

Abstract
Omega‐6 (n‐6) arachidonic acid (AA) and its pro‐inflammatory metabolites, including prostaglandin E2 (PGE 2), are known to promote tumorigenesis. Delta‐6 desaturase (D6D) is the rate‐limiting enzyme for converting n‐6 linoleic acid (LA) to AA. Our objective was to determine if AA synthesis, specifically D6D activity, and PGE 2 levels are increased in cancerous breast tissue, and whether these variables differ between estrogen receptor positive (ER+) and negative (ER−) breast cancers. Gas chromatography was performed on surgical breast tissue samples collected from 69 women with breast cancer. Fifty‐four had ER+ breast cancer, and 15 had ER− breast cancer. Liquid chromatography‐mass spectrometry was used to determine PGE 2 levels. Lipid analysis revealed higher levels of LA metabolites (C18:3 n‐6, C20:3 n‐6, and AA) in cancerous tissue than in adjacent noncancerous tissue (P < 0.01). The ratio of LA metabolites to LA, a measure of D6D activity, was increased in cancerous tissue, suggesting greater conversion of LA to AA (P < 0.001), and was higher in ER− than in ER+ patients, indicating genotype‐related trends. Similarly, PGE 2 levels were increased in cancerous tissue, particularly in ER− patients. The results showed that the endogenous AA synthetic pathway, D6D activity, and PGE 2 levels are increased in breast tumors, particularly those of the ER− genotype. These findings suggest that the AA synthetic pathway and the D6D enzyme in particular may be involved in the pathogenesis of breast cancer. The development of drugs and nutritional interventions to alter this pathway may provide new strategies for breast cancer prevention and treatment.
Omega‐6 (n‐6) and omega‐3 (n‐3) polyunsaturated fatty acids (PUFAs) are crucial for human development and normal biological functions, including brain development and inflammatory responses.1, 2, 3, 4 Linoleic acid (LA), an 18‐carbon n‐6 PUFA, and alpha‐linolenic acid (ALA), an 18‐carbon n‐3 PUFA, are essential fatty acids that must be obtained through the diet and cannot be synthesized de novo in mammals. Linoleic acid and ALA share the same metabolic enzymes, and, through a series of desaturation and elongation reactions, are converted into their respective 20‐carbon products: arachidonic acid (AA) and eicosapentaenoic acid (EPA). Arachidonic acid and EPA are then further metabolized into eicosanoids such as prostaglandins (PGs) and leukotrienes (LTs) in additional reactions. The eicosanoids derived from n‐6 and n‐3 PUFAs are functionally distinct and often have opposing physiological effects; for example, AA‐derived eicosanoids are pro‐inflammatory whereas the EPA‐derived eicosanoids are anti‐inflammatory.1, 5, 6, 7 It is well known that AA and its downstream metabolites, especially prostaglandin E2 (PGE2), promote tumorigenesis by supporting processes such as inflammation and angiogenesis.6, 8, 9 The regulatory pathways of the downstream AA metabolites have been extensively studied,10 but little research has addressed the possibility of limiting AA formation as a method for blocking tumorigenesis. Delta‐6‐desaturase (D6D), also known as fatty acid desaturase 2, is the first and rate‐limiting step in the metabolic conversion of LA to AA and ALA to EPA.1, 5, 11, 12, 13, 14 Logically, targeting an enzyme that is upstream of AA in this metabolic pathway should prevent the formation of AA and its pro‐inflammatory eicosanoids. Studies using the highly selective D6D inhibitor SC‐26196 in cancer models have revealed that it prevents the synthesis of AA and that it also has anti‐inflammatory properties that can be reversed with the dietary addition of AA.15, 16, 17
Breast cancer (BC) is one of the most frequently occurring cancers; approximately 1 million new cases are diagnosed each year throughout the world.18, 19, 20, 21, 22 Estrogens are known to influence the pathogenesis of many breast cancers.22, 23, 24, 25, 26 Specifically, it is recognized that alterations in the estrogen signaling pathway combined with various genetic and environmental factors (e.g. diet and exercise) contribute to the development of BC.18, 19, 20, 24, 26, 27 Still, BC is a heterogeneous disease that can be grouped into two main genotypes with distinct treatment options and outcomes: estrogen‐receptor positive (ER+) and estrogen‐receptor negative (ER−) breast cancers. ER+ BC is extremely common (about 70–80% of all diagnoses), often initially detected by mammogram, and typically responds well to endocrine treatment.21, 22, 23, 24 Tamoxifen, a selective estrogen receptor antagonist, or aromatase inhibitors are the backbone of treatment (and prevention) of ER+ BC.20, 21, 22, 23 However, treating ER− patients remains a major challenge because this aggressive pathology does not respond to hormone‐directed drugs and lacks any other targeted treatment options. Current therapy for patients with ER− BC is cytotoxic chemotherapy, but the overall 5‐year survival rate is still lower in ER− than in ER+ patients.18, 19, 27, 28, 29 Thus, there is an urgent need to develop a better understanding of, and more effective treatment options for, ER− BC. Given that AA and its downstream metabolites promote tumorigenesis by supporting processes such as inflammation and angiogenesis, one possibility is that fatty acid metabolism is dysregulated in BC patients and may represent a new target for cancer treatment. However, it remains unknown whether AA production or the D6D pathway are altered in breast cancer patients, or according to BC phenotype.
