antiproliferation effects of HY‐1 and VP‐16 on human colon carcinoma HCT116 cells by BrdU ELISA. All data represent the mean ± SD from three independent experiments. *P < 0.05 versus blank control; **P < 0.01 versus blank control; #P < 0.05 versus positive control; ##P < 0.01 versus positive control (n = 5).
Effects of HY‐1 on proliferation in HCT116 cells
A BrdU ELISA was used to determine the newly synthesized DNA to indicate the effect of compounds on HCT116 cell proliferation. The results showed that there was a significant decrease in BrdU incorporation in a dose‐dependent manner following 48 h of incubation with HY‐1 (Fig. 3b). After treatment with 10 μM HY‐1 or VP‐16, the percentage of decrease in BrdU incorporation was approximately 30% and 10%, respectively. The effect of HY‐1 to inhibit BrdU incorporation in different concentrations was better than that of VP‐16. On the basis of the above results, further studies were focused on the HCT116 cell line.
Cell cycle analysis
We examined the effects of HY‐1 on the cell cycle by flow cytometry in HCT116 cells. Interestingly, a concentration‐dependent change was observed in the cell cycle pattern (Fig. 4a). Treatment of HCT116 cells for 24 h with HY‐1 at 0, 1.25, 2.5, 5, and 10 μM significantly increased the percentage of cells at G2/M phase from 35.68% to 94.23%. HY‐1 arrested the cell cycle at a concentration of 1.25 μM, with 93.77% of cells at G2/M phase. The flow cytometric data clearly indicated that HY‐1 caused arrest at G2/M phase, following the same pattern as VP‐16.
Figure 4.
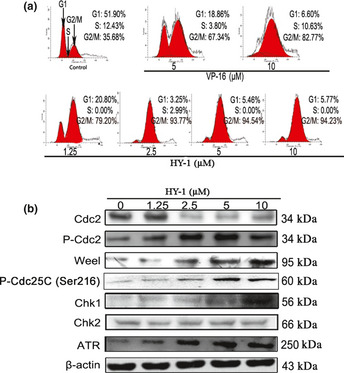
(a) Effects of compound 4β‐(benzoyl‐thioureido)‐4‐deoxypodophyllotoxin (HY‐1) on the cell cycle of human colon carcinoma HCT‐116 cells. Cells were incubated with 0, 1.25, 2.5, 5, or 10 μM HY‐1 for 24 h and stained with propidium iodide (PI). Their DNA content was analyzed by fluorescence flow cytometry. (b) Effect of HY‐1 on G2/M cell cycle regulator proteins. HCT116 cells treated with 0, 1.25, 2.5, 5, or 10 μM HY‐1 for 24 h. The protein levels of Cdc2, p‐Cdc2, Weel, P‐Cdc25C, Chk1, Chk2, and ATR were assessed by Western blotting.
Effect of HY‐1 on expression of G2/M cell cycle regulatory proteins
To elucidate the mechanism for G2/M arrest in HY‐1 treated cells, we determined its effect on expression of cell cycle regulatory proteins that are critical to G2/M transition, including ATR, Chk1, Chk2, cdc2, P‐cdc2, and P‐cdc25c. Western blot results indicated that the expression levels of cdc2 were decreased in a dose‐dependent manner, whereas the expression levels of P‐cdc2, P‐cdc25c, Weel, Chk1, and ATR were increased (Fig. 4b). These results suggested the predominant changes in the expression of G2/M regulatory proteins were induced through the ATR‐Chk1‐Cdc25C and Weel pathways, which finally resulted in G2/M phase arrest in HCT116 cells.
Effect of HY‐1 on cell apoptosis
The morphological changes in HCT116 cells after treatment by different concentrations of HY‐1 were measured by fluorescence staining. Cells were stained with Hoechst 33 342 and visualized using a fluorescent microscope. After treatment with HY‐1, the number of cells was significantly reduced, and most cells showed characteristics typical of apoptotic cells, like plasma membrane blebbing (Fig. 5a).
Figure 5.
