

Abstract
Clofarabine (CAFdA) is incorporated into leukemic cells by human equilibrative nucleoside transporters (hENT) 1 and 2 and human concentrative nucleoside transporter (hCNT) 3. CAFdA is then phosphorylated to the active metabolite CAFdA triphosphate (CAFdATP) by deoxycytidine kinase (dCK) and deoxyguanosine kinase (dGK). Two novel CAFdA‐resistant variants were established and their mechanism of resistance was elucidated. The two variants (HL/CAFdA20, HL/CAFdA80) were 20‐fold and 80‐fold more CAFdA‐resistant than HL‐60, respectively. mRNA levels of hENT1, hENT2 and hCNT3 were 53.9, 41.8 and 17.7% in HL/CAFdA20, and 30.8, 13.9 and 7.9% in HL/CAFdA80, respectively, compared with HL‐60. Thus, the total nucleoside transport capacity of CAFdA was reduced in both variants. dCK protein levels were 1/2 in HL/CAFdA20 and 1/8 in HL/CAFdA80 of that of HL‐60. dGK protein levels were 1/2 and 1/3, respectively. CAFdATP production after 4‐h incubation with 10 μM CAFdA was 20 pmol/107cells in HL/CAFdA20 and 3 pmol/107cells in HL/CAFdA80 compared with 63 pmol/107cells in HL‐60. The decreased CAFdATP production attenuated drug incorporation into both mitochondrial and nuclear DNA. In addition, the two variants were resistant to CAFdA‐induced apoptosis due to Bcl2 overexpression and decreased Bim. A Bcl2 inhibitor, ABT737, acted synergistically with CAFdA to inhibit the growth with combination index values of 0.27 in HL/CAFdA20 and 0.23 in HL/CAFdA80, compared with 0.65 in HL‐60. Thus, the mechanism of resistance primarily included not only reduced CAFdATP production, but also increased antiapoptosis. The combination of CAFdA and ABT737 may be effective against CAFdA resistance.
The mainstay of leukemia chemotherapy has included purine and pyrimidine nucleoside analogs for nearly 50 years. Clofarabine (2‐chloro‐2′‐arabinofluoro‐2′‐deoxyadenosine, CAFdA) is a relatively new purine nucleoside analog.1, 2, 3, 4, 5 The rationale behind its design was to combine the structural features of cladribine and fludarabine.5, 6 CAFdA is toxic to non‐proliferating human lymphocytes and rapidly proliferating cells.5 Preclinical studies showed that CAFdA had a high degree of efficacy. In 2004, the US Food and Drug Administration approved CAFdA (Clolar, Genzyme, Cambridge, MA, USA) for the treatment of pediatric patients with acute lymphoblastic leukemia (ALL). The anti‐cancer activities of CAFdA toward various types of tumors are thus being investigated in other age groups of leukemia.7, 8, 9, 10
On administration, CAFdA is transported into leukemic cells through two types of nucleoside transporters: human equilibrative nucleoside transporters (hENT) 1 and 2 and human concentrative nucleoside transporter (hCNT) 3.11 The transport via hENT is a sodium‐independent mechanism involving primarily bidirectional facilitative diffusion driven by a gradient in nucleoside concentration. In contrast, the hCNTs are sodium‐dependent active transporters for their ability against a concentration gradient. Inside the cell, CAFdA is phosphorylated to its monophosphate derivative, not only by the cytoplasmic enzyme deoxycytidine kinase (dCK) but also by the mitochondrial enzyme deoxyguanosine kinase (dGK). Further intracellular phosphorylation results in the production of the active metabolite CAFdA triphosphate (CAFdATP). CAFdATP is incorporated into DNA, thereby terminating DNA elongation and eventually inducing apoptosis.12, 13, 14, 15, 16, 17 CAFdA also induces apoptosis via direct mitochondrial damage.13
Pharmacological understanding of action of CAFdA is crucial to its optimal administration. Moreover, the biochemical and molecular mechanisms of drug resistance may be a clue to overcoming treatment failure. In the present study, two resistant to CAFdA were developed and their mechanisms of resistance were investigated. The study focused on factors that were involved in intracellular CAFdATP production and in drug‐induced apoptosis. An effective strategy to sensitize the cells to CAFdA was also proposed.
Materials and Methods
Drugs and chemicals
CAFdA was kindly provided by Genzyme. Cytarabine (ara‐C), gemcitabine, cladribine, doxorubicin, etoposide, vincristine and nitrobenzylmercaptopurine ribonucleoside were purchased from Sigma Aldrich (St. Louis, MO, USA). Bcl2 inhibitor ABT737 was obtained from Selleck Chemical Company (Houston, TX, USA). [3H]‐clofarabine was purchased from the Japanese Isotope Association (Tokyo, Japan).
Cell culture and establishment of two CAFdA‐resistant HL‐60 variants
The human acute leukemia HL‐60 cells were cultured in RPMI 1640 media supplemented with 10% FCS at 37°C. The parental HL‐60 cells were treated with escalating concentrations of CAFdA. The initial concentration was 1/100 the concentration required to inhibit 50% growth of the cells (IC50). CAFdA was added stepwise increasingly. After repeated passages, one cell line resistant to CAFdA (HL/CAFdA20) was cloned by the limiting dilution method. A portion of this clone was further maintained to develop another variant (HL/CAFdA80) that exhibited greater CAFdA resistance than the first variant, HL/CAFdA20.
