

Abstract
The sorting nexin (SNX) family is a diverse group of cytoplasmic and membrane‐associated proteins that are involved in membrane‐trafficking steps within the endocytotic network. SNX1 and SNX2 are components of the mammalian retromer complex and they also play critical roles in the membrane trafficking of growth factor receptors including epidermal growth factor receptor (EGFR) and c‐Met. The human lung cancer cell lines, which harbor activating mutations in the kinase domain of EGFR gene, are sensitive to EGFR‐targeted drugs gefitinib or erlotinib. However, a lung cancer cell line harboring gene amplification of c‐Met is sensitive to the c‐Met‐targeted drug SU11274 but not to EGFR‐targeted drugs. C‐Met overexpression is identified as one of the bypass mechanisms for acquired resistance to EGFR‐targeted drugs. Here we show that the siRNA‐mediated knockdown of SNX2 decreases the cell‐surface localization of c‐Met, but not that of EGFR, resulting in lysosomal degradation of the c‐Met protein. SNX2 specifically interacts with c‐Met and treatment with lysosomal inhibitors almost completely annihilates downregulation of c‐Met protein by SNX2 knockdown. Therefore, silencing of SNX2 markedly alters sensitivity to anticancer drugs targeted to c‐Met (SU11274) and EGFR (gefitinib and erlotinib) through promotion of compensatory activation of the EGFR pathway in lung cancer cells. These findings suggest that development of drugs targeting SNX2 could be useful in overcoming drug resistance to EGFR‐targeted drugs in lung cancer cells harboring c‐Met gene amplification.
Lung cancer is the most commonly diagnosed malignancy and the most common cause of cancer‐related death worldwide.1 Many tumor types, including non‐small‐cell lung cancer (NSCLC), demonstrate activated epidermal growth factor receptor (EGFR) expression. The EGFR‐targeted drugs, such as gefitinib and erlotinib, have therefore been approved for use in NSCLC treatment. The susceptibility of NSCLC to EGFR‐targeted drugs can be regulated by activating somatic mutations of the EGFR kinase domain.2, 3, 4 Moreover, some acquired resistance to EGFR‐targeted drugs has been closely associated with overexpression of the receptor tyrosine kinase c‐Met through gene amplification,5 whereas c‐Met modifies the sensitivity of cancer cells to gefitinib through human epidermal growth factor receptor (HER) 3‐dependent activation of the phosphatidylinositol 3‐kinase (PI3K)/protein kinase B (Akt) pathway.6 Overexpression of hepatocyte growth factor (HGF), which is a ligand for c‐Met, also markedly alters drug sensitivity to gefitinib in lung cancer cells,7 whereas c‐Met plays an important role in determining the therapeutic efficacy of EGFR‐targeted drugs against NSCLC.
The sorting nexin (SNX) family is a diverse group of cytoplasmic and membrane‐associated proteins with a common phospholipid‐binding motif: the phox homology (PX) domain.8 The SNX family comprises approximately 33 proteins with a range of biological functions, which play specialized and/or generalized roles in the regulation of protein trafficking, including endosomal trafficking of membrane receptors and transporters.9 SNX2 shares 63% sequence identity with SNX1 and forms heteromeric complexes with SNX1 and SNX4, whereas SNX1, SNX2, SNX5 and SNX6 together form the mammalian retromer complex.10, 11 Unlike SNX1, SNX2 binds to phosphatidylinositol‐3, 4, 5‐trisphosphate (PtdIns[3, 4, 5]P3), with the SNX2 PX domain binding preferentially to PtdIns(3)P.12 SNX1, SNX2 and SNX6 also interact with receptors for epidermal growth factor (EGF), platelet‐derived growth factor (PDGF), insulin and leptin.9 SNX1 and SNX2 modulate intracellular membrane trafficking and degradation of EGFR8, 12, 13, 14 and SNX6 also promotes its degradation.15 However, it remains unclear how the membrane trafficking of EGFR and other membrane growth factor receptors is regulated through these SNX.
Gullapalli and colleagues examined the relationship between the cellular expression levels of EGFR or c‐Met and SNX and found that activated EGFR co‐localizes preferentially with SNX2‐positive rather than SNX1‐positive endosomes.14 However, knockdown of SNX1 and SNX2 did not alter EGF/transforming growth factor‐α (TGF‐α)‐induced downregulation of EGFR, suggesting non‐essential roles of SNX1 and/or SNX2 in the EGFR degradation pathway.14 Yeast two‐hybrid analysis and co‐immunoprecipitation analysis identified SNX2 as an interaction partner of c‐Met;16 however, it is not clear which SNX specifically affects the expression of EGFR and c‐Met and how SNX‐mediated downregulation or upregulation of growth factor receptors specifically alters the sensitivity of cancer cells to molecular‐targeting drugs. Therefore, we investigated whether SNX2 could regulate the expression and localization of c‐Met or EGFR family proteins and also could affect the sensitivity of the cells to c‐Met‐ or EGFR‐targeted drugs.
Materials and Methods
Cells and reagents
The characteristics of the human lung cancer cell lines (EBC‐1, H1993 PC‐9 and 11–18) used in the present study have been published previously.17 EBC‐1 and H1993 harbor wild‐type EGFR, PC‐9 harbors activating mutant EGFR (delE746‐A750) and 11–18 harbors activating mutant EGFR (L858R). Also, GR5 harboring c‐Met amplification was established as a gefitinib‐resistant cell line from HCC827 harboring activating mutant EGFR (exon 19 deletion mutation) as described previously.18 EBC‐1 was cultured in DMEM supplemented with 10% FBS and PC‐9, 11–18, H1993, HCC827 and GR5 were cultured in RPMI supplemented with 10% FBS. Anti‐lysosomal‐associated membrane protein (LAMP) 1 was provided by Dr K. Furuta (National Cancer Center Research Institute, Tokyo, Japan). Gefitinib was provided by AstraZeneca Inc. (Macclesfield, UK) and erlotinib was from F. Hoffmann‐La Roche Ltd (Basel, Switzerland). Anti‐EGFR, pEGFR, pHER3, Akt, pAkt, Erk1/2 and pErk1/2 antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti‐c‐Met and anti‐HER3 antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti‐HER2, pHER2 and c‐Met (for reacting with the amino‐terminal domain of c‐Met for immunohistochemical analysis) antibodies were from Millipore Corp. (Billerica, MA, USA). Anti‐SNX1 and anti‐SNX2 antibodies were from BD Bioscience (San Jose, CA, USA). Anti‐GAPDH antibody was from Trevigen (Gaithersburg, MD, USA). Anti‐transferrin receptor (TfR) antibody was from Invitrogen (Carlsbad, CA, USA). Anti‐cathepsin D antibody was from Dako Cytomation (Glostrup, Denmark). The secondary goat anti‐rabbit antibody conjugated with Cy3 was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The Alexa 488‐labelled goat anti‐mouse secondary antibody was from Molecular Probes (Eugene, OR, USA). SU11274 and MG132 were purchased from Calbiochem (Darmstadt, Germany) and hydroxychloroquine and protease inhibitor cocktail were from Sigma‐Aldrich Co. (St Louis, MO, USA). Leupeptin, pepstatin A and E64d were from Peptide Institute, Inc. (Osaka, Japan) and Protein‐A coupled Sepharose 4B was purchased from Pharmacia Biotech (Uppsala, Sweden).