In this study, we tested the hypothesis that activity of the D6D pathway is augmented in the tumor tissues of BC patients. Using both cancerous and noncancerous breast tissue from each patient, we evaluated samples from 109 women. Sixty‐nine women had enough cancerous and adjacent noncancerous tissue to determine their lipid profiles. We also performed liquid chromatography‐mass spectrometry (LC‐MS) for 16 of these women to measure their levels of PGE2, a pro‐inflammatory, AA‐derived eicosanoid. Additionally, the tumors were classified by genotype: 54 women had ER+ tumors and 15 had ER− tumors. The lipid profiles and LC‐MS data were compared to assess whether D6D activity is higher in women with ER− breast cancer.
Materials and Methods
Reagents
Methanol and chloroform were purchased from Fisher Scientific (Pittsburgh, PA, USA). Boron trifluoride–methanol reagent (14%,), butylated hydroxytoluene, petroleum ether, diethyl ether, ethyl acetate, and acetic acid were purchased from Sigma‐Aldrich (St. Louis, MO, USA). High performance liquid chromatography‐grade pure hexane (>99%) was purchased from Acros Organics (Geel, Belgium). Fatty acid standards were purchased from Nu‐chek Prep (Elysian, MN, USA). High performance liquid chromatography‐grade acetonitrile and HPLC‐grade water were purchased from VWR International (Radnor, PA, USA). The PGE2 standard and internal standard D4‐PGE2 were purchased from Cayman Chemical (Ann Arbor, MI, USA).
Human samples
The Institutional Review Boards at North Shore Medical Center, Salem, Massachusetts and Partners Health Care, Boston, approved the use of human subjects. Beginning in February of 2009, women diagnosed with invasive breast cancers who had tumor sizes larger than 1‐cm were approached about participating in the study. The women were asked in the order in which they were seen at the clinic. Each participant consented to have 200 mg of cancerous tissue and 200 mg of adjacent noncancerous tissue removed and frozen at the time of surgery. If the pathologist felt that the entire specimen was needed for histological analysis, no tissue was collected; thus, of the 109 patients who consented to participation (mean age, 64 years; mean BMI, 28.6), 69 pairs of cancerous and noncancerous tissue samples were obtained. Frozen tissues were stored transiently in dry ice until it was determined that the tissue was not needed for margin analysis or other clinical decision making, after which the samples were transferred to the lab and stored at −80°C. Demographic data gathered from the medical records were age at diagnosis, BMI, and characteristics of the tumor including ER status, PR and H2N status, and grade and stage of the tumor.