Proliferation assay
To evaluate growth inhibition effects, the XTT assay was performed according to the manufacturer's instructions (Roche, Indianapolis, IN, USA), with slight modifications.18
Quantitative RT‐PCR
To evaluate mRNA levels of hENT1, hENT2 and hCNT3, real‐time RT‐PCR was performed using the ABI Prism 7900 sequence detection system (Applied Biosystems, Foster City, CA, USA).19 The hENT1 primers were: forward, 5′‐TGTTTCCAGCCGTAGACT‐3′ and reverse, 5′‐CAGGCCACATGAATACAG‐3′. The hENT2 primers were: forward, 5′‐CAGGAATCTGCATGTTCATCC‐3′ and reverse, 5′‐ACCTAGGCCCGAAAACACTGT‐3′. The hCNT3 primers were: forward, 5′‐TGCCAATATCGGGTCCCAT‐3′ and reverse, 5′‐GAGGCGATATC ACGCTTTCTG‐3′. The β2 micro globulin primers were: forward, 5′‐CTAGGAGGGCTGGCAACTTA‐3′ and reverse, 5′‐CCCCCAAATT CTAAGCAGAG‐3′. The thymidine kinase 1 (TK1) primers were: forward, 5′‐GTGTGCATGGAGTGCTTCC‐3′ and reverse, 5′‐TGTGTCGGCTCTGCTACTTCAA‐3′. The thymidine kinase 2 (TK2) primers were: forward, 5′‐GAGGAGTGGCTCATCAAAGG‐3′, and reverse, 5′‐TGAACAAAATCGGGATCGA‐3′. All primers were purchased from Invitrogen (San Diego, CA, USA). RNA from HL60 cells was used as a reference control to normalize each of the nucleoside transporter/β2 microglobulin amplification ratios.
Nucleoside transport capacity
To determine cellular transport capacity, nucleoside analog uptake was quantified in all cell lines using the method of Wiley et al.20, with slight modifications. The cells were pulsed for 20, 40 s with tritiated CAFdA, solubilized and subjected to scintillation counting. Transport capacity was determined in the absence and presence of nitrobenzylmercaptopurine ribonucleoside (NBMPR).
Western blot analysis
To determine protein levels of dCK, dGK, the members of the Bcl2 family and caspase 3, western blotting was performed. The membranes were probed using standard techniques. The membrane was visualized using Image Quant LAS4000 mini apparatus (GE Healthcare, Uppsala, Sweden). The antibody against dCK was provided by Dr Hori.21 Mouse polyclonal anti‐dGK (Abcam, Cambridge, UK), rabbit polyclonal anti‐Bcl‐2, rabbit polyclonal anti‐Bim, rabbit polyclonal anti‐Bax, rabbit polyclonal anti‐caspase 3 and rabbit polyclonal anti‐Cox IV antibody, rabbit polyclonal anti‐BclxL, rabbit polyclonal anti‐Mcl‐1, rabbit polyclonal anti‐Bak, rabbit polyclonal anti‐Bid, rabbit polyclonal anti‐Bad, rabbit polyclonal anti‐Bik, rabbit polyclonal anti‐Puma (all from Cell Signaling Technology, Beverly, MA, USA) and rabbit polyclonal anti‐actin antibody (Sigma, St Louis, MO, USA) were used as primary antibodies. dCK in the cytoplasmic fraction and dGK in the mitochondrial fraction were evaluated after both fractions had been separated using the Calbiochem Proteo Extract Subcellular Proteome Extraction Kit (EMD Biosciences, Darmstadt, Germany).
Determination of CAFdATP
To quantify intracellular CAFdATP concentrations, the HPLC method previously established in our laboratory was used.22 Cells (1 × 106/mL, 20 mL) were incubated with CAFdA at different concentrations for various time periods at 37°C. The intracellular concentrations of CAFdATP were expressed as pmol/107cells. The limit of detection was 10 pmol.
Mitochondrial membrane potential
To elucidate CAFdA‐mediated mitochondrial damage, mitochondrial membrane potential was determined. The cells were treated for 24 h with 1 or 10 μM CAFdA, were stained with near infrared (NIR) dye and underwent flowcytometric analysis.
Double stain of nuclei and mitochondria
The cells were treated for 24 h with 1 or 10 μM CAFdA, and were incubated with NIR dye and 1 μM Hoechist 33342 dye for 30 min at 37°C. The images were analyzed using Confocal Microscopy (Leica TCS SP2, Wetzler, Germany). Cells were classified into three groups: live cells (mitochondria were intact), mitochondria‐damaged cells (mitochondrial particles were aggregated), and apoptotic cells (mitochondria disappeared). We counted approximately 40 cells in four different fields for each sample to classify them in the three groups. The data showed the average percentage of all of the cells in three cell lines at the indicated concentration.
Distribution of CAFdA inside the cell
To determine the localization of CAFdA inside the cell, nuclear and mitochondrial fractions were separated after treatment for 24 h with tritiated CAFdA (specific activity: 3.3 μCi/mL at 0.2 μM). The radioactivity of each fraction was calculated using a scintillation counter.
Quantification of CAFdA‐induced apoptosis
To measure the cytotoxicity of CAFdA, drug‐induced apoptosis was calculated. Treated with different concentrations for 24 h, the cells were stained with propidium iodide and subjected to cell cycle analysis using flowcytometry. The sub‐G1 fraction was quantified. Alternatively, the cells were labeled with annexin‐V and propidium iodide simultaneously and evaluated for their positivity using flowcytometry.23 Moreover, apoptosis was detected by evidence of caspase 3 cleavage.
Calculation of the combination index
Combination index (CI) analysis provides quantitative information on the nature of drug interaction. CI was calculated as follows:
CAx and CBx are the concentrations of drug A and drug B used in combination to achieve x% drug effect. ICxA and ICxB are the concentrations for single agents to achieve the same effect. CI values of less than, equal to and more than 1 indicate synergy, additivity and antagonism, respectively.24
Statistical analyses
All graphs and curves were generated with GraphPad Prism software (GraphPad Software, San Diego, CA, USA). All statistical analyses were performed using Microsoft Excel (Microsoft Corporation, Redmond, WA, USA). P‐values were calculated using the unpaired t‐test with two‐tailed analysis.