Silencing of SNX2 and western blotting
SiRNA corresponding to the nucleotide sequences of SNX1 (5′‐AAG AAC AAG ACC AAG AGC CAC‐3′) and SNX2#1 (5′‐AAG UCC AUC AUC UCC ACC AAG AGC CAC‐3′) and negative control siRNA were purchased from Genix Talk (Osaka, Japan). SNX2#2 (HSS110066) and SNX2#3 (HSS186027) were purchased from Invitrogen. C‐Met stealth RNAi (HSS106477) was from Invitrogen. SiRNA duplexes were transfected using Lipofectamine RNAiMAX and Opti‐MEM medium (Invitrogen) according to the manufacturer's recommendations. At 48 h after siRNA transfection, cells were rinsed with ice‐cold PBS and lysed in a lysis buffer (pH 7.5) containing 50 mmol/L Tris‐HCl, 0.1% Nonidet P‐40 (NP‐40), 350 mmol/L NaCl, 50 mmol/L NaF, 1 mmol/L Na3VO4, 5 mmol/L EDTA, 1 mmol/L PMSF and 10 μg/mL each of aprotinin and leupeptin. Lysates were subjected to SDS‐PAGE and blotted onto Immobilon membranes (Millipore Corp.). After transfer, membranes were incubated with the primary antibody and visualized with the secondary antibody coupled to horseradish peroxidase and ECL western blotting detection reagents (GE Healthcare, Piscataway, NJ, USA). The intensity of luminescence was quantified using a charge‐coupled device camera combined with an image analysis system (LAS‐4000; Fujifilm, Tokyo, Japan).
Immunoprecipitation
EBC‐1 cells were lysed in TBS‐T buffer (50 mmol/L Tris‐HCl buffer [pH 7.5], 0.15 mol/L NaCl, 1% Triton‐X100 and 0.5% deoxycholic acid) containing a protease inhibitor cocktail. After centrifugation for 15 min at 21 500 g, the supernatant was used as total cell lysate for either immunoblotting or immunoprecipitation. Protein‐A Sepharose 4B was pre‐incubated for 2 h at 4°C with appropriate antibodies. The total cell lysate was incubated with the antibody‐coupled sepharose for 20 h at 4°C and then washed five times with TBS‐T buffer. Immunoprecipitated proteins were washed with lysis buffer several times, eluted with SDS sample buffer and subjected to SDS‐PAGE.
Cell viability assay
Exponentially growing cell suspensions (3 × 103 cells/100 μL) were seeded into each well of 96‐well microtiter plates. The following day, various concentrations of gefitinib, erlotinib or SU11274 were added. After incubation for 72 h at 37°C, 20 μL of Cell Count Reagent SF (Nacalai tesque, Kyoto, Japan) was added to each well and the plates were incubated for a further 1–2 h at 37°C. Absorbance was measured at 450 nm with a 96‐well plate reader. Triplicate wells were tested at each drug concentration. The IC50 value (defined as the concentration giving a 50% reduction in absorbance) was calculated from the survival curves.
Real‐time quantitative polymerase chain reaction (qPCR)
RNA was reverse transcribed using random hexamers and avian myeloblastosis virus reverse transcriptase (Promega, Madison, WI, USA). Real‐time qPCR was performed using the Real‐time PCR system 7300 (Applied Biosystems, Foster city, CA, USA). In brief, the PCR amplification reaction mixtures (20 μL) contained cDNA, primer pairs, the dual‐labelled fluorogenic probe and the Taq Man Universal PCR Master Mix (all from Applied Biosystems). The thermal cycling conditions were 50°C for 2 min, 95°C for 10 min, and then 40 cycles of 95°C for 15 s and 60°C for 1 min. The relative gene expression for each sample was determined using the formula , which reflected the target gene expression normalized to GAPDH levels.
Immunofluorescene microscopy and antibody internalization
EBC‐1 cells transfected with siRNA were cultured for 48 h. The cells were treated with the lysosomal protease inhibitors leupeptin (40 μmol/L), pepstatin A (40 μmol/L) and E64d (40 μg/mL) during the final 20 h, fixed in −20°C methanol for 20 min and processed for immunofluorescene analysis with antibodies against c‐Met and LAMP1. The secondary goat anti‐mouse and anti‐rabbit antibodies conjugated with Cyanine Dyes (Cy) 3 or Cy5 were used at 5 mg/mL. Confocal images were obtained through Zeiss 510 meta microscopy (Carl Zeiss Microscopy GmbH, Oberkochen, Germany). For antibody internalization, EBC‐1 cells transfected with 20 μmol/L of the indicated siRNA were cultured for 48 h. The cells were then incubated in medium containing anti‐c‐Met antibody (diluted to 1/10) for 90 min in the presence of lysosomal protease inhibitors, as described above. The cells were then fixed in −20°C methanol for 20 min and processed for immunofluorescene analysis with anti‐LAMP1 antibody (green). The internalized antibody to c‐Met was detected with a Cy3‐conjugated secondary antibody (red).
Results
Expression of growth factor receptors and sensitivity to molecular‐targeted drugs in human lung cancer cells
We initially examined the expression levels of growth factor receptors and their phosphorylated forms in the following cultured human lung cancer cell lines: 11–18, harboring the activating mutation L858R; PC‐9, harboring the activating EGFR mutation delE746‐A750; and EBC‐1 and H1993, harboring c‐Met gene amplification. The cells showed various expression levels of receptors and their downstream signaling molecules including Akt and Erk 1/2 (Fig. 1A). C‐Met was highly expressed in EBC‐1 and H1993 cells.
Figure 1.