Fatty acid analysis
Phospholipid extraction and consequent methylation were performed as previously described.30 Briefly, approximately 50 mg of breast tissue sample was crushed in liquid nitrogen to produce a powder. A volume of 5 mL of extraction reagent (2:1 chloroform/methanol solution, 0.005% butylated hydroxytoluene as an antioxidant) was added to the powder, and the contents were sealed under nitrogen. After a 30‐min incubation at room temperature, 1 mL of 0.9% NaCl was added, and the sample was vortexed and then centrifuged at 1800g for 5 min. The bottom chloroform phase (containing the lipids) was extracted, dried under nitrogen, and re‐dissolved in 50 μL of extraction reagent. Thin layer chromatography (TLC) was performed to isolate the phospholipid fraction under the following conditions: the silica‐gel‐coated plate was activated at 80°C for 1 h, and the development buffer was an 80:20:1 mixture of petroleum ether: diethyl ether: acetic acid. After development (30–45 min), the TLC plate was sprayed with anilino naphthalene sulfonic acid (ANSA) and visualized under UV light. The phospholipid layer (where the samples were originally spotted onto the plate) was removed and subjected to methylation.
As previously described, 1.5 mL of hexane and 1.5 mL of 14% boron trifluoride–methanol reagent were added to the TLC plate scrapings, the contents were sealed under nitrogen, and the sample was heated at 100°C for 1 h (with vortexing every 15 min).30 Following the heating procedure, samples were cooled at room temperature for 15 min. Next, 1 mL of double‐distilled water was added, and the samples were vortexed and then centrifuged at 1800g for 10 min. The top hexane layer, which contained the fatty acid methyl esters, was removed and dried under nitrogen. Samples were reconstituted in 40 μL of hexane.
Fatty acid profiles were determined using gas chromatography (GC). The fatty acid methyl esters were injected onto a fully automated Agilent 6890N Network GC system with a 7683 Series Injector and a flame ionization detector (Agilent Technologies, Santa Clara, CA, USA). An Omegawax 250 capillary column (30.0 m × 250 μm × 0.25 μm nominal; Sigma‐Aldrich) was used. Samples were injected at a volume of 2.0 μL and a split ratio of 15:1. The total run time was 57 min and took place under the following conditions: an initial oven temperature of 130°C, hold for 3 min, ramp at 5°C/min up to 180°C, ramp at 2.5°C/min up to 200°C, hold for 15 min, ramp at 1°C/min up to 210°C, hold for 5 min, and ramp at 5°C/min up to 240°C. Peaks of the resolved fatty acids were identified by comparing retention time with reference standards (GLC‐461; Nu‐chek‐Prep). The relative percent areas for all resolved peaks were analyzed with a Perkin‐Elmer M1 integrator (Boston, MA, USA).
Measurement of PGE2
Approximately 0.15 g of tissue was homogenized in 2.55 mL of water prior to the addition of 20 μL of internal standard (D4‐PGE2, 5 μg/mL, in ethanol). Methanol was added (0.45 mL) and the sample was vortexed for 1 min before placing on ice for 1 h. The sample was centrifuged for 15 min at 1200g and the supernatant was collected and adjusted with 0.2 N HCl to a pH of 3.0. A high‐performance C‐18 Solid Phase Extraction column (Agilent Technologies) was preconditioned with 2 mL of water and 2 mL of methanol. The sample solution was loaded onto the column, washed with 1 mL of water, and eluted with 2 mL of ethyl acetate. The sample was dried under nitrogen and re‐dissolved in 100 μL of ethanol for LC/MS analysis.
The LC/MS parameters were as follows: mobile phase: A: 0.01% HOAc‐H2O, B: 0.01% HOAc‐ACN; gradient: 0–14 min, 32% B, 16–20 min, 95% B, 22–25 min, 32% B; flow rate: 0.8 mL/min; injection: 20 μL; electrospray ionization in negative mode; full scan from m/z 50 to m/z 500; target: m/z 351; nebulizer pressure: 15.0 psi; dry gas: 5.0 L/min; dry temperature: 325°C; compound stability: 20%; and average: 50. The amount of PGE2 (ng/g) was quantified using an internal standard curve (concentration of PGE2 versus the peak area ratio of PGE2 to D4‐PGE2).