Results
Establishment of two new leukemic cell lines resistant to CAFdA
The growth inhibitory effects of CAFdA were compared among HL‐60, HL/CAFdA20 and HL/CAFdA80. The IC50 values indicated that HL/CAFdA20 and HL/CAFdA80 were 20‐fold (P < 0.01) and 80‐fold (P < 0.01) more CAFdA‐resistant than HL‐60, respectively (Table 1). HL/CAFdA20 and HL/CAFdA80 showed cross‐resistance against similar nucleoside analogs, including ara‐C, gemcitabine, cladribine and fludarabine nucleoside, and against anthracycline doxorubicin. HL/CAFdA80 was more resistant to these analogs than HL/CAFdA20. However, no cross‐resistance was seen with vincristine and etoposide (Table 1). The results thus indicated that two CAFdA‐resistant cell lines with different degrees of resistance were successfully developed, and they were cross‐resistant to similar nucleoside analogs.
Table 1.
Drug sensitivities of HL‐60, HL/CAFdA20 and HL/CAFdA80
| IC50 (nM) | |||
|---|---|---|---|
| HL‐60 | HL/CAFdA20 | HL/CAFdA80 | |
| Clofarabine | 22.5 | 451.4 (20.0) | 1775.0 (79.0) |
| Cytarabine | 17.6 | 10 001.0 (568.0)a | ND |
| Gemcitabine | 3.3 | 35.3 (11.0)a | 1124.0 (341.0)a |
| Cladribine | 4.1 | 373.0 (91.0)a | 1825.0 (445.0)a |
| F‐ara‐A | 113.0 | 217.0 (1.9)n.s. | 489.0 (4.3)a |
| Doxorubicin | 57.4 | 98.1 (1.7)n.s. | 323.0 (5.6)a |
| Vincristine | 28.3 | 33.3 (1.2)n.s. | 30.9 (1.1)n.s. |
| Etoposide | 79.7 | 93.3 (1.2)n.s. | 153.0 (1.9)n.s. |
| ABT737 | 63.8 | 294.0 (4.6)n.s. | 1135.0 (18.0)n.s. |
*P < 0.01. Exponential growing cells (HL‐60, HL/CAFdA20 and HL/CAFdA80) were exposed to increasing concentrations of a given anticancer agent as indicated for 72 h. The number in the parentheses is the relative resistance, which was obtained by dividing the IC50 value of HL/CAFdA20 or HL/CAFdA80 by that of HL‐60. F‐ara‐A, fludurabine nucleoside; ND, not determined because the IC50 values were beyond the upper detection limit.
Decreased production of intracellular CAFdATP
The intracellular active metabolite CAFdATP was quantified in the three cell lines using HPLC.22 When HL‐60 was incubated with various concentrations of CAFdA, CAFdATP production increased concentration‐dependently and reached a plateau at concentrations between 10 and 20 μM (Fig. 1a). Compared with the HL‐60, the variants yielded decreased CAFdATP, with a greater reduction in HL/CAFdA80 than in HL/CAFdA20. The CAFdATP level after 4‐h incubation with 20 μM CAFdA was 28 pmol/107cells in HL/CAFdA20, 3 pmol/107cells in HL/CAFdA80 and 54 pmol/107cells in HL‐60. The results suggested that the decrease in the intracellular active metabolite was the major mechanism of CAFdA resistance, with a greater reduction in the more resistant cell line.
Figure 1.
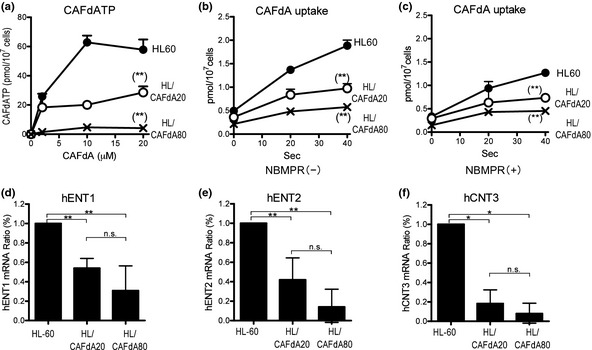
(a) Measurement of intracellular CAFdATP. The cells were incubated for 4 h with different concentrations (0, 2, 10 and 20 μM) of CAFdA, followed by quantification of CAFdATP using HPLC. (b,c) Cellular uptake of tritiated CAFdA. The capacity of the transporters was determined as the initial drug uptake. The cells were pulsed with tritiated CAFdA for 0, 20 and 40 s in the absence of nitrobenzylmercaptopurine ribonucleoside (NBMPR) (b) and in the presence of NBMPR (c). The experiments were performed at least three times in each cell line. (d–f) Cellular uptake of CAFdA. mRNA expression of human equilibrative nucleoside transporters (hENT) 1, hENT2 and hCNT3 in HL‐60 cells and the resistant variants determined using quantitative RT‐PCR. The data shown are the mean and SD of at least three independent experiments. *P ≤ 0.05; **P ≤ 0.01.
Membrane transporters and drug uptake capacity
Membrane transporters and dCK and dGK are most closely associated with the intracellular CAFdATP production.25, 26 Quantitative RT‐PCR demonstrated that mRNA levels of hENT1, hENT2 and hCNT3 were 53.9, 41.8 and 17.7% in HL/CAFdA20, and 30.8, 13.9 and 7.9% in HL/CAFdA80, respectively, compared with HL‐60 (Fig. 1d–f). The nucleoside uptake capacity was also determined as these transporters' function. The values of the initial uptake of CAFdA after 40‐s pulse treatment with tritiated CAFdA were 0.97 ± 0.10 pmol/107cells in HL/CAFdA20 and 0.57 ± 0.05 pmol/107cells in HL/CAFdA80, compared with 1.88 ± 0.11 pmol/107cells in HL60 (Fig. 1b). The results indicated a decreased capacity of CAFdA uptake, which correlates with the reduced CAFdATP production. Greater suppression was found in the more resistant cell line. Thus, these results suggested that the decreased drug uptake was due to the reduced transporters in variant cell lines. Of note, the reduction in hCNT3 was first reported in cancer cells resistant to CAFdA.