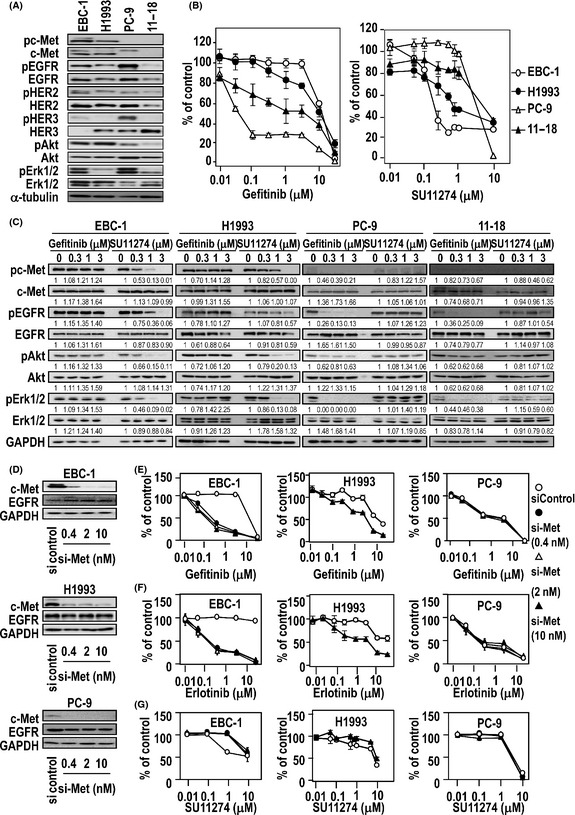
Expression of growth factor receptors and drug sensitivity to gefitinib (epidermal growth factor receptor [EGFR] tyrosine kinase inhibitor) or SU11274 (c‐Met tyrosine kinase inhibitor) in human lung cancer cell lines and altered sensitivities to gefitinib, erlotinib and SU11274 following treatment with c‐Met siRNA. (A) Expression of phosphorylated c‐Met (pc‐Met), c‐Met, phosphorylated EGFR (pEGFR), EGFR, phosphorylated human epidermal growth factor receptor (pHER) 2, human epidermal growth factor receptor (HER) 2, pHER3, HER3, phosphorylated protein kinase B (pAkt), protein kinase B (Akt), pErk1/2 and Erk1/2 determined using immunoblotting of protein lysates. Loading control, α‐tubulin. (B) Sensitivity of lung cancer cell lines to gefitinib or SU11274 determined using a cell proliferation assay. (C) Effect of gefitinib and SU11274 on expression of growth factor receptors and their downstream signaling molecules determined using immunoblotting. (D) Knockdown of c‐Met by c‐Met siRNA as determined using immunoblotting. (E–G) Sensitivity of c‐Met siRNA‐treated lung cancer cell lines to gefitinib (E), erlotinib (F) and SU11274 (G), as determined using a cell proliferation assay.
We then compared the sensitivity of these cell lines to gefitinib, which is an inhibitor of EGFR tyrosine kinase, or to SU11274, which is an inhibitor of c‐Met tyrosine kinase. Consistent with our previous study,17 both PC‐9 and 11–18 cells were highly sensitive to the cytotoxic effect of gefitinib, with IC50 values of 0.03 μmol/L and 1.86 μmol/L, respectively (Fig. 1B), whereas EBC‐1 and H1993 were highly sensitive to SU11274, with IC50 values of 0.20 and 0.97 μmol/L, respectively. Gefitinib markedly suppressed the phosphorylation of EGFR, Akt and Erk1/2 in PC‐9 and 11–18 but not EBC‐1 and H1993 cells (Fig. 1C), consistent with our previous study.17 In contrast, phosphorylation of c‐Met, Akt and Erk1/2 was markedly suppressed by SU11274 in EBC‐1 and H1993 cells (Fig. 1C).
We examined whether c‐Met knockdown could affect sensitivity to gefitinib, erlotinib or SU11274 in these cell lines. Treatment with c‐Met siRNA led to a marked decrease in the expression of c‐Met protein in EBC‐1, H1993 and PC‐9 cells (Fig. 1D). C‐Met siRNA sensitized cell proliferation in EBC‐1 and H1993 cells to EGFR‐targeted drugs (Fig. 1E,F). In contrast, c‐Met siRNA decreased sensitivity of EBC‐1 and H1993 cells to SU11274 (Fig. 1G), but did not affect the sensitivity of PC‐9 cells to these drugs.
Altered expression of growth factor receptor and downstream signaling molecules by SNX2 knockdown in lung cancer cells
Next we examined whether SNX2 knockdown affected the expression of growth factor receptors in the four lung cancer cell lines. Treatment with various doses of SNX2 siRNA (SNX2 siRNA#1) effectively suppressed the expression of SNX2 protein in all four cell lines (Fig. 2A,B). In both EBC‐1 and H1993 cells, SNX2 knockdown induced a marked decrease in c‐Met expression, but did not decrease expression of EGFR, HER2 or HER3. Also, SNX2 siRNA moderately decreased the expression of c‐Met in both PC‐9 and 11–18 cells (Fig. 2A,B). However, treatment of EBC‐1 cells with SNX2 siRNA did not alter TfR expression (Fig. 2C). Treatment with two other SNX2 siRNA (SNX2#2 and SNX2#3) also suppressed expression of both SNX2 protein and c‐Met protein in EBC‐1 cells (Fig. 2D). Therefore, we used SNX2 siRNA#1 in all experiments and herein describe it as SNX2 siRNA.
Figure 2.
