

Abstract
Cytarabine (ara‐C) is the key agent for treating acute myeloid leukemia. After being transported into leukemic cells, ara‐C is phosphorylated, by several enzymes including deoxycytidine kinase (dCK), to ara‐C triphosphate (ara‐CTP), an active metabolite, and then incorporated into DNA, thereby inhibiting DNA synthesis. Therefore, the cytotoxicity of ara‐C depends on the production of ara‐CTP and the induction of apoptosis. Here, we established a new ara‐C‐resistant acute myeloid leukemia cell line (HL‐60/ara‐C60) with dual resistance characteristics of the anti‐antimetabolic character of decreased ara‐CTP production and an increase in the antiapoptotic factors Bcl‐2 and Bcl‐XL. We further attempted to overcome resistance by augmenting ara‐CTP production and stimulating apoptosis. A relatively new nucleoside analog, 9‐β‐d‐arabinofuranosylguanine (ara‐G), and the small molecule Bcl‐2 antagonist YC137 were used for this purpose. HL‐60/ara‐C60 was 60‐fold more ara‐C‐resistant than the parental HL‐60 cells. HL‐60/ara‐C60 cells exhibited low dCK protein expression, which resulted in decreased ara‐CTP production. HL‐60/ara‐C60 cells were also refractory to ara‐C‐induced apoptosis due to overexpression of Bcl‐2 and Bcl‐XL. Combination treatment of ara‐C with ara‐G augmented the dCK protein level, thereby increasing ara‐CTP production and subsequent cytotoxicity. Moreover, the combination of ara‐C with YC137 produced a greater amount of apoptosis than ara‐C alone. Importantly, the three‐drug combination of ara‐C, ara‐G and YC137 provided greater cytotoxicity than ara‐C+ara‐G or ara‐C+YC137. These findings suggest possible combination strategies for overcoming ara‐C resistance by augmenting ara‐CTP production and reversing refractoriness against the induction of apoptosis in ara‐C resistant leukemic cells.
Cytarabine (1‐β‐d‐arabinofuranosylcytosine; ara‐C), a pyrimidine nucleoside analog, is key in the treatment of acute myeloid leukemia (AML).1, 2, 3 Standard induction therapy, which consists of conventional doses of ara‐C for 7 days plus anthracycline for 3 days, provides remission rates of over 70% for younger patients with AML.1, 2, 3, 4, 5 Nevertheless, only 40% of patients are long‐term survivors and most relapse with the development of drug resistance. Thus, overcoming ara‐C resistance is essential to improve clinical outcomes.1, 2, 3, 4, 5, 6
Ara‐C is transported into leukemic cells by membrane transporters including the human equilibrative nucleoside transporter 1 (hENT1).7 Inside the cell, ara‐C is phosphorylated to ara‐C 5′‐monophoshate by the rate‐limiting enzyme deoxycytidine kinase (dCK) and subsequently to ara‐C 5′‐triphosphate (ara‐CTP), an active metabolite of ara‐C. Ara‐CTP is then incorporated into DNA strands in the S phase of the cell cycle, resulting in the inhibition of DNA synthesis and the consequent induction of apoptosis.1, 8, 9, 10, 11
We have extensively investigated the mechanisms of resistance to ara‐C to improve therapeutic efficacy.12, 13, 14, 15, 16, 17, 18 Because the toxicity of ara‐C depends on its cellular activation to ara‐CTP and the induction of apoptosis, cellular factors that can regulate ara‐C activation and the apoptotic pathway may be critical in ara‐C resistance. Thus, we hypothesized that strategies that could enhance ara‐CTP production and stimulate apoptosis would augment ara‐C's cytotoxicity and overcome resistance to it in leukemic cells.