Cytoplasmic deoxycytidine kinase and mitochondrial deoxyguanosine kinase levels
Quantitative RT‐PCR demonstrated that HL/CAFdA20 and HL/CAFdA80 exhibited 43.5 ± 10.5% (P < 0.01) and 30.0 ± 13.0% (P < 0.01), respectively, of dCK mRNA levels compared with HL‐60 (Fig. 2a). Moreover, these two variants showed 78.1 ± 7.2% (P < 0.05) and 62.9 ± 13.8% (P < 0.05) of dGK mRNA expression, respectively, compared with HL‐60 (Fig. 2b). Greater reduction in both transcripts was found in the more resistant cell line. The transcript level of cytosolic 5′‐nucleotidase II was not increased in the resistant cells (Fig. 2c), suggesting that dephosphorylation of CAFdA was not involved in the development of resistance. TK1 and TK2 mRNA levels, both of which were not associated with the intracellular metabolism of CAFdA, were not changed significantly (Fig. 2d,e). Similarly, western blotting revealed that cytoplasmic dCK and mitochondrial dGK proteins were downregulated (Fig. 2f–h). Importantly, the reduction in the dGK level has not been reported previously.25 Thus, the results suggested that decreased levels of dCK and dGK were responsible for the attenuated intracellular CAFdATP production, thereby compromising the cytotoxicity in the two resistant variants.
Figure 2.
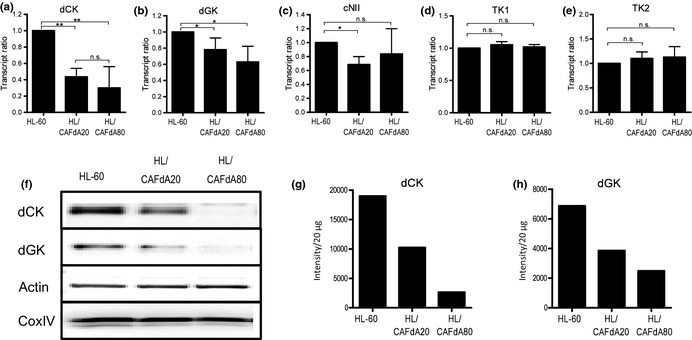
(a–e) mRNA expression of deoxycytidine kinase (dCK), deoxyguanosine kinase (dGK), cytosolic 5′‐nucleotidase II (cN‐II), thymidine kinase 1 (TK1) and thymidine kinase 2 (TK2) in HL‐60, HL/CAFdA20 and HL/CAFdA80 determined using quantitative RT‐PCR. RNA from the HL60 was used as a reference control to normalize each determinant/β2 microglobulin amplification ratio. The data are the means and SD of at least three independent experiments. (f) Protein levels of dCK and dGK. The cells were fractionated to cytosol and mitochondria. dCK protein was determined in the cytosolic fraction, while dGK was in the mitochondrial fraction in each cell line. (g,h) Intensity levels of dCK and dGK were quantified. Data are representative of at least three biological replicates. *P ≤ 0.05; **P ≤ 0.01.
Incorporation of CAFdA into nuclear and mitochondrial DNA
The incorporation of CAFdA into nuclear and mitochondrial DNA was measured after incubation with tritiated CAFdA and fractionated, followed by scintillation counting. The incorporation of CAFdA into both nuclear and mitochondrial DNA was decreased with a greater reduction in the more resistant cell line (Fig. 3a,b). The results thus suggested that the decreased CAFdATP production led to the attenuated incorporation of the drug into both mitochondrial and nuclear DNA.
Figure 3.
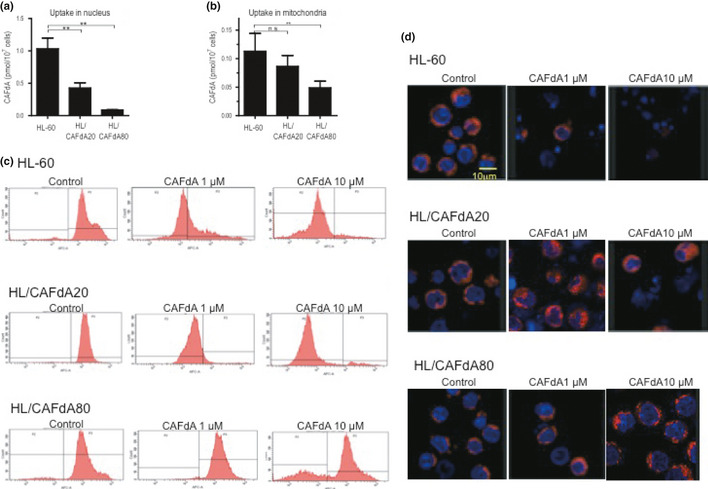
(a,b) The cells (HL‐60, HL/CAFdA20 and HL/CAFdA80) were incubated with tritiated CAFdA at 0.2 μM for 24 h, and fractionated into cytoplasm, mitochondria and nuclei, followed by scintillation counting of the radioactivity of each fraction. Data shown are the means and SD of three independent experiments. (c) CAFdA‐mediated mitochondrial damage. Mitochondrial membrane potentials were determined using near infrared (NIR) stain treated with 1 or 10 μM CAFdA for 24 h. Representative flow cytometry is shown. (d) Double stains were also performed in the cells at 24 h after administration of 1 or 10 μM CAFdA. Nuclei were dyed by Hoechst 33342, while mitochondria were dyed by NIR.