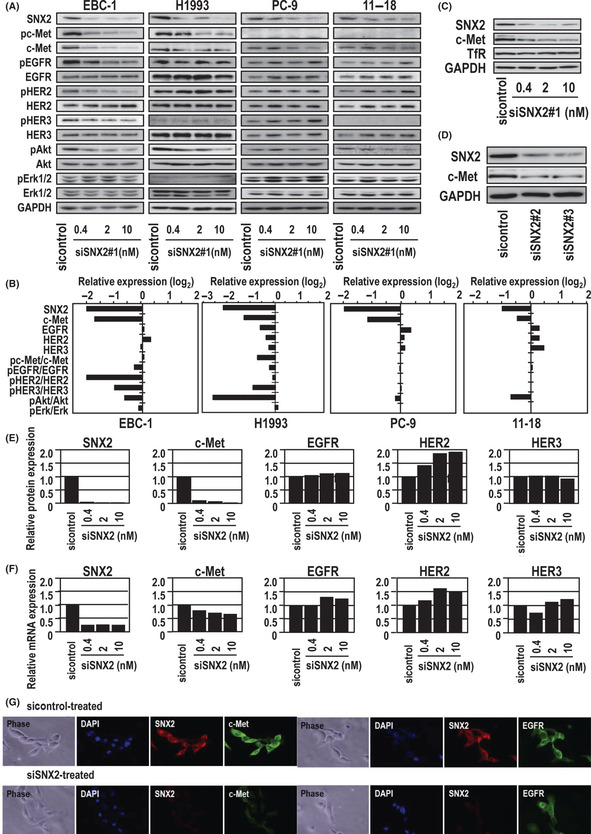
Effect of sorting nexin (SNX) 2 knockdown on expression of growth factor receptors and downstream signaling molecules. (A) Knockdown of SNX2 by SNX2 siRNA as determined using immunoblotting. (B) Relative protein expression levels normalized to GAPDH. Activities of phosphorylated c‐Met, epidermal growth factor receptor (EGFR), human epidermal growth factor receptor (HER) 2, HER3, protein kinase B (Akt) and Erk1/2 normalized to non‐phosphorylated forms. (C) Effect of SNX2 knockdown on c‐Met and transferrin receptor (TfR) expression in EBC‐1 cells as determined using immunoblotting. (D) Effect of SNX2 knockdown using two other SNX2 siRNA on c‐Met expression in EBC‐1 cells as determined using immunoblotting. (E,F) Comparison of protein and mRNA levels of growth factor receptors in EBC‐1 cells treated with SNX2 siRNA. (G) Immunofluorescence analysis of SNX2, c‐Met and EGFR expression in EBC‐1 treated with SNX2 siRNA. Nuclear‐specific staining using DAPI is also shown.
In EBC‐1 cells, SNX2 knockdown reduced the expression of SNX2 protein and mRNA by at least 80% compared with the control (Fig. 2E,F). Surprisingly, SNX2 siRNA treatment blocked the expression of c‐Met protein almost completely (Fig. 2E), whereas the expression of c‐Met mRNA was blocked by only 20–30% (Fig. 2F). However, there was no apparent effect on either the mRNA or protein expression of the EGFR family. These results suggest that the suppression of c‐Met expression by SNX2 knockdown is mainly attributable to the loss of c‐Met protein rather than mRNA expression. Immunofluorescene analysis confirmed that SNX2 siRNA almost completely inhibited c‐Met but not EGFR expression (Fig. 2G).
Effect of SNX2 knockdown on cell proliferation and sensitivity to molecular‐targeted drugs in lung cancer cells
Next we examined whether SNX2 knockdown affected cell proliferation in EBC‐1, H1993 and PC‐9 cells. SNX2 siRNA treatment strongly inhibited the proliferation of EBC‐1 cells, but had little or no effect on PC‐9 cells (Fig. 3A). Sensitivity to gefitinib was increased by SNX2 siRNA in EBC‐1 and H1993 cells, but not in PC‐9 cells (Fig. 3B). Similarly, the sensitivity of EBC‐1 and H1993 cells to erlotinib was increased by SNX2 knockdown (Fig. 3C). In contrast, SNX2 knockdown decreased the sensitivity of EBC‐1 and H1993 cells to SU11274, but did not affect the sensitivity of PC‐9 cells (Fig. 3D). However, the effect of SNX2 knockdown on cellular sensitivity to molecular‐targeted drugs was more marked in EBC‐1 cells than H1993 cells and that in 11–18 cells was almost similar to PC‐9 cells (data not shown). Taken together, these results suggest that the proliferation of EBC‐1 and H1993 cells is more dependent on the c‐Met pathway than that of PC‐9 cells.
Figure 3.

Altered drug sensitivity to gefitinib, erlotinib and SU11274 after treatment with sorting nexin (SNX) 2 siRNA. (A) Effects of SNX2 knockdown on cell proliferation. (B–D) Sensitivity of SNX2 siRNA‐treated lung cancer cell lines to gefitinib (B), erlotinib (C) and SU11274 (D), as determined using a cell viability assay.
We then compared protein expression with or without gefitinib under SNX2 knockdown conditions. Compared with the control siRNA, phosphorylation of Akt and Erk1/2 was markedly suppressed by gefitinib when treated with SNX2 siRNA (Fig. 4). Activation of downstream regulatory molecules thus seems to be highly susceptible to EGFR‐targeted drugs when the c‐Met‐driven signaling pathway is blocked by SNX2 knockdown.
Figure 4.
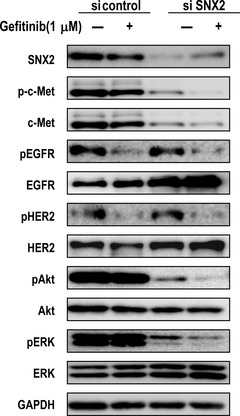
Effect of gefitinib on activation of growth factor receptors and the downstream signaling molecules in sorting nexin (SNX) 2‐silenced EBC‐1 cells. Effects of gefitinib on expression of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor (HER) 2, protein kinase B (Akt) and Erk1/2 and the phosphorylated molecules in SNX2‐silenced cells determined using immunoblotting analysis. HER, human epidermal growth factor receptor; pAkt, phosphorylated protein kinase B; pc‐Met, phosphorylated c‐Met; pEGFR, phosphorylated EGFR; pHER2, phosphorylated human epidermal growth factor receptor 2.
The structure and protein‐trafficking function of SNX2 are similar to those of SNX1. Therefore, we investigated whether SNX1 affects c‐Met expression and sensitivity to EGFR‐targeted or c‐Met‐targeted drugs in the same way as SNX2. Treatment with SNX1 siRNA effectively decreased the expression of SNX1 but not SNX2 in EBC‐1 cells (Fig. S1A). Knockdown of SNX1 did not affect the expression of c‐Met or EGFR in either EBC‐1 or PC‐9 cells (Fig. S1B) or the sensitivity to EGFR‐targeted drugs or SU11274 (Fig. S1C–E).
Inhibition of lysosomal function restores c‐Met downregulation by SNX2 knockdown
Next we examined whether the SNX2 protein directly interacts with the c‐Met protein. In EBC‐1 cells, co‐immunoprecipitation of c‐Met and SNX2 proteins was revealed (Fig. 5A), consistent with the findings of a previous study by Schaaf et al.16 In contrast, no co‐immunoprecipitation of c‐Met proteins and SNX1 was observed following immunoblotting with a SNX1‐specific antibody (Fig. 5A).
Figure 5.