Here, we report that we have established a unique new ara‐C‐resistant AML cell line with coexpression of anti‐antimetabolic and antiapoptotic factors. We further attempted to overcome ara‐C resistance by augmenting ara‐CTP production and stimulating apoptosis. A relatively new nucleoside analog, 9‐β‐d‐arabinofuranosylguanine (ara‐G, the metabolite of nelarabine), and the small molecule apoptosis inhibitor YC137 were used for this purpose. Ara‐G is a purine nucleoside analog that is similar to ara‐C but that has a slightly different activation pathway.19, 20, 21, 22 YC137 disrupts the function of the antiapoptotic molecule Bcl‐2, inducing cytochrome c release from mitochondria and activating caspase‐9. YC137 has been shown to induce apoptosis of hematopoietic progenitor cells overexpressing Bcl‐2.23, 24
Materials and Methods
Chemicals and reagents
Ara‐C was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Ara‐G was purchased from RI Chemical (Orange, CA, USA). YC137 was purchased from Calbiochem (Darmstadt, Germany). All other chemicals were of analytical grade.
Development of an ara‐C‐resistant leukemic cell line
Human leukemia HL‐60 cells were cultured in RPMI1640 media supplemented with 10% heat‐inactivated FBS at 37°C in humidified air containing 5% carbon dioxide. To develop an ara‐C‐resistant HL‐60 variant, parental HL‐60 cells were cultured in media containing ara‐C. The cultures were observed daily and allowed to grow. Drug concentrations were gradually increased on subsequent passages, and an ara‐C‐resistant subclone (HL‐60/ara‐C60) was isolated using the limiting‐dilution method. HL‐60/AD cells were established to be ara‐C‐resistant and possess Bcl‐2 overexpression in our previous study.13
Growth inhibition assay
HL‐60 cells and HL‐60/ara‐C60 cells (1 × 105/mL) were incubated for 72 h with different concentrations of anticancer agents. The growth‐inhibitory effects were evaluated using either the trypan blue dye exclusion assay or the sodium 3′‐(1‐[(phenylamino)‐carbonyl‐3,4‐tetrazolium])‐bis(4‐methoxy‐6‐nitro)benzene sulfonic acid hydrate (XTT) assay, according to the manufacturer's instructions (Roche Diagnostics, Indianapolis, IN, USA) with slight modifications.18
Determination of intracellular production of ara‐CTP and ara‐GTP
The acid‐soluble fraction (nucleotide pool) was extracted from HL‐60 cells or HL‐60/ara‐C60 cells (1 × 106/mL, 10 mL), treated or untreated. High performance liquid chromatography was then used to determine intracellular ara‐CTP and ara‐GTP as described in previous studies.12, 15, 25
Nucleoside transport capacity
To evaluate the capacity of membrane nucleoside transporters, ara‐C uptake was quantified using the method of Wiley et al. with slight modifications.7, 16 Non‐facilitated drug uptake was determined in the presence of 3 μM nitrobenzylthioinosine (Sigma‐Aldrich), which interferes with the function of membrane nucleoside transporters. The capacity of the transporter was determined as the difference between drug uptake in the absence and presence of nitrobenzylthioinosine.
Determination of dCK and apoptosis‐related protein expression using Western blotting
Protein expression levels of dCK and apoptosis‐related factors were determined using Western blot analysis as described previously.18 Mouse monoclonal anti‐dCK, which was developed in the Department of Pediatrics, Mie University School of Medicine26 and rabbit polyclonal anti‐Bad, rabbit polyclonal anti‐Bcl‐2, rabbit polyclonal anti‐Bcl‐XL antibodies (Cell Signaling Technology, Beverly, MA, USA) and anti‐actin antibody (Sigma‐Aldrich) were used as the primary antibodies. Anti‐mouse IgG antibody and anti‐rabbit IgG‐horseradish peroxidase‐conjugated antibody (Amersham Biosciences, Bucks, UK) were used as the secondary antibody. The band density was measured using a Dolphin‐View Band Tool software (Kurabo Industries, Osaka, Japan).