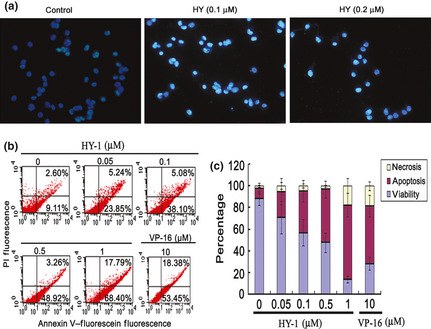
(a) Morphological study of human colon carcinoma HCT‐116 cells treated with 4β‐(benzoyl‐thioureido)‐4‐deoxypodophyllotoxin (HY‐1) for 72 h, assessed by Hoechst 33342 staining assay. Cells were treated with 0, 0.1, or 0.2 μM HY‐1 for 72 h, then visualized using a fluorescence microscope. The cells became rounded and contained fragmented nuclei, typical morphological features of apoptosis. (b) HY‐1‐induced HCT116 cell apoptosis was detected by annexin V/propidium iodide (PI). Cells were treated with various concentrations of HY‐1 for 72 h, then incubated with annexin V and PI dyes. Apoptotic cells were analyzed by flow cytometry. (c) Determination of the rates of apoptosis induced by HY‐1. All data represent the mean ± SD from three independent experiments.
Annexin V/PI staining assay showed that HY‐1 induced apoptosis of HCT116 cells in a dose‐dependent manner. VP‐16 caused 53.45% cells into apoptosis at 10 μM, whereas HY‐1 induced 68.40% cells into apoptosis at 1 μM (Fig. 5b,c). Taken together, these results show that HY‐1 induced cell death primarily through apoptosis.
Discussion
Podophyllotoxin derivatives have important therapeutic value in the treatment of human cancers.8 The synthesis and preliminary screen of aroylthiourea derivatives of podophyllotoxin bearing different benzyl moieties have already been described in published reports.3 Aroylthioureas are known as good ligands for metal ions, and these metal ions play important roles in tumorigenesis. Moreover, aroylthioureas possess good inhibitory activity against protein tyrosine kinases. We found that coupling aroylthiourea with podophyllotoxin could significantly improve its antiproliferative acitivity.
The results of the TopoII‐mediated kDNA decatenation assay proved that HY‐1 could completely inhibited the catalysis activity of TopoII at 100 μM, which indicated HY‐1 was a potent TopoII inhibitor with the same mechanism of action as VP‐16.
Treated with TopoII inhibitors, cells may undergo an intricate network of multiple pathways, such as DNA damage, cell cycle arrest, and apoptosis. We found that HY‐1 caused arrest at G2/M phase in a dose‐dependent manner, but the mechanisms were not investigated in depth.
Progression of the cell cycle is tightly controlled through the actions of various classes of cyclins and cyclin‐dependent protein kinases.9 The G2/M transition is controlled, at least in part, by cdc2‐cyclin B1 activity through two mechanisms.10 On one hand, ATM/ATR are two of the most important kinases that respond to DNA double‐strand breaks induced by TopoII inhibitors and are involved in subsequent cell cycle arrest. On the other hand, Weel and Myt1 could phosphorylate cdc2 on Thr‐14 and Tyr‐15, thereby inactivating it.11, 12, 13 Our results confirmed that the mechanism of action for HY‐1 in HCT116 cells involved these two processes. First, by investigating the expression level of cdc2, we found it was decreased in HY‐1 treated cells, and a significant increase in phosphorylation of cdc2 was observed at the same time. The phosphorylation of cdc2 is carried out primarily by Weel. The present study found that the expression level of Weel was increased in HY‐1‐treated HCT116 cells. Second, increases in the expressions levels of Chk1, ATR, and P‐cdc25c were detected (Fig. 4b). These results strongly suggested the predominant changes in the expression of G2/M regulatory proteins were induced through the ATR‐Chk1‐Cdc25C and Weel pathways, which finally resulted in phosphorylation of cdc2 and a decrease of cdc2 kinase activity (Fig. 6).
Figure 6.
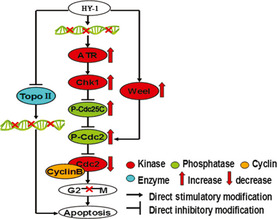
Schematic explanation of the contribution of cell cycle regulatory proteins to the development of G2/M phase arrest in human colon carcinoma HCT116 cells treated with 4β‐(benzoyl‐thioureido)‐4‐deoxypodophyllotoxin (HY‐1).