Statistical analyses
The data were compiled using Microsoft Office Excel 2007 (Microsoft, Redmond, WA, USA), and Student's t‐tests (P < 0.05) were used to detect statistically significant differences between comparison groups. We used both the intermediate metabolites (C18:3 n‐6 and C20:3 n‐6) and AA to calculate D6D activity:
Results
Lipid biomarker of D6D activity
There were significant differences in the lipid profiles of cancerous and noncancerous tissues. Based on GC results, we calculated the percentage of each of the relevant fatty acids by breast tissue type (cancerous versus noncancerous), overall, and according to genotype. There is a lower percentage of LA in the cancerous tissue samples compared to the noncancerous samples (10.71% vs 12%, respectively, P < 0.05). In contrast, the sum of the relative percentage of LA metabolites (C18:3 n‐6 + C20:3 n‐6 + AA) was significantly larger in the cancerous than in the noncancerous samples (10.2% vs 8.6%, respectively, P < 0.01; Fig. 1). The ratio of the LA metabolites to LA is significantly larger in the cancerous samples than in the noncancerous samples (1.18 vs 0.78, respectively, P < 0.001, Fig. 2), implying that there is more D6D enzymatic conversion of LA in breast cancer tumors.
Figure 1.
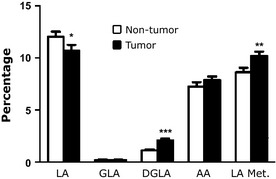
Fatty acid profile of the D6D pathway in tumor and non‐tumor breast tissue. Percentage of individual fatty acids of the delta‐6‐desaturase (D6D) pathway, including linoleic acid (LA, C18:2 n‐6), gamma‐linolenic acid (GLA, C18:3 n‐6), dihomo‐gamma‐linolenic acid (DGLA, C20:3 n‐6), and arachidonic acid (AA, C20:4 n‐6), as well as the sum of the metabolites that are downstream of LA (“LA Met.” = GLA + DGLA + AA). (n = 69 patients; *P < 0.05, **P < 0.003, ***P < 0.0001).
Figure 2.
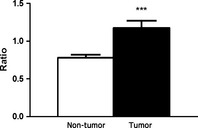
D6D activity in tumor and non‐tumor breast tissue. D6D activity is expressed as the ratio of the sum of linoleic acid (LA) metabolites to LA. The ratio is significantly higher in tumor tissue than in non‐tumor tissue. (n = 69 patients; ***P < 0.001).
We also looked for differences in D6D activity between breast cancer genotypes. As shown in Figure 3, D6D activity is significantly higher in the cancerous tissues of both ER+ and ER− patients compared to their respective noncancerous tissues. The ratio of LA metabolites/LA in the noncancerous tissue was very similar between ER+ and ER− samples (0.75 and 0.90, respectively). However, this ratio greatly differed between ER+ and ER− in cancerous tissues (0.98 and 1.87, respectively, P < 0.001), illustrating that D6D activity is significantly augmented in ER− cancerous tissue versus ER+ cancerous tissue. Collectively, these data show that there is a significantly higher amount of D6D activity in cancerous breast tissue than noncancerous breast tissue, and that this trend is more pronounced in the more clinically aggressive ER− genotype.
Figure 3.
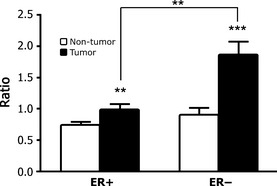
Differential D6D activity in estrogen receptor positive (ER+) and negative (ER−) genotypes. D6D activity is significantly higher in tumor tissue than in non‐tumor tissue for both the ER+ and ER− genotypes, and the ratio is more profound in the ER− than ER+ genotype. (n = 15 for ER+ samples and n = 54 for ER− samples; **P < 0.01; ***P < 0.001).
PGE2 measurement
Liquid chromatography‐mass spectrometry was performed to determine the PGE2 levels in samples of cancerous and noncancerous breast tissue. Values of PGE2 were measured as ng/g and are shown in Figures 4 and 5. There was significantly more PGE2 in the cancerous samples than in their matched noncancerous samples (30.81 vs 6.33 ng/g, respectively, n = 16, P < 0.001; Fig. 4). This trend was also present when we compared PGE2 levels of tissues between genotypes. Though not significant, PGE2 levels were higher in the cancerous tissues of women with ER− breast cancer than in the women with ER+ breast cancer (36.26 vs 25.36 ng/g, respectively, m = 8, P = 0.39; Fig. 5) This is consistent with the finding that there was a higher percentage of AA detected in the ER− cancerous samples than in the ER+ cancerous samples (8.81% vs 7.63%, respectively, P = 0.28). Finally, the PGE2 values for the noncancerous tissue did not significantly differ between the genotypes (6.09 ng/g for ER− vs 6.58 ng/g for ER+).