CAFdA‐mediated mitochondrial damage
CAFdA‐mediated damage of mitochondria was evaluated by determining the membrane potential in the cells after treatment with 1 and 10 μM CAFdA for 24 h. Each cell showed 82.5 ± 5.9%, 63.2 ± 8.3% and 15.3 ± 7.9% with 1 μM and 94.5 ± 6.2%, 96.6 ± 4.5% and 34.8 ± 5.6% (mean ± SD) with 10 μM reduction in the potential, respectively, suggesting CAFdA‐mediated mitochondrial damage in HL‐60 and HL/CAFdA20 (Fig. 3c). The cells were further double‐stained with NIR for mitochondria and Hoechst 33342 for nuclei (Fig. 3d). HL‐60 exhibited nuclear condensation and fragmentation, which suggested the induction of apoptosis. HL/CAFdA20 was refractory to the insult by 1 μM CAFdA, but underwent apoptosis by 10 μM CAFdA. HL/CAFdA80 maintained clear staining with 10 μM CAFdA, indicating the refractoriness of the cells to CAFdA's cytotoxicity. We counted live cells, mitochondria damaged cells and dead cells in four different fields, and calculated the ratio (%) in each cell lines. At 24 h after CAFdA administration, live cells (mitochondria intact), damaged cells (mitochondria particle aggregation) and dead cells (mitochondria collapsed and disappeared) were as follows: 90.3 ± 4.8%, 7.6 ± 2.8% and 2.1 ± 0.8% (CAFdA 0 μM), 1.9 ± 0.9%, 21.1 ± 7.7% and 77.0 ± 14.8% (CAFdA 1 μM), 0%, 10.6 ± 5.8% and 89.4 ± 8.9% (CAFdA 10 μM) in HL60, 91.4 ± 4.8%, 7.1 ± 4.5% and 1.5 ± 1.0% (CAFdA 0 μM), 15.4 ± 6.6%, 62.1 ± 15.2% and 22.5 ± 10.8% (CAFdA 1 μM), 9.6 ± 4.5%, 51.1 ± 13.0% and 39.3 ± 12.7% (CAFdA 10 μM) in HL/CAFdA20, 93.6 ± 3.3%, 5.1 ± 2.9% and 1.3 ± 0.8% (CAFdA 0 μM), 66.7 ± 14.8%, 19.4 ± 8.5% and 13.9 ± 5.9% (CAFdA 1 μM), 58.3 ± 14.4%, 14.1 ± 9.8% and 27.6 ± 8.4% (CAFdA 10 μM) in HL/CAFdA80, respectively. Thus, these results suggested that CAFdA damaged mitochondria and induced apoptosis dose‐dependently in HL‐60, but the two variants were resistant to the insult.
Induction of apoptosis by CAFdA
To investigate drug‐induced apoptosis, cells were evaluated for the cleavage of caspase 3 (Fig. 4a). When the cells were incubated with 1 μM CAFdA, cleaved caspase 3 was detected in HL‐60 at 24 h. Caspase 3 cleavage was induced less in HL/CAFdA20 by the same treatment. HL/CAFdA80 was refractory to CAFdA‐induced apoptosis. The induction of apoptosis was further quantitated by using annexin V stain. Treated with CFAdA at 1 or 10 μM for 24 h, apoptotic cell death accounted for 91.5 ± 4.8% and 91.7 ± 2.3% in HL‐60, 35.0 ± 7.6% and 75.6 ± 7.9% in HL/CAFdA20, and 4.0 ± 1.9% and 36.5 ± 8.4% in HL/CAFdA80, respectively (Fig. 4b). Alternatively, apoptosis was measured as induction of the sub‐G1 population (Fig. 4c). The results were compatible with those demonstrated using annexin V stain. Thus, the two variants were refractory to CAFdA‐induced apoptosis and HL/CAFdA80 was more refractory than HL/CAFdA20.
Figure 4.
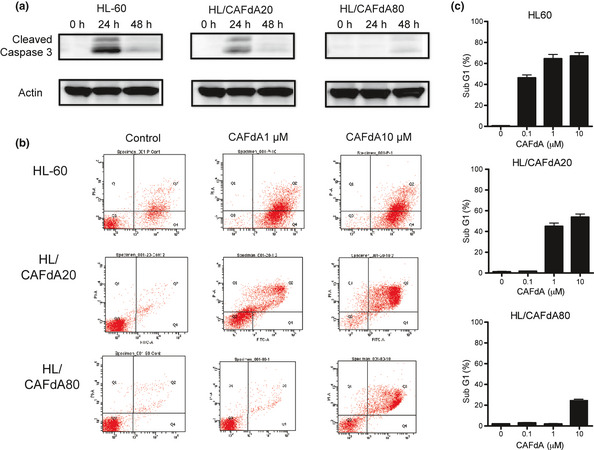
Induction of apoptosis. (a) Time course of cleaved caspase 3. Cleaved caspase 3 was detected in the cells at 24 and 48 h after 1 μM CAFdA treatment. Data are representative of at least three biological replicates. (b) The induction of apoptosis detected after 24‐h incubation with CAFdA (1 and 10 μM), using annexin V and propidium iodide stain. Annexin V‐positive cells are considered apoptotic. Data are representative of at least three replicates. (c) The ratio of sub‐G1 was determined by flowcytometric analysis in the cells after 24‐h incubation with CAFdA (0.1, 1 and 10 μM).
Apoptosis‐related proteins
To reveal the mechanisms of the resistance to CAFdA‐induced apoptosis, apoptosis‐related proteins were determined (Fig. 5a). Prosurvival Bcl2 protein was overexpressed and proapoptotic Bim was reduced in the resistant variants, while Bax, BclxL, Mcl‐1, Bak, Bid, Bad, Bik and Puma were unchanged. Thus, these results suggested the contribution of Bcl2 and Bim to antiapoptosis.
Figure 5.