In summary, we have shown that HY‐1 is able to inhibit cell proliferation, target TopoII, induce apoptosis, and arrest the cell cycle at G2/M phase in HCT116 cells. The G2/M phase arrest is associated with inhibited acitivity of cdc2 kinase and increase in cdc2 phosphorylation. Taken together, our data support the superiority of HY‐1 to the parent compound VP‐16, indicating that HY‐1 is a potential anticancer drug candidate that should be further developed.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
The studies in the laboratories were supported by the natural science foundation of Jiangsu Province (BK2011390), Natural Science Foundation of Nantong City (K2010024), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), National Natural Science Foundation of China (Grant No. 30971197 & 31171143), 973 Program (2011CB510004).
(Cancer Sci 2013; 104: 1062–1066)
References
- 1. Schrecker AW, Hartwell JL. Communications‐components of podophyllin. XX. The absolute configuration of podophyllotoxin and related lignans. J Org Chem 1956; 21: 381–2. [Google Scholar]
- 2. Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer 1998; 34: 1514–21. [DOI] [PubMed] [Google Scholar]
- 3. Zhao Y, Wang CN, Wu ZH, Fang JH, Zhu L. Synthesis and antitumor activity of novel aroylthiourea derivatives of podophyllotoxin. Invest New Drugs 2012; 30: 17–24. [DOI] [PubMed] [Google Scholar]
- 4. Choi HJ, Fukui M, Zhu BT. Role of cyclin B1/Cdc2 up‐regulation in the development of mitotic prometaphase arrest in human breast cancer cells treated with nocodazole. PLoS ONE 2011; 6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dai XJ, Yamasaki KS, Yang LJ et al Keratinocyte G2/M growth arrest by 1,25‐dihydroxyvitamin D3 Is caused by Cdc2 phosphorylation through Wee1 and Myt1 regulation. J Invest Dermatol 2004; 122: 1356–64. [DOI] [PubMed] [Google Scholar]
- 6. Yu LJ, Orlandi L, Wang P et al UCN‐01 abrogates G2 arrest through a Cdc2‐dependent pathway that is associated with inactivation of the Wee1 Hu kinase and activation of the Cdc25C phosphatase. J Biol Chem 1998; 273: 33455–64. [DOI] [PubMed] [Google Scholar]
- 7. Tanabe K, Ikegami Y, Ishida R, Andoh T. Inhibition of topoisomerase II by antitumor agents bis (2,6‐dioxopiperazine) derivatives. Cancer Res 1991; 51: 4903–7. [PubMed] [Google Scholar]
- 8. Gordaliza M, Garcia PA, Corral JM, Castro MA, Gomez‐Zurita MA. Podophyllotoxin: distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004; 44: 441–59. [DOI] [PubMed] [Google Scholar]
- 9. Corellou F, Brownlee C, Kloareg B, Bouget F. Cell cycle‐dependent control of polarised development by a cyclin‐dependent kinase‐like protein in the Fucus zygote. Development 2001; 128: 4383–92. [DOI] [PubMed] [Google Scholar]
- 10. Moon DO, Kim MO, Nam TJ, Kim SK, Choi YH, Kim GY. Pectenotoxin‐2 induces G2/M phase cell cycle arrest in human breast cancer cells via ATM and Chk1/2‐mediated phosphorylation of cdc25C. Oncol Rep 2010; 24: 271–6. [DOI] [PubMed] [Google Scholar]
- 11. Lu XB, Nguyen TA, Donehower LA. Reversal of the ATM/ATR‐ mediated DNA damage response by the oncogenic phosphatase PPM1D. Cell Cycle 2005; 4: 1060–4. [PubMed] [Google Scholar]
- 12. Tyagi A, Singh RP, Agarwal C, Siriwardana S, Sclafani RA, Agarwal R. Resveratrol causes Cdc2‐tyr15 phosphorylation via ATM/ATR–Chk1/2– Cdc25C pathway as a central mechanism for S phase arrest in human ovarian carcinoma Ovcar‐3 cells. Carcinogenesis 2005; 26: 1978–87. [DOI] [PubMed] [Google Scholar]
- 13. Park I, Avraham HK. Cell cycle‐dependent DNA damage signaling induced by ICRF‐193 involves ATM, ATR, CHK2, and BRCA1, Exp. Cell Res 2006; 312: 1996–2008. [DOI] [PubMed] [Google Scholar]