Figure 4.
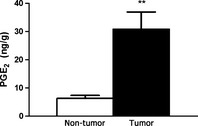
PGE 2 levels in tumor and non‐tumor breast tissue. Tumor samples had a significantly higher amount of prostaglandin E2 (PGE 2). (n = 16 for each group; **P < 0.01).
Figure 5.
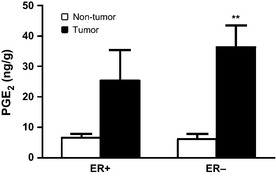
Differential PGE 2 levels in estrogen receptor positive (ER+) and negative (ER−) genotypes. PGE 2 levels are higher in the tumor tissue for each genotype, though this effect was only significant for the ER− genotype. (n = 8 for each group, **P < 0.01).
Discussion
Determining the fatty acid profiles for the breast tumor and adjacent tissue samples from BC patients showed that the activity of the AA synthetic pathway is indeed augmented in BC. We detected less LA and more AA in the cancerous samples, implying that LA had been converted to AA and that the D6D pathway is more active in breast cancer. D6D activity was higher in cancerous tissue from the BC patients with the more clinically aggressive ER− genotype. The fact that the levels of D6D activity were very similar between the noncancerous tissues from the two genotypes and that only the cancerous tissues differed in D6D activity may imply that the pathogenesis of breast cancer is impacted by these changes in the fatty acid profile.
The results obtained from the LC‐MS analysis of PGE2 levels in the tissue samples were also consistent with the fatty acid profiling data and the notion that the AA synthesis pathway is augmented in cancerous tissue, particularly from ER− patients. A higher percentage of AA was detected in the cancerous samples than in the noncancerous samples though this difference was not significant. However, as AA can be metabolized to further downstream metabolites such as PGE2, and consequently higher PGE2 levels were detected in the cancerous samples relative to the noncancerous samples. When we examined the data by genotype, this trend was present in both ER+ and ER− tissues but only significant for the ER− tissues, consistent with the result that D6D activity is most augmented in ER− breast cancer. These data support our hypothesis that the more aggressive ER− breast cancer genotype is associated with higher D6D activity and higher levels of PGE2.
One limitation of this study is that we were unable to measure D6D gene and protein expression in the breast tissue because we lacked sufficient sample. However, three recent studies support our evidence by showing that there are significant differences in D6D mRNA levels between tumor and normal tissues (>2‐fold higher in tumors than normal tissue) in patients with breast cancer, brain tumors, or cervical cancer.31, 32, 33 Another issue to consider is the small sample size of the present study. Over 100 women enrolled in the study, but only 69 women had sufficient tissue for lab analyses. Fifty‐four of these women were further classified as having ER+ breast cancer, with the remaining 15 women having ER− cancers. This breakdown of 78% ER+ and 22% ER− is consistent with national genotypic diagnostic rates.21, 22 However, the ER− group was very small, and analyses were limited because tissues from only eight of these women were sufficient for PGE2 measurement. Future studies should actively recruit more ER− women to increase the genotype‐specific group sizes and reduce variation. Still, although the number of subjects in our study was small, the impact of variation was minimized by the use of a within‐subjects design.
Regarding methodology, two aspects of our study can be further clarified. First, breast tissue is adipose tissue and thus not directly comparable to a breast tumor derived of epithelial cells; for example, adipose tissue cells contain high levels of triglycerides. For this reason, when we performed lipid analysis we only considered the phospholipids, which are present in the cell membranes of all human cells. By comparing only the phospholipid layer between cancerous and noncancerous tissues we were able to minimize variation in fatty acid content due to tissue type. Second, it is important to emphasize that the definition of D6D activity lacks consistency throughout the literature. We chose to incorporate both the intermediate metabolites (C18:3 n‐6 and C20:3 n‐6) and AA into our ratio for enzymatic conversion of LA to AA. However, this method leaves D6D activity as the sum of these fatty acids and does not account for LA in its ratio.34 We felt that it was important to include all of the fatty acids in the LA→AA pathway in our definition of D6D activity. Unless one is using radioisotope‐labeled studies, which is not always feasible or ethical in human subjects, it is important to have an equation as a proxy measure of enzyme activity.