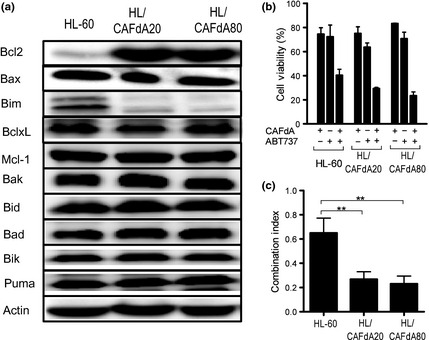
(a) Western blot analysis of apoptosis‐related proteins. Bcl2, Bax, Bim, BclxL, Mcl‐1, Bak, Bid, Bad, Bik and Puma are compared between HL60 and the CAFdA‐resistant variants. (b) The combination treatment of CAFdA with Bcl2 inhibitor ABT737. The cells were incubated for 72 h with CAFdA or ABT737 or both in combination at the concentration of 1/3 IC 50 for each agent. Cell viability was determined using the XTT assay. Data are representative of at least three independent experiments. (c) The combination index in HL60 and the two CAFdA‐resistant cell lines.
Bcl2 inhibitor ABT737 partially overcame CAFdA resistance
Because Bcl2 was increased in the resistant cells, the Bcl2 inhibitor ABT737 was used to see if this could reverse the cellular resistance against CAFdA.27, 28, 29 In each cell line, the combination treatment of CAFdA and ABT737 produced a synergistic effect (Fig. 5b). The CI values were 0.650 ± 0.123, 0.268 ± 0.063 and 0.230 ± 0.065 (Fig. 5c). Thus, these results suggested that ABT737 and CAFdA acted synergistically in their cytotoxicity against leukemic cells with greater synergism in the resistant cell lines. It was also suggested that the development of CAFdA resistance is partly attributable to upregulated Bcl2.
Discussion
In the present study, two CAFdA‐resistant leukemic clones were successfully developed. The mechanism of drug resistance was multifactorial, but it was ultimately associated with reduction in intracellular CAFdATP production and antiapoptosis. The decreased CAFdATP production was attributable to reduced transport capacity (Fig. 1b,c) and decreased dCK and dGK (Fig. 2f–h). The extent of the resistance appeared to be associated with the level of CAFdATP production (Fig. 1a). The reduced CAFdATP production attenuated the incorporation of CAFdA into the nuclei and mitochondria (Fig. 3a,b). In contrast, antiapoptosis was associated with increased Bcl2 and decreased Bim (Fig. 5a). Importantly, the alterations of hCNT3, dGK, Bcl2 and Bim have not been previously reported in the context of CAFdA resistance. In these respects, and in contrast to previous studies, the present study investigated the mechanism of CAFdA resistance intensively and extensively.10, 11, 12, 13, 14
The resistant variants developed here exhibited cross‐resistance against similar nucleoside analogs (Table 1). Because the activation pathway is basically the same,30 such cross‐resistance may be caused by the decrease in transport capacity and the reduced dCK. Two variants were also resistant to doxorubicin. This might be due to the overexpression of Bcl2, because doxorubicin‐induced apoptosis was reported to be inhibited in cancer cells showing Bcl2 and/or Bcl‐xL overexpression.31, 32 However, the cells were not resistant to etoposide. Etoposide‐induced apoptosis is mediated by proapoptotic protein Bax, which was unchanged in the resistant variants.33 It is suggested that the mechanism of the cross‐resistance shown would be specific in part to nucleoside analogs and in part to their antiapoptotic nature.
The cellular uptake is mediated via hENT 1, 2 and hCNT 3. King et al.11 report that CAFdA is carried into oocytes by hCNT3, which attributes >5 times more than hENT1 and hENT2, suggesting that hCNT3 is crucial to the cellular uptake of CAFdA. Our results of uptake ability using tritited CAFdA also suggested that hCNT3 would attribute significantly to transport nucleoside analogs. Transcript levels of all three transporters were reduced in the two CAFdA‐resistant cell lines (Fig. 1d–f). Nevertheless, the reduction in hCNT3 mRNA appeared to be more profound compared with the reductions in hENT1 and 2. Thus, the results suggested the importance of hCNT3 in relation to CAFdA resistance, which has not been reported previously.
In the two variants, the reduction in dGK was demonstrated at protein and transcript levels (Fig. 2b,f,h). Ara‐C and cladribine are phosphorylated exclusively by dCK, while dGK also participates in the phosphorylation of CAFdA and 9‐beta‐d‐arabinofuranosyl guanine.34, 35 It was previously shown that, in cladribine‐resistant cells, the activity of dCK decreased, but the activity of dGK was unchanged.34 However, Månsson et al.35 demonstrated that the cell line (MOLT‐4) that was developed to be resistant to 9‐beta‐d‐arabinofuranosylguanine showed a 42% decrease in dCK enzyme activity and a 26% decrease in dGK enzyme activity, the results of which indicated a more crucial role of dGK to metabolize 9‐beta‐d‐arabinofuranosyl guanine than dCK. The present results demonstrated (Fig. 2f–h) were compatible with their findings, which strongly suggested that dGK has an important role in the phosphorylation pathway of CAFdA.
The cellular activation mechanism of ara‐C is similar to that of CAFdA.30 Ara‐C is influxed into leukemic cells mainly through hENT1, not using hCNT3, and phosphorylated to the active metabolite ara‐C triphosphate (ara‐CTP) by dCK, but not by dGK. Ara‐CTP is incorporated into DNA, thereby terminating DNA synthesis. Ara‐C does not inhibit ribonucleotide reductase nor induce apoptosis directly. The combination of CAFdA and the Bcl2 inhibitor ABT737 gave synergistic effects in three cell lines (Fig. 5b,c). Bcl2 protein expresses less in HL60 cells than in resistant cell lines, but ABT737 is effective in three cell lines. ABT737 has an effect on both BclxL and Bcl2. The CI was lower in both resistant variants, which might correspond to Bcl2 protein (Fig. 5a), suggesting the contribution of this increased antiapoptotic factor to the development of CAFdA resistance. In a literature search, a combination of Bcl2 inhibitor and nucleoside analog has been suggested for targeting solid tumor and hematological malignancies.36, 37, 38, 39, 40 Hann et al. report that ABT737 induced apoptosis and had synergistic effects with etoposide against primary small cell lung cancer xenografts. The combination of ABT737 and etoposide caused significant decreases in tumor growth rates.39 Ugarenko et al. reported that ABT737 was used in combination with doxorubicin to overcome doxorubicin‐resistant, Bcl2‐overexpressing HL‐60 cells. The addition of ABT‐737 enhanced the cytotoxicity of doxorubicin‐induced DNA adducts and subsequently induced classical apoptosis.40 The present study clearly demonstrated that ABT737 also enhanced the cytotoxicity of a nucleoside analog against cancer cells.