Determination of apoptotic cell death using nuclear staining with Hoechst 33342 and flow cytometry
Treated or untreated cells (1 × 105/mL) were incubated with 20 μg/mL Hoechst 33342 at room temperature for 15 min. The samples were observed by fluorescence microscopy, and cells with apoptotic morphology were determined by counting 100 cells per treatment. Apoptosis was also analyzed using an Annexin‐V‐FLUOS Staining Kit (Roche Diagnostics, Indianapolis, IN, USA) using FACSCanto II (BD Bioscience, Franklin Lakes, NJ, USA).
Statistical analyses
All statistical analyses were performed using Microsoft Excel 2007 software (Microsoft, Redmond, WA, USA). All graphs, curves and columns were generated using GraphPad Prism software (version 5.0) (GraphPad Software, San Diego, CA, USA). Values of P ≤ 0.05 were considered statistically significant.
Results
Establishment of ara‐C‐resistant leukemic cells
The XTT assay demonstrated that HL‐60/ara‐C60 cells were about 60‐fold more resistant to ara‐C than HL‐60 cells (Table 1). HL‐60/ara‐C60 cells exhibited cross resistance to the similar nucleoside analogs gemcitabine (dFdC) and cladribine (2CdA) (Table 1). Thus, the ara‐C‐resistant subclone (HL‐60/ara‐C60) was successfully established.
Table 1.
Sensitivity of HL‐60 cells and HL‐60/ara‐C60 cells to anticancer agents
| IC50 (μM) | |||
|---|---|---|---|
| HL‐60 | HL‐60/ara‐C60 | RR | |
| Ara‐C | 0.0800 ± 0.0030 | 4.9100 ± 0.2300 | 61.0 |
| dFdC | 0.0040 ± 0.0002 | 0.1200 ± 0.0300 | 30.0 |
| 2CdA | 0.0700 ± 0.0060 | 17.10 ± 2.8200 | 244.0 |
| Ara‐G | >1000 | >1000 | – |
| Ara‐C + Ara‐G | 0.1000 ± 0.0800 | 0.2300 ± 0.0500 | 2.3 |
| CPT‐11 | 0.0080 ± 0.0002 | 0.0400 ± 0.0040 | 5.0 |
Cells were treated with each agent for 72 h, followed by the determination of IC50 using the XTT assay. RR, relative resistance calculated as the ratio of the IC50 of HL‐60/ara‐C60 cells relative to that of HL‐60 cells. Ara‐C, cytarabine; dFdC, gemcitabine; 2CdA, cladribine; CPT‐11, irinotecan; ara‐G, 9‐β‐d‐arabinofuranosylguanine; ND, not determined due to insensitivity to ara‐G. Values for IC50 are the means ± SD.
Intracellular ara‐CTP production and ara‐C‐related factors
Intracellular ara‐CTP is a surrogate marker of ara‐C‐induced cytotoxicity, so the production of ara‐CTP was evaluated in both cell lines. When cells were incubated for 6 h with different concentrations of ara‐C, the production of ara‐CTP increased in a concentration‐dependent manner (Fig. 1a). However, the ara‐CTP level was significantly lower in HL‐60/ara‐C60 cells than HL‐60 cells, suggesting that ara‐CTP is critical to the sensitivity of cells to ara‐C.
Figure 1.