Of the PUFAs, only LA and ALA are considered essential fatty acids. It has been estimated that LA accounts for 90% of all PUFAs consumed in North America, and that Westerners consume at least 10 times the minimum daily requirement.1, 16, 35, 36 This disparity can be attributed to high intake of oils rich in LA, such as soybean, corn, sunflower, and safflower oils.1 Interestingly, n‐6 and n‐3 PUFA compete for the D6D enzyme and it is well documented that ALA is the preferred substrate for D6D; specifically, the affinity for ALA is two to three times greater than for LA.11, 36, 37, 38, 39 However, n‐6 often outcompetes n‐3 for D6D because the amount of LA in our Western diet is so high, making the conversion of ALA quite low. Even in the absence of competing n‐6 substrates, greater dietary intake of ALA unfortunately does not correlate with increased phospholipid DHA levels.40 Our data indicate that in breast cancer, D6D is overactive in the n‐6 PUFA pathway and that a D6D inhibitor may have therapeutic potential. Recently, we showed in mice that D6D activity is upregulated during melanoma and lung tumor growth and that suppressing D6D activity, either by RNAi knockdown or a specific D6D inhibitor, dramatically reduces tumor growth without any detectable negative effects on the health of the animal,17 supporting the notion that D6D is upregulated in cancer cells and increased D6D may be critical for tumor development.
Our data show that the D6D pathway is augmented in breast cancer, and more specifically, in the clinically aggressive ER− genotype. This is an interesting result with potential applications for the field of breast cancer treatment and prevention. Given that D6D is overactive in breast cancer, D6D inhibition may be a useful method of treatment. Because we rely largely on diet to obtain adequate levels of n‐3 PUFAs rather than on the conversion of ALA to EPA and DHA, inhibiting D6D should not greatly impact n‐3 levels in the body. However, EPA and DHA supplementation in conjunction with pharmaceutical D6D inhibition would likely prevent any deficit in EPA and DHA. Given the limited number of subjects in this study, we were unable to investigate correlations with other clinicopathological factors. Future studies examining the relationship between the D6D pathway and other clinicopathological factors, such as patient age, lifestyle factors, menopausal status, histological grade, or other genotypes, are therefore warranted.
Because ER− breast cancer is aggressive and cannot be treated effectively, understanding the pathways that drive this disease is extremely important. In this study we have shown that D6D activity and PGE2 levels are higher in cancerous tissue as compared to noncancerous tissue, and this difference is most pronounced in ER− breast cancer. These are potentially important biomarkers to monitor, particularly in women who are at risk of developing breast cancer. Drugs to alter this pathway may also be useful in the treatment or possibly prevention of breast cancer. Finally, future work should consider the development of non‐pharmaceutical intervention methods to address D6D activity, focusing on n‐3 PUFA rich products for breast cancer prevention. These suggestions could also be made on the basis of genotype, targeting women with ER− breast cancer.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
We thank the READ Charitable Trust (Salem, MA, USA) for their generous financial support of this study and Erin D. Gleason for manuscript edits and preparation.