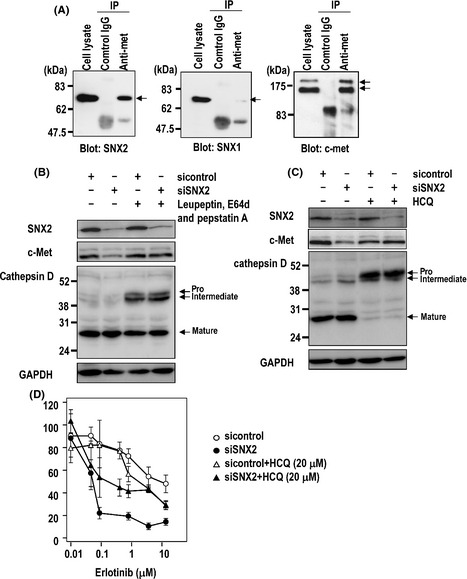
Co‐immunoprecipitation (IP) of sorting nexin (SNX) 2 and c‐Met and the effect of lysosomal inhibitors on expression of c‐Met by SNX2 knockdown in EBC‐1 cells. (A) Immunoprecipitation with anti‐c‐Met antibody and blotting with anti‐SNX1, SNX2 and c‐Met antibody. (B,C) Effect of lysosome inhibitors on c‐Met expression with or without SNX2 siRNA. (B) EBC‐1 cells were transfected with siRNA, cultured for 48 h and treated with lysosome inhibitors during the final 20 h. (C) Cells were transfected with siRNA, cultured for 48 h and treated with hydroxychloroquine (HCQ) during the final 42 h. Protein extracts were resolved using immunoblotting and probed with various antibodies. (D) Cells were transfected with siRNA, cultured for 24 h and treated with erlotinib and HCQ. After 72 h, the number of viable cells was counted.
We examined whether various inhibitors of membrane trafficking could restore the downregulation of c‐Met by SNX2 siRNA. Treatment with the calcium ionophore A23187 and the matrix metalloproteinase inhibitor GM6001 did not restore SNX2 siRNA‐induced downregulation of c‐Met protein (data not shown). The proteasome inhibitor MG132 partially restored the expression of SNX2 protein without affecting that of c‐Met protein (data not shown). As shown in Figure 5(B,C), treatment with the endosome/lysosome inhibitor leupeptin, pepstatin A, E64d or hydroxychloroquine markedly restored c‐Met but not SNX2 expression; cathepsin D expression was also restored using this treatment because maturation of cathepsin D is known to be processed in lysosome.
We further examined whether an endosome/lysosome inhibitor could annihilate the inhibitory effect of an EGFR‐targeted drug on cell proliferation. Treatment with hydroxychloroquine was found to reduce the inhibitory effect of erlotinib under SNX2 knockdown conditions in EBC‐1 cells compared with control conditions (Fig. 5D). Under SNX2 knockdown conditions, the inhibition of lysosome function thus restored c‐Met expression, resulting in cancellation of the sensitization to erlotinib.
Localization of c‐Met in the lysosomal compartment by SNX2 knockdown in the presence of lysosome inhibitors
We further examined which compartment was responsible for the degradation of c‐Met under SNX2‐knockdown conditions. As c‐Met expression was almost completely inhibited following SNX2 knockdown, we used the lysosomal protease inhibitors leupeptin, pepstatin A and E64d to examine the localization of c‐Met using confocal microscopy (Fig. 6). Under control conditions (control siRNA), c‐Met exclusively localized to the cell surface, but could not readily be detected in the intracellular compartments such as late endosomes and lysosomes, as indicated by LAMP1 even in the presence of lysosomal protease inhibitors (Fig. 6A). In contrast, following SNX2 siRNA transfection, c‐Met was not expressed on cell‐surface membranes but was localized to the late endosomes and lysosomes in the presence of lysosome inhibitors (Fig. 6A, arrows). Incubation with the anti‐c‐Met antibody following control siRNA resulted in exclusive retention of the antibody at the cell surface, suggesting that there was little internalization and trafficking of c‐Met to late endosomes and lysosomes (Fig. 6B). However, under SNX2‐silencing conditions, the internalized antibody co‐localized with LAMP1 (Fig. 6B, arrows). Together, these results suggest that the c‐Met protein is internalized from the cell surface and transported to late endosomes and lysosomes when SNX2 is depleted.
Figure 6.
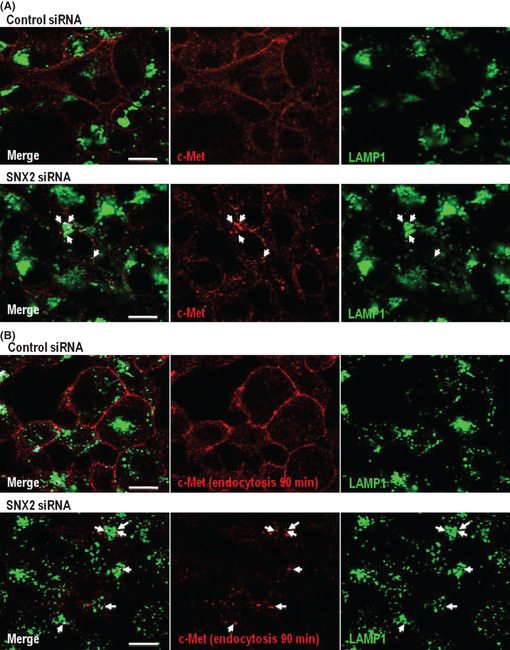
Localization of c‐Met protein in late endosome and/or lysosome by sorting nexin (SNX) 2 knockdown. Effect of SNX2 knockdown on c‐Met expression in late endosome and/or lysosome. (A) Under SNX2 knockdown in the presence of lysosomal protease inhibitors, c‐Met co‐localized with lysosomal‐associated membrane protein 1 (LAMP1) (arrows). (B) Under SNX2 knockdown in the presence of anti‐c‐Met antibody with lysosomal protease inhibitors for another 90 min, internalized antibody against c‐Met also co‐localized with LAMP1 (arrows).
Overcoming the effect of gefitinib resistance in a lung cancer cell line harboring both activated EGFR mutation and c‐Met amplification by SNX2 knockdown
Last, we examined whether SNX2 knockdown overcomes drug resistance in lung cancer cell line GR5 cells harboring c‐Met amplification and activation mutation of EGFR.18 As shown in Figure 7(A), GR5 cells showed higher drug resistance to gefitinib and also to SU11274, while parental HCC827cells showed higher sensitivity to gefitinib and higher resistance to SU11274. Also, GR5 cells showed enhanced expression of c‐Met and HER3 compared with HCC827 cells (Fig. 7B). Treatment with SNX2 siRNA decreased expression of c‐Met but not expression of EGFR and HER3 (Fig. 7C). However, gefitinib markedly suppressed Akt phosphorylation under SNX2 knockdown conditions in GR5 cells. Cellular sensitivity to gefitinib was increased by SNX2 siRNA and also by c‐Met siRNA in GR5 cells (Fig. 7D). Thus, SNX2 knockdown seems to overcome drug resistance to gefitinib in cancer cells harboring c‐Met amplification.