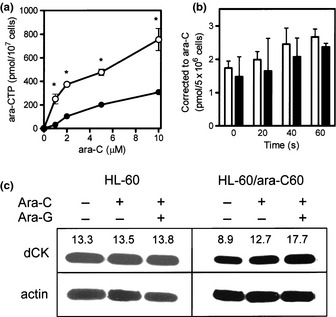
(a) Production of intracellular ara‐C triphosphate (ara‐CTP). Cells (1 × 106 cells/mL, 10 mL) were incubated for 4 h with 1, 2, 5, 10 μM ara‐C. The acid‐soluble fraction was extracted from each sample and applied to high performance liquid chromatography (HPLC). O, HL60, •; HL60/ara‐C60. Each value represents the mean ± SD of at least three independent experiments. *P ≤ 0.05 (b) Transport of ara‐C into parental and ara‐C‐resistant cells. Membrane nucleoside transport of ara‐C was assessed by pulsing the cells with 0.32 μM tritiated ara‐C for 0, 20, 40, and 60 s, followed by quantification of cellular drug uptake by scintillation counting. Open bars, HL‐60 cells; closed bars, HL‐60/ara‐C60 cells. Each value represents the mean ± SD of at least three independent experiments. (c) dCK protein expression. The cells (1 × 106 cells/mL, 10 mL) were incubated with 10 μM ara‐G for 2 h (or not), followed by washing in media and incubation with 10 μM ara‐C for 3 h. dCK protein expression was determined using Western blot analysis. The value of densitometer added to above dCK protein band.
For successful production of ara‐CTP, ara‐C must be transported into cells and phosphorylated to the ara‐C nucleotide. When cells were pulsed with ara‐C, the drug was rapidly incorporated in both cell lines (Fig. 1b). However, the analog uptake was a little lower in HL‐60/ara‐C60 cells than HL‐60 cells. Moreover, HL‐60/ara‐C60 cells exhibited lower expression of the rate‐limiting enzyme dCK than HL‐60 cells (Fig. 1c). Therefore, these results suggest that the development of ara‐C resistance in HL‐60/ara‐C60 cells was in part attributable to altered ara‐C‐related factors that resulted in the cells' low capability for ara‐CTP production.
Sensitization of the ara‐C‐resistant variant by ara‐G
One strategy that increases the intracellular ara‐CTP concentration is pre‐treatment with a purine nucleoside analog such as fludarabine (9‐β‐d‐arabinofuranosyl‐2‐fluoroadenine‐5′‐monophosphate, F‐ara‐AMP).27, 28 Theoretically, the intracellular active metabolite F‐ara‐A triphosphate inhibits ribonucleotide reductase, thereby reducing the dCTP pool. This stimulates dCK activity, which then enhances the production of ara‐CTP. Here, a relatively new purine nucleoside analog, ara‐G, and conventional F‐ara‐A were similarly evaluated. Combination of a minimally toxic concentration (10 μM) of either ara‐G or F‐ara‐A with 10 μM ara‐C similarly enhanced ara‐CTP production in both cell lines compared with treatment with ara‐C alone (Fig. 2a,b). The combination effect was more potent for ara‐G than F‐ara‐A in HL‐60/ara‐C60 cells (Fig. 2b). This increase in ara‐CTP production was in accordance with the augmentation of dCK protein expression by the addition of ara‐G to ara‐C (Fig. 1c). Moreover, we demonstrated small‐scale production of ara‐GTP in both HL‐60 and HL‐60/ara‐C60 cells on treatment with ara‐G (Fig. 2c). The combination also enhanced ara‐C's cytotoxicity in both cell lines (Fig. 2d). Thus, these results suggest that ara‐G augmented dCK expression, thereby increasing ara‐CTP production and as a result enhancing ara‐C cytotoxicity.
Figure 2.
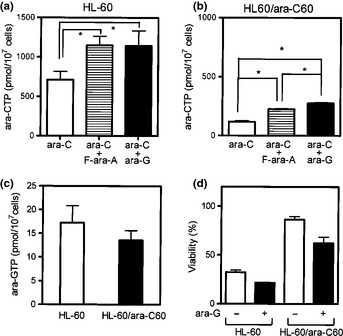
(a,b) Enhancement of ara‐C triphosphate (ara‐CTP) production by ara‐G. Cells (1 × 106 cells/mL, 10 mL) were incubated with 10 μM F‐ara‐A or ara‐G for 2 h (or not), followed by washing in media and incubation with 10 μM ara‐C for 3 h. (c) Cells (1 × 106 cells/mL, 10 mL) were incubated with 10 μM ara‐G for 4 h. The intracellular ara‐GTP was measured using high performance liquid chromatography (HPLC) as described in Materials and Methods (a–c). (d) Growth inhibition was determined using the trypan blue dye exclusion assay after the cells had been treated with ara‐C plus ara‐G as described above. Open bars, without ara‐G; closed bars, with ara‐G. Each value represents the mean ± SD of at least three independent experiments. *P ≤ 0.05.