(Cancer Sci, doi: 10.1111/cas.12129, 2013)
References
- 1. Simopoulos AP. Genetic variants in the metabolism of omega‐6 and omega‐3 fatty acids: their role in the determination of nutritional requirements and chronic disease risk. Exp Biol Med 2010; 235: 785–95. [DOI] [PubMed] [Google Scholar]
- 2. Innis SM. Dietary (n‐3) fatty acids and brain development. J Nutr 2007; 137: 855–9. [DOI] [PubMed] [Google Scholar]
- 3. Simopoulos AP. Omega‐3 fatty acids in health and disease and in growth and development. Am J Clin Nutr 1991; 54: 438–63. [DOI] [PubMed] [Google Scholar]
- 4. Zurier B. Essential fatty acids and inflammation. Ann Rheum Dis 1991; 50: 745–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schaeffer L, Gohlke H, Müller M et al Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum Mol Genet 2006; 15: 1745–56. [DOI] [PubMed] [Google Scholar]
- 6. Petrik MB, McEntee MF, Chiu CH, Whelan J. Antagonism of arachidonic acid is linked to the antitumorigenic effect of dietary eicosapentaenoic acid in Apc(Min/+) mice. J Nutr 2000; 130: 1153–8. [DOI] [PubMed] [Google Scholar]
- 7. Li B, Birdwell C, Whelan J. Antithetic relationship of dietary arachidonic acid and eicosapentaenoic acid on eicosanoid production in vivo . J Lipid Res 1994; 35: 1869–77. [PubMed] [Google Scholar]
- 8. Bartsch H, Nair J, Owen RW. Dietary polyunsaturated fatty acids and cancers of the breast and colorectum: emerging evidence for their role as risk modifiers. Carcinogenesis 1999; 20: 2209–18. [DOI] [PubMed] [Google Scholar]
- 9. Agatha G, Hafer R, Zintl F. Fatty acid composition of lymphocyte membrane phospholipids in children with acute leukemia. Cancer Lett 2001; 173: 139–44. [DOI] [PubMed] [Google Scholar]
- 10. Levy GN. Prostaglandin H synthases, nonsteroidal anti‐inflammatory drugs, and colon cancer. FASEB J 1997; 11: 234–47. [PubMed] [Google Scholar]
- 11. Rodriguez A, Sarda P, Nessmann C, Boulot P, Leger CL, Descomps B. Delta6‐ and delta5‐desaturase activities in the human fetal liver: kinetic aspects. J Lipid Res 1998; 39: 1825–32. [PubMed] [Google Scholar]
- 12. D'andrea S, Guillou H, Jan S et al The same rat Delta6‐desaturase not only acts on 18‐ but also on 24‐carbon fatty acids in very‐long‐chain polyunsaturated fatty acid biosynthesis. Biochem J 2002; 364: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Horrobin DF. Fatty acid metabolism in health and disease: the role of delta‐6‐desaturase. Am J Clin Med 1993; 57: 732S–7S. [DOI] [PubMed] [Google Scholar]
- 14. .Cho HP, Nakamura MT, Clarke SD. Cloning, expression, and nutritional regulation of the mammalian Delta‐6 desaturase. J Bio Chem 1999; 274: 471–7. [DOI] [PubMed] [Google Scholar]
- 15. Hansen‐Petrik MB, McEntee MF, Johnson BT et al Selective inhibition of Delta‐6 desaturase impedes intestinal tumorigenesis. Cancer Lett 2002; 175: 157–63. [DOI] [PubMed] [Google Scholar]
- 16. Obukowicz M, Welsch D, Salsgiver W et al Novel, selective delta6 or delta5 fatty acid desaturase inhibitors as antiinflammatory agents in mice. Lipids 1999; 34: S149. [DOI] [PubMed] [Google Scholar]
- 17. He C, Qu X, Wan J et al Inhibiting delta‐6 desaturase activity suppresses tumor growth in mice. PLoS ONE 2012; 7: e47567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nanda R. “Targeting” triple‐negative breast cancer: the lessons learned from BRCA1‐associated breast cancers. Semin Oncol 2011; 38: 254–62. [DOI] [PubMed] [Google Scholar]
- 19. Pal SK, Childs BH, Pegram M. Triple negative breast cancer: unmet medical needs. Breast Cancer Res Treat 2011; 125: 627–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cuzick J, DeCensi A, Arun B et al Preventive therapy for breast cancer: a consensus statement. Lancet Oncol 2011; 12: 496–503. [DOI] [PubMed] [Google Scholar]