Figure 7.
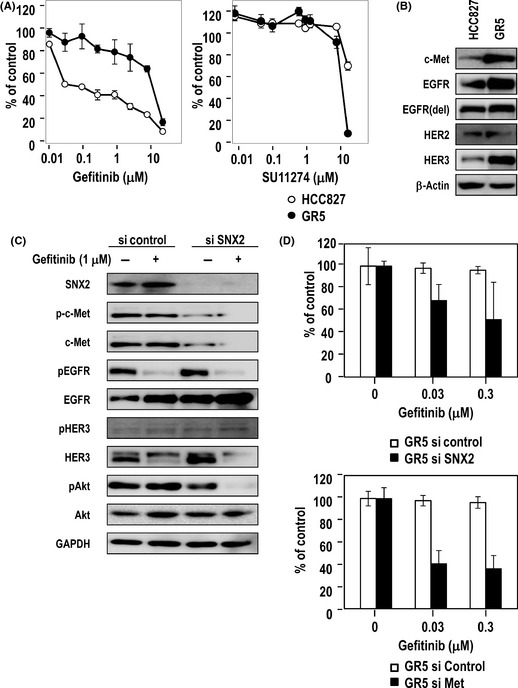
Reversal effect of sorting nexin (SNX) 2 knockdown on gefitinib resistance in a lung cancer cell line harboring both activated epidermal growth factor receptor (EGFR) mutation and c‐Met amplification. (A) Sensitivity of HCC827 and its gefitinib‐resistant counterpart GR5 cells to gefitinib or SU11274 using a cell viability assay. (B) Expression of growth factor receptors determined using immunoblotting. Loading control, β‐actin. (C) Effect of gefitinib on expression of phosphorylated c‐Met (pc‐Met), phosphorylated EGFR (pEGFR), phosphorylated human epidermal growth factor receptor (pHER) 2, pHER3, phosphorylated protein kinase B (pAkt) and pErk1/2 in SNX2‐silenced GR5 cells determined using immunoblotting. (D) Altered drug sensitivity to gefitinib after treatment with SNX2 siRNA or c‐Met siRNA, as determined using a cell viability assay.
Discussion
The present study demonstrated that expression of c‐Met was specifically downregulated by SNX2 knockdown in four human lung cancer cell lines. This was accompanied by suppression of the activation of PI3K/Akt in EBC‐1 and H1993 but not PC‐9 or 11–18 cells (Fig. 2A). Of these four lung cancer cell lines, c‐Met was amplified with wild‐type EGFR in two cell lines, EBC‐1 and H1993, which is consistent with a previous study.17 The results from our present study also show that growth of EBC‐1 or H1993 cells is closely dependent on the c‐Met pathway, because they were highly susceptible to SNX2 siRNA‐induced downregulation of c‐Met. This also explains why EBC‐1 and H1993 cells show high sensitivity to SU11274 but not to gefitinib or erlotinib (Fig. 8). In contrast, the growth of PC‐9 cells was most closely dependent on the EGFR pathway, due to the presence of an activating mutation in exon 19 of the EGFR gene that sensitizes the cells to gefitinib.3, 17 Consistent with these previous reports, PC‐9 cells showed higher sensitivity to gefitinib and lower sensitivity to SU11274 than EBC‐1 or H1993 cells. Furthermore, 11–18 cells, which harbor an activating mutation in exon 21 of the EGFR gene, also showed higher sensitivity to gefitinib and lower sensitivity to SU11274 than EBC‐1 cells. The growth of PC‐9 or 11–18 cells was not affected by SNX2 siRNA, which suppressed the expression of c‐Met but not that of EGFR.
Figure 8.
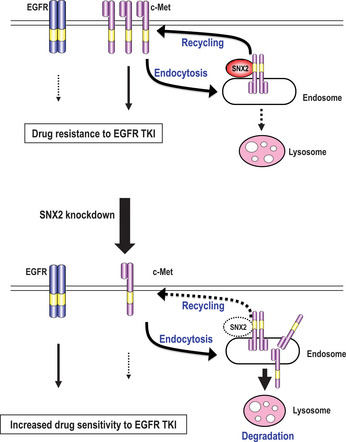
Schematic model of sensitization to epidermal growth factor receptor (EGFR)‐targeted drugs by sorting nexin (SNX) 2 knockdown in lung cancer cells. SNX2 promotes membrane trafficking of c‐Met from recycling or early endosomes to the plasma membrane, resulting in activation of the c‐Met signaling pathway and in drug resistance to EGFR tyrosine kinase inhibitors (TKI). Under SNX2 knockdown, expression of c‐Met is downregulated through blockage of the SNX2‐mediated recycling pathway together with promotion of the lysosome‐induced degradation pathway. C‐Met degradation by SNX2 knockdown is accompanied by compensatory activation of EGFR, resulting in a gain of drug sensitivity to EGFR TKI.
C‐Met amplification is one of the resistance mechanisms in EGFR‐TKI. To demonstrate the importance of SNX2 in EGFR‐TKI resistance, we used GR5 cells that harbor both EGFR mutation and c‐Met amplification. As drug resistance to gefitinib in GR5 cells is expected to be mediated partially through c‐Met amplification, SNX2 knockdown reduced expression of c‐Met in GR5 cells accompanied by increased sensitivity to gefitinib. Akt phosphorylation was also suppressed by gefitinib under SNX2 knockdown (Fig. 7C). This overcoming effect of gefitinib resistance by SNX2 knockdown strongly suggests an important role for SNX2 in acquiring drug resistance to EGFR‐targeted drugs via c‐Met amplification.