There has been one report of an investigation of the mechanisms of CAFdA resistance by Månsson et al.41; however, they mainly demonstrate a decrease in dCK and do not deal with dGK, transporters and apoptosis‐related factors.15, 41 The present study showed conclusively that the mechanism of cellular resistance to CAFdA in the two variants was multifactorial, but primarily involved reduced intracellular CAFdATP and antiapoptosis. Moreover, CAFdA and the Bcl2 inhibitor ABT737 showed synergistic effects in the two variants with Bcl2 overexpression, which suggests that this combination treatment has clinical potential.
Disclosure Statement
The authors have no conflict of interest to declare.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (23501307, 2010) and by a Grant from the Gout Research Foundation (2010). The authors thank Dr Hori, who provided the antibody against dCK. The antibody was developed in the Department of Pediatrics, Mie University School of Medicine.
(Cancer Sci 2013; )
References
- 1. Gaynon PS, Trigg ME, Heerema NA et al Children's Cancer Group trials in childhood acute lymphoblastic leukemia: 1983–1995. Leukemia 2000; 14: 2223–33. [DOI] [PubMed] [Google Scholar]
- 2. Jeha S, Gandhi V, Chan KW et al Clofarabine, a novel nucleoside analog, is active in pediatric patients with advanced leukemia. Blood 2004; 103: 784–9. [DOI] [PubMed] [Google Scholar]
- 3. Appelbaum FR, Gundacker H, Head DR et al Age and acute myeloid leukemia. Blood 2006; 107: 3481–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burnett AK, Mohite U. Treatment of older patients with acute myeloid leukemia – new agents. Semin Hematol 2006; 43: 96–106. [DOI] [PubMed] [Google Scholar]
- 5. Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev 2009; 109: 2880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parker WB, Shaddix SC, Chang CH et al Effects of 2‐chloro‐9‐(2‐deoxy‐2‐fluoro‐/3‐d‐arabinofuranosyl) adenine on K562 cellular metabolism and the inhibition of human ribo‐nucleotide reductase and DNA polymerases by its 5′‐triphosphate. Cancer Res 1991; 51: 2386–94. [PubMed] [Google Scholar]
- 7. Karp JE, Ricklis RE, Balakrishnan K et al A phase 1 clinical‐laboratory study of clofarabine followed by cyclophosphamide for adults with refractory acute leukemias. Blood 2007; 110: 1762–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kantarjian H, Gandhi V, Cortes J et al Phase 2 clinical and pharmacologic study of clofarabine in patients with refractory or relapsed acute leukemia. Blood 2003; 102: 2379–86. [DOI] [PubMed] [Google Scholar]
- 9. Kantarjian HM, Gandhi V, Kozuch P et al Phase I clinical and pharmacology study of clofarabine in patients with solid and hematologic cancers. J Clin Oncol 2003; 21: 1167–73. [DOI] [PubMed] [Google Scholar]
- 10. Cooper T, Kantarjian H, Plunkett W, Gandhi V. Clofarabine in adult acute leukemias: clinical success and pharmacokinetics. Nucleosides Nucleotides Nucleic Acids 2004; 23: 1417–23. [DOI] [PubMed] [Google Scholar]
- 11. King KM, Damaraju VL, Vickers MF et al A comparison of the transportability, and its role in cytotoxicity, of clofarabine, cladribine, and fludarabine by recombinant human nucleoside transporters produced in three model expression systems. Mol Pharmacol 2006; 69: 346–53. [DOI] [PubMed] [Google Scholar]
- 12. Zhenchuk A, Lotfi K, Juliusson G, Albertioni F. Mechanisms of anticancer action and pharmacology of clofarabine. Biochem Pharmacol 2009; 78: 1351–9. [DOI] [PubMed] [Google Scholar]
- 13. Genini D, Adachi S, Chao Q et al Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood 2000; 96: 3537–43. [PubMed] [Google Scholar]
- 14. Sampath D, Rao VA, Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene 2003; 22: 9063–74. [DOI] [PubMed] [Google Scholar]
- 15. Lotfi K, Månsson E, Spasokoukotskaja T et al Biochemical pharmacology and resistance to 2‐chloro‐2′‐arabino‐fluoro‐2′‐deoxyadenosine, a novel analogue of cladribine in human leukemic cells. Clin Cancer Res 1999; 5: 2438–44. [PubMed] [Google Scholar]
- 16. Parker WB, Shaddix SC, Rose LM et al Comparison of the mechanism of cytotoxicity of 2‐chloro‐9‐(2‐deoxy‐2‐fluoro‐b‐d‐arabinofuranosyl) adenine, 2‐chloro‐9‐(2‐deoxy‐2‐fluoro‐b‐d‐ribofuranosyl) adenine, and 2‐chloro‐9‐(2‐deoxy‐2,2‐difluoro‐b‐d‐ribofuranosyl) adenine in CEM Cells. Mol Pharmacol 1999; 55: 515–20. [PubMed] [Google Scholar]
- 17. Xie C, Plunkett W. Metabolism and actions of 2‐chloro‐9‐(2‐deoxy‐2‐fluoro‐β‐d‐arabinofuranosyl)‐adenine in human lymphoblastoid cells. Cancer Res 1995; 55: 2847–52. [PubMed] [Google Scholar]
- 18. Negoro E, Yamauchi T, Urasaki Y, Nishi R, Hori H, Ueda T. Characterization of cytarabine‐resistant leukemic cell lines established from five different blood cell lineages using gene expression and proteomic analyses. Int J Oncol 2011; 38: 911–9. [DOI] [PubMed] [Google Scholar]
- 19. Yamauchi T, Negoro E, Kishi S et al Intracellular cytarabine triphosphate production correlates to deoxycytidine kinase/cytosolic 5′‐nucleotidase II expression ratio in primary acute myeloid leukemia cells. Biochem Pharmacol 2009; 77: 1780–6. [DOI] [PubMed] [Google Scholar]