Expression of Bcl‐2‐family proteins in leukemic cells
Many anticancer agents exert their cytotoxicity by inducing apoptotic cell death.29, 30 The Bcl‐2 family contains key regulators of the mitochondrial pathway of apoptosis. Of these, Bcl‐2 has been found to be overexpressed in many cancer cells, including B‐cell‐derived lymphomas. Western blot analysis demonstrated increased protein levels of antiapoptotic Bcl‐2 and Bcl‐XL in HL‐60/ara‐C60 cells compared with HL‐60 cells (Fig. 3). Thus, inhibition of apoptotic function was suggested to be involved in the mechanisms of resistance to ara‐C in HL‐60/ara‐C60 cells.
Figure 3.
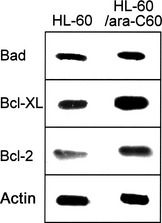
Protein expression levels of Bad, Bcl‐XL, Bcl‐2 in HL‐60 cells and HL‐60/ara‐C60 cells; Western blot analysis.
Sensitization of ara‐C‐resistant cells by Bcl‐2 inhibitor
Antiapoptotic Bcl‐2 might represent a target for the treatment of cancers, typically those in which Bcl‐2 is overexpressed. The Bcl‐2 inhibitor YC137 was designed to inhibit the binding of the proapoptotic Bid BH3 peptide to Bcl‐2, thus disrupting an interaction essential for antiapoptotic activity.24 When ara‐C‐resistant HL‐60/ara‐C60 cells were treated with ara‐C plus YC137, the induction of apoptosis was more prominent than in cells treated with ara‐C alone (Fig. 4b,e). The combination also induced apoptosis in ara‐C‐sensitive HL‐60 cells, and the enhancement was apparent (Fig. 4a,d). The addition of YC137 augmented the growth‐inhibition effects of nucleoside analogs including ara‐C, dFdC and 2CdA on HL‐60/ara‐C60 cells (Fig. 5, Table 2). These results suggest inhibition of Bcl‐2 as a possible strategy to overcome the resistance of leukemia cells to ara‐C.
Figure 4.
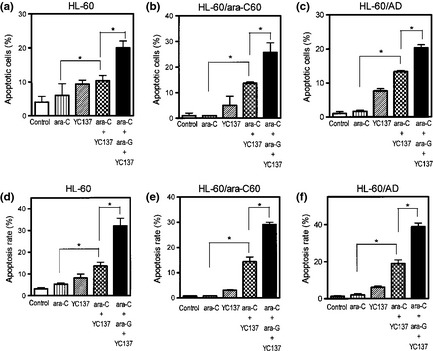
Enhancement of apoptotic cell death by the combination of ara‐C and the Bcl‐2 inhibitor YC137. Cells were treated with 0.005 μM ara‐C (a,d), 0.05 μM ara‐C (b,c,e,f) or 2.5 μM YC137, or both in combination for 72 h. Induction of apoptosis was determined using nuclear staining with Hoechst 33342 (a–c) and was analyzed by flow cytometry (d–f).
Figure 5.
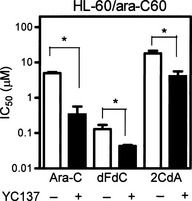
Cells were incubated with a given nucleoside analog with or without co‐incubation with 2.5 μM YC137 for 72 h. The IC50 was determined using the XTT assay. Ara‐C, cytarabine; dFdC, gemcitabine, 2CdA, cladribine. Open bars, HL‐60 cells; closed bars, HL‐60/ara‐C60 cells. Each value represents the mean ± SD of at least three independent experiments. *P ≤ 0.05.