It was reported that SNX1 interacts with EGFR and EGFR degradation is enhanced in cells overexpressing SNX1,8 suggesting its role for late endosome‐to‐lysosome trafficking.8 However, in the present study we did not observe any apparent change in the expression of c‐Met and EGFR or in the sensitivity to gefitinib in either EBC‐1 or PC‐9 cells following SNX1 knockdown (Fig. S1). This suggests that SNX1 does not play an essential role in the expression of EGFR and c‐Met in our lung cancer cell lines. Gullapalli and colleagues previously reported that both SNX1 and SNX2 are involved in endosome‐to‐lysosome trafficking of EGFR and that EGFR degradation is also blocked by SNX2 knockdown, which enhances the expression of HER2 but not that of EGFR or HER3.14 Together, these observations indicate that regulation of EGFR and its family proteins by SNX2 depletion is cell‐type dependent and possibly involves other molecules.9
The specific control of c‐Met by SNX2 is of particular interest. On stimulation by HGF, c‐Met is rapidly internalized and transported through an early endosome followed by a late endosome and then it is degraded by the proteasome or non‐proteasome pathway.19, 20, 21 Schaaf et al.16 first identified SNX2 as one of the various molecules that interact with c‐Met. SNX2 is often localized in the early endosome and co‐localizes with growth factor receptors such as EGFR and c‐Met.14, 22 It was reported that intracellular degradation of c‐Met by HGF stimulation was almost completely blocked by proteasome inhibitors such as concanamycin and lactacystin/MG132.19, 20 In the present study, downregulation of c‐Met by SNX2 knockdown was not restored by MG132 (data not shown). Therefore, it seems less likely that the proteasome degradation pathway plays a key role in c‐Met downregulation by SNX2 knockdown.
Western blot analysis showed that SNX2 siRNA induced downregulation of c‐Met, but not EGFR, HER2, HER3 or TfR, suggesting selective downregulation of c‐Met expression (Fig. 2). Although treatment with SNX2 siRNA resulted in a marked decrease in c‐Met at the cell surface and degradation in late endosomes and lysosomes, localization of both EGFR and TfR was not affected (data not shown). Marked restoring of c‐Met protein expression occurred with treatment by lysosomal inhibitors following SNX2 knockdown. As the SNX2 silencing conditions led to c‐Met accumulation in late endosomes and lysosomes (Fig. 5A,B), the specific interaction between SNX2 and c‐Met might be responsible for the rapid recycling of the c‐Met protein to the cell surface from early endosomes (Fig. 8). Consistent with these biological cell findings, a cell proliferation assay demonstrated an apparent reduction of sensitivity to erlotinib when treated with a lysosomal/endosomal inhibitor (Fig. 5D), suggesting an important role for SNX2‐driven c‐Met trafficking in cellular sensitivity to EGFR‐targeted drugs. In contrast, Casitas B lineage lymphoma (Cbl) protein was previously shown to limit the initial endocytotic step of EGFR and other growth factor receptors,23 while the activity of the EGFR signaling complex decreased in a coordinate manner with Cbl after the exogenous addition of EGF/TGF‐α.24 Cbl might therefore be involved in the downregulation of c‐Met by SNX2 knockdown. However, our previous study found no expression of Cbl protein in EBC‐1 cells,17 suggesting that Cbl is less likely to play any critical role. Further study is required to understand the SNX2/c‐Met interaction and how this complex acts in close context with the endosome‐lysosome pathway.
In both EBC‐1 and H1993 cells, knockdown of SNX2 promotes compensatory activation of the EGFR pathway, suggesting that downregulation of c‐Met results in a gain of sensitivity to gefitinib. Knockdown of c‐Met also renders EBC‐1 and H1993 cells sensitive to gefitinib (Fig. 8). Engelman and Jänne reported acquired resistance to gefitinib or erlotinib in lung cancer patients through gain of c‐Met overexpression.5 Bean and colleagues reported that NSCLC patients refractory to EGFR‐targeted drugs harbor a representative secondary EGFR mutation of T790M, as well as c‐Met gene amplification.25 Furthermore, one could ask how the underlying mechanism of SNX2 knockdown‐induced c‐Met downregulation is correlated with increased sensitivity to gefitinib or erlotinib. SNX2 knockdown blocked activation of PI3K/Akt and also phosphorylation of EGFR, HER2 and HER3 in EBC‐1 and H1993 cells (Fig. 2A,B). In both cell lines, cell proliferation and drug sensitivity to gefitinib, erlotinib and SU11274 were affected by SNX2 knockdown (Fig. 3). Figure 4 showed more marked inhibition of Akt and Erk1/2 phosphorylation by gefitinib when SNX2 was silenced. Taken together, SNX2 knockdown‐induced c‐Met downregulation might activate the alternative EGFR/HER2/HER3‐driven PI3K/Akt signaling pathway resulting in sensitization of cancer cells to EGFR‐targeted drugs.
Compartmentalization of signals by endocytosis of growth factors has been recently reported to determine various cell functions of normal and cancer cells.26, 27, 28 In the concept of ‘signaling endosome’, the endocytic process might play a key role in cell migration, cell survival and malignant transformation.29, 30 Overexpression of a clathrin‐associated protein named Huntingtin interacting protein 1 altered EGFR trafficking and also tumorigenesis by cancer cells.31 A recent study by Joffre et al.32 has first presented a close link between malignant transformation and impaired endosomal degradation. C‐Met activating mutations such as M1268T and D1246N exhibited increased endocytosis or recycling activity and decreased levels of degradation. These two activating mutations in the kinase domain of c‐Met lead to increased levels of c‐Met endocytosis/recycling and reduced levels of c‐Met degradation, resulting in malignant transformation, and the blocking of endocytotic degradation of mutant c‐Met suppresses cell migration and metastasis by cancer cells.32 Membrane trafficking of c‐Met protein might therefore play an important role not only in malignant transformation and metastasis32 but also in determination of drug sensitivity to molecular‐targeted drugs (the present study).
In conclusion, SNX2 was shown to control expression and localization of c‐Met, possibly through the endosome‐lysosome pathway. Downregulation of c‐Met expression by SNX2 knockdown resulted in a marked loss of sensitivity to a c‐Met‐targeted drug, leading to activation of alternatives to the EGFR signaling pathway. These results indicate that SNX2 could be a novel molecular target in the development of novel optimized anticancer treatments.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Drug sensitivities to gefitinib, erlotinib and SU11274 under sorting nexin (SNX) 1 knockdown. (A) Effect of SNX1 knockdown by SNX1 siRNA, as determined using immunoblotting. (B) Effect of SNX1 knockdown on expression of c‐Met and EGFR in EBC‐1 and PC‐9 cells, as determined using immunoblotting. (C–E) Drug sensitivity of SNX1 siRNA‐treated EBC‐1 and PC‐9 cells to gefitinib (C), erlotinib (D) and SU11274 (E), as determined using a cell viability assay.
Acknowledgments
This research is supported by the 3rd Term Comprehensive Control Research for Cancer from the Ministry of Health, Labour and Welfare, Japan (M. Kuwano). We thank Ms Eri Kajita for editorial support.