- 20. Wiley JS, Jones SP, Sawyer WH, Paterson ARP. Cytosine arabinoside influx and nucleoside transport sites in acute leukemia. J Clin Invest 1982; 69: 479–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshio N, Kawai Y, Hori H, Ueda T. Resistance to 9‐beta‐d‐arabinofuranosyl‐2‐fluoroadenine due to reduced incorporation into DNA from competition by excess deoxyadenosine triphosphate: implications for different sensitivities to nucleoside analogues. Int J Hematol 2005; 81: 405–12. [DOI] [PubMed] [Google Scholar]
- 22. Yamauchi T, Ueda T. Determination of clofarabine triphosphate concentrations in leukemia cells using sensitive, isocratic high‐performance liquid chromatography. Anticancer Res 2011; 31: 2863–7. [PubMed] [Google Scholar]
- 23. Yamauchi T, Nowak BJ, Keating MJ, Plunkett W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4‐hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin Cancer Res 2001; 7: 3580–9. [PubMed] [Google Scholar]
- 24. Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res 2004; 10: 7994–8004. [DOI] [PubMed] [Google Scholar]
- 25. Zhu C, Johansson M, Karlsson A. Incorporation of nucleoside analogs into nuclear or mitochondrial DNA is determined by the intracellular phosphorylation site. J Biol Chem 2000; 275: 26727–67. [DOI] [PubMed] [Google Scholar]
- 26. Damaraju VL, Damaraju S, Young JD et al Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003; 22: 7524–36. [DOI] [PubMed] [Google Scholar]
- 27. Sartorius UA, Krammer PH. Upregulation of Bcl2 is involved in the mediation of chemotherapy resistance in human small cell lung cancer cell lines. Int J Cancer 2002; 97: 584–92. [DOI] [PubMed] [Google Scholar]
- 28. van Delft MF, Wei AH, Mason KD et al The BH3 mimetic ABT‐737 targets selective Bcl‐2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl‐1 is neutralized. Cancer Cell 2006; 10: 389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trudel S, Stewart AK, Li Z et al The Bcl‐2 family protein inhibitor, ABT‐737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res 2007; 13: 621–9. [DOI] [PubMed] [Google Scholar]
- 30. Garcia‐Carbonero R, Ryan DP, Chabner BA. Cytidine analogs In: Chabner BA, Longo DL, eds. Cancer Chemotherapy and Biotherapy. Philadelphia, PA: Lippincott‐Raven Publishers, 1996; 265–94. [Google Scholar]
- 31. Huigsloot M, Tijdens IB, Mulder GJ, van de Water B. Differential regulation of doxorubicin‐induced mitochondrial dysfunction and apoptosis by Bcl‐2 in mammary adenocarcinoma (MTLn3) cells. J Biol Chem 2002; 277: 35869–79. [DOI] [PubMed] [Google Scholar]
- 32. Takahashi M, Saito H, Atsukawa K, Ebinuma H, Okuyama T, Ishii H. Bcl‐2 prevents doxorubicin‐induced apoptosis of human liver cancer cells. Hepatol Res 2003; 25: 192–201. [DOI] [PubMed] [Google Scholar]
- 33. Karpinich NO, Tafani M, Schneider T, Russo MA, Farber JL. The course of etoposide‐induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J Cell Physiol 2002; 19: 16547–52. [DOI] [PubMed] [Google Scholar]
- 34. Månsson E, Spasokoukotskaja T, Sällström J, Eriksson S, Albertioni F. Molecular and biochemical mechanisms of fludarabine and cladribine resistance in a human promyelocytic cell line. Cancer Res 1999; 59: 5956–63. [PubMed] [Google Scholar]
- 35. Månsson E, Liliemark E, Söderhäll S, Gustafsson G, Eriksson S, Albertioni F. Real‐time quantitative PCR assays for deoxycytidine kinase, deoxyguanosine kinase and 5′‐nucleotidase mRNA measurement in cell lines and in patients with leukemia. Leukemia 2002; 16: 386–92. [DOI] [PubMed] [Google Scholar]
- 36. Bhat UG, Gartel AL. Nucleoside analog ARC targets Mcl‐1 to induce apoptosis in leukemia cells. Leukemia 2010; 24: 851–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kohl TM, Hellinger C, Ahmed F et al BH3 mimetic ABT‐737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3‐independent expression of BCL2 in primary AML blasts. Leukemia 2007; 21: 1763–72. [DOI] [PubMed] [Google Scholar]
- 38. Whitecross KF, Alsop AE, Leonie A et al Defining the target specificity ofABT‐737 and synergistic antitumor activities in combination with histone deacetylase inhibitors. Blood 2009; 113: 1982–91. [DOI] [PubMed] [Google Scholar]
- 39. Hann CL, Daniel VC, Sugar EA et al Therapeutic efficacy of ABT‐737, a selective inhibitor of BCL‐2, in small cell lung cancer. Cancer Res 2008; 68: 2321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ugarenko M, Nudelman A, Rephaeli A et al ABT‐737 overcomes Bcl‐2 mediated resistance to doxorubicin‐DNA adducts. Biochem Pharmacol 2010; 79: 339–49. [DOI] [PubMed] [Google Scholar]
- 41. Månsson E, Flordal E, Liliemark J et al Down‐regulation of deoxycytidine kinase in human leukemic cell lines resistant to cladribine and clofarabine and increased ribonucleotide reductase activity contributes to fludarabine resistance. Biochem Pharmacol 2003; 65: 237–47. [DOI] [PubMed] [Google Scholar]