- 21. Adelson K, Germain D, Raptis G, Biran N. Hormonal modulation in the treatment of breast cancer. Endocrinol Metab Clin North Am 2011; 40: 519–32. [DOI] [PubMed] [Google Scholar]
- 22. Ali S, Buluwela L, Coombes RC. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu Rev Med 2011; 62: 217–32. [DOI] [PubMed] [Google Scholar]
- 23. Arun B, Dunn BK, Ford LG, Ryan A. Breast cancer prevention trials: large and small trials. Semin Oncol 2010; 37: 367–83. [DOI] [PubMed] [Google Scholar]
- 24. Perks CM, Holly JM. Hormonal mechanisms underlying the relationship between obesity and breast cancer. Endocrinol Metab Clin North Am 2011; 40: 485–507. [DOI] [PubMed] [Google Scholar]
- 25. Lonning PE. The potency and clinical efficacy of aromatase inhibitors across the breast cancer continuum. Ann Oncol 2011; 22: 503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clemons M, Goss P. Estrogen and the risk of breast cancer. N Engl J Med 2001; 344: 276–85. [DOI] [PubMed] [Google Scholar]
- 27. Dawood S. Triple‐negative breast cancer: epidemiology and management options. Drugs 2010; 70: 2247–58. [DOI] [PubMed] [Google Scholar]
- 28. Elias AD. Triple‐negative breast cancer: a short review. Am J Clin Oncol 2010; 33: 637–45. [DOI] [PubMed] [Google Scholar]
- 29. Oakman C, Viale G, Di Leo A. Management of triple negative breast cancer. Breast 2010; 19: 312–21. [DOI] [PubMed] [Google Scholar]
- 30. Kang JX, Wang J. A simplified method for analysis of polyunsaturated fatty acids. BMC Biochem 2005; 6: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scotto L, Narayan G, Nandula SV et al Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosom Cancer 2008; 47: 755–65. [DOI] [PubMed] [Google Scholar]
- 32. Sun L, Hui AM, Su Q et al Neuronal and glioma‐derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006; 9: 287–300. [DOI] [PubMed] [Google Scholar]
- 33. Zhao H, Langerød A, Ji Y et al Different gene expression patterns in invasive lobular and ductal carcinomas of the breast. Mol Biol Cell 2004; 15: 2523–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grønn M, Christensen E, Hagve TA, Christophersen BO. Effects of dietary purified eicosapentaenoic acid (20:5 (n‐3)) and docosahexaenoic acid (22:6(n‐3)) on fatty acid desaturation and oxidation in isolated rat liver cells. Biochim Biophys Acta 1992; 1125: 35–43. [DOI] [PubMed] [Google Scholar]
- 35. Burdge G. Alpha‐linolenic acid metabolism in men and women: nutritional and biological implications. Curr Opin Clin Nutr Metab Care 2004; 7: 137–44. [DOI] [PubMed] [Google Scholar]
- 36. Huang YS, Smith RS, Redden PR, Cantrill RC, Horrobin DF. Modification of liver fatty acid metabolism in mice by n‐3 and n‐6 delta 6‐desaturase substrates and products. Biochim Biophys Acta 1991; 1082: 319–27. [DOI] [PubMed] [Google Scholar]
- 37. Garg ML, Sebokova E, Thomson AB, Clandinin MT. Delta 6‐desaturase activity in liver microsomes of rats fed diets enriched with cholesterol and/or omega 3 fatty acids. Biochem J 1988; 249: 351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Portolesi R, Powell BC, Gibson RA. Competition between 24:5n‐3 and ALA for Delta 6 desaturase may limit the accumulation of DHA in HepG2 cell membranes. J Lipid Res 2007; 48: 1592–8. [DOI] [PubMed] [Google Scholar]
- 39. Christiansen EN, Lund JS, Rørtveit T, Rustan AC. Effect of dietary n‐3 and n‐6 fatty acids on fatty acid desaturation in rat liver. Biochim Biophys Acta 1991; 1082: 57–62. [DOI] [PubMed] [Google Scholar]
- 40. Brenna JT, Salem N Jr, Sinclair AJ et al Alpha‐Linolenic acid supplementation and conversion to n‐3 long‐chain polyunsaturated fatty acids in humans. Prostaglandins Leukot Essent Fatty Acids 2009; 80: 85–91. [DOI] [PubMed] [Google Scholar]