(Cancer Sci 2013; 104: 573–583)
References
- 1. Spiro SG, Silvestri GA. One hundred years of lung cancer. Am J Respir Crit Care Med 2005; 172: 523–9. [DOI] [PubMed] [Google Scholar]
- 2. Lynch TJ, Bell DW, Sordella R et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 3. Paez JG, Jänne PA, Lee JC et al EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 4. Pao W, Miller V, Zakowski M et al EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non‐small cell lung cancer. Clin Cancer Res 2008; 14: 2895–9. [DOI] [PubMed] [Google Scholar]
- 6. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 7. Yano S, Wang W, Li Q et al Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res 2008; 68: 9479–87. [DOI] [PubMed] [Google Scholar]
- 8. Kurten RC, Cadena DL, Gill GN. Enhanced degradation of EGF receptors by a sorting nexin, SNX1. Science 1996; 272: 1008–10. [DOI] [PubMed] [Google Scholar]
- 9. Worby CA, Dixon JE. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol 2002; 3: 919–31. [DOI] [PubMed] [Google Scholar]
- 10. Haft CR, de la Luz Sierra M, Barr VA, Haft DH, Taylor SI. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol Cell Biol 1998; 18: 7278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Griffin CT, Trejo J, Magnuson T. Genetic evidence for a mammalian retromer complex containing sorting nexins 1 and 2. Proc Natl Acad Sci U S A 2005; 102: 15173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cozier GE, Carlton J, McGregor AH et al The phox homology (PX) domain‐dependent, 3‐phosphoinositide‐mediated association of sorting nexin‐1 with an early sorting endosomal compartment is required for its ability to regulate epidermal growth factor receptor degradation. J Biol Chem 2002; 277: 48730–6. [DOI] [PubMed] [Google Scholar]
- 13. Carlton J, Bujny M, Peter BJ et al Sorting nexin‐1 mediates tubular endosome‐to‐TGN transport through coincidence sensing of high‐curvature membranes and 3‐phosphoinositides. Curr Biol 2004; 14: 1791–800. [DOI] [PubMed] [Google Scholar]
- 14. Gullapalli A, Garrett TA, Paing MM, Griffin CT, Yang Y, Trejo J. A role for sorting nexin 2 in epidermal growth factor receptor down‐regulation: evidence for distinct functions of sorting nexin 1 and 2 in protein trafficking. Mol Biol Cell 2004; 15: 2143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cavet ME, Pang J, Yin G, Berk BC. An epidermal growth factor (EGF)‐dependent interaction between GIT1 and sorting nexin 6 promotes degradation of the EGF receptor. FASEB J 2008; 22: 3607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schaaf CP, Benzing J, Schmitt T et al Novel interaction partners of the TPR/MET tyrosine kinase. FASEB J 2005; 19: 267–9. [DOI] [PubMed] [Google Scholar]
- 17. Ono M, Hirata A, Kometani T et al Sensitivity to gefitinib (Iressa, ZD1839) in non‐small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal‐regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther 2004; 3: 465–72. [PubMed] [Google Scholar]
- 18. Yoshida T, Okamoto I, Okamoto W et al Effects of Src inhibitors on cell growth and epidermal growth factor receptor and MET signaling in gefitinib‐resistant non‐small cell lung cancer cells with acquired MET amplification. Cancer Sci 2010; 101: 167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hammond DE, Urbé S, Vande Woude GF, Clague MJ. Down‐regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001; 20: 2761–70. [DOI] [PubMed] [Google Scholar]
- 20. Kermorgant S, Zicha D, Parker PJ. Protein kinase C controls microtubule‐based traffic but not proteasomal degradation of c‐Met. J Biol Chem 2003; 278: 28921–9. [DOI] [PubMed] [Google Scholar]
- 21. Foveau B, Ancot F, Leroy C et al Down‐regulation of the met receptor tyrosine kinase by presenilin‐dependent regulated intramembrane proteolysis. Mol Biol Cell 2009; 20: 2495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carlton JG, Bujny MV, Peter BJ et al Sorting nexin‐2 is associated with tubular elements of the early endosome, but is not essential for retromer‐mediated endosome‐to‐TGN transport. J Cell Sci 2005; 118: 4527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer 2001; 37: 3–8. [DOI] [PubMed] [Google Scholar]
- 24. Ettenberg SA, Magnifico A, Cuello M, et al Cbl‐b‐dependent coordinated degradation of the epidermal growth factor receptor signaling complex. J Biol Chem 2001; 276: 27677–84. [DOI] [PubMed] [Google Scholar]
- 25. Bean J, Brennan C, Shih JY et al MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA 2007; 104: 20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Teis D, Wunderlich W, Huber LA. Localization of the MP1‐MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev Cell 2002; 3: 803–14. [DOI] [PubMed] [Google Scholar]
- 27. Palamidessi A, Frittoli E, Garré M et al Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell 2008; 134: 135–47. [DOI] [PubMed] [Google Scholar]
- 28. Schenck A, Goto‐Silva L, Collinet C et al The endosomal protein App l1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 2008; 133: 486–97. [DOI] [PubMed] [Google Scholar]
- 29. Le Roy C, Wrana JL. Signaling and endocytosis: a team effort for cell migration. Dev Cell 2005; 9: 167–8. [DOI] [PubMed] [Google Scholar]
- 30. Polo S, Di Fiore PP. Endocytosis conducts the cell signaling orchestra. Cell 2006; 124: 897–900. [DOI] [PubMed] [Google Scholar]
- 31. Rao DS, Bradley SV, Kumar PD et al Altered receptor trafficking in Huntingtin Interacting Protein 1‐transformed cells. Cancer Cell 2003; 3: 471–82. [DOI] [PubMed] [Google Scholar]
- 32. Joffre C, Barrow R, Ménard L, Calleja V, Hart IR, Kermorgant S. A direct role for Met endocytosis in tumorigenesis. Nat Cell Biol 2011; 13: 827–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Drug sensitivities to gefitinib, erlotinib and SU11274 under sorting nexin (SNX) 1 knockdown. (A) Effect of SNX1 knockdown by SNX1 siRNA, as determined using immunoblotting. (B) Effect of SNX1 knockdown on expression of c‐Met and EGFR in EBC‐1 and PC‐9 cells, as determined using immunoblotting. (C–E) Drug sensitivity of SNX1 siRNA‐treated EBC‐1 and PC‐9 cells to gefitinib (C), erlotinib (D) and SU11274 (E), as determined using a cell viability assay.