Table 2.
YC137 augmented growth inhibition effects of nucleoside analogs on HL‐60/ara‐C60 cells
| Drugs | IC50 (μM) | RR |
|---|---|---|
| YC137 | 5.80 ± 0.34 | – |
| Ara‐C + YC137 | 0.30 ± 0.23 | 8.1 |
| dFdC + YC137 | 0.05 ± 0.02 | 4.7 |
| 2CdA + YC137 | 3.90 ± 1.80 | 78.0 |
| Ara‐C + Ara‐G + YC137 | 0.10 ± 0.06 | 2.0 |
Cells were incubated with a given nucleoside analog with or without co‐incubation with 2.5 μM YC137 for 72 h. The IC50 was determined using the XTT assay. RR, relative resistance calculated as the ratio of the IC50 of HL‐60/ara‐C60 cells relative to that of HL‐60 cells. Ara‐C, cytarabine; Ara‐G, 9‐β‐d‐arabinofuranosylguanine; dFdC, gemcitabine; 2CdA, cladribine. Values for IC50 are the means ± SD.
Sensitization of ara‐C‐resistant cells by the combination of ara‐G and Bcl‐2 inhibitor
The effect of the combination of ara‐C with ara‐G and/or Bcl‐2 inhibitor was evaluated in HL‐60/ara‐C60 cells. However, the addition of ara‐G was marginally effective for inducing apoptosis in HL‐60/ara‐C60 cells. Therefore, the cells were treated with ara‐G, YC137 and ara‐C in combination. The three drugs in combination provided greater cytotoxicity than ara‐C+YC137 in terms of the growth inhibition and the induction of apoptosis (Table 2, Fig. 4b,e). To examine whether this enhanced cytotoxicity is specific to HL‐60/ara‐C60 cell line, HL‐60/AD cells13 were similarly examined. HL‐60/AD cells were established to be ara‐C‐resistant and possess Bcl‐2 overexpression in our previous study.13 Ara‐C, ara‐G and YC137 in combination exerted greater cytotoxicity against HL‐60/AD cells than ara‐C+YC137 (Table 3, Fig. 4c,f). Thus, ara‐C resistance was effectively overcome by the addition of ara‐G and YC137 to ara‐C.
Table 3.
Ara‐G and YC137 augmented growth inhibition effects of ara‐C on HL‐60/AD cells
| IC50 (μM) | ||
|---|---|---|
| HL‐60/AD | RR | |
| Ara‐C | 2.70 ± 0.59 | 34.0 |
| Ara‐C + Ara‐G | 0.40 ± 0.14 | 4.0 |
| Ara‐C + YC137 | 0.17 ± 0.19 | 4.3 |
| Ara‐C + Ara‐G + YC137 | 0.07 ± 0.01 | 1.4 |
Cells were incubated with a given nucleoside analog with or without co‐incubation with 10 μM ara‐G or 2.5 μM YC137 for 72 h. The IC50 was determined using the XTT assay. RR, relative resistance calculated as the ratio of the IC50 of HL‐60/AD cells relative to that of HL‐60 cells. Ara‐C, cytarabine; Ara‐G, 9‐β‐d‐arabinofuranosylguanine. Values for IC50 are the means ± SD.
Discussion
Ara‐C is one of the most effective chemotherapeutic agents used in the treatment of AML. However, the resistance of leukemic cells to ara‐C remains a major drawback. We and others have extensively investigated the mechanisms of ara‐C resistance in vitro and in vivo.12, 13, 14, 15, 16, 17, 18 Deficient dCK activity, decreased nucleoside transporter content, decreased DNA polymerase sensitivity, overexpression of the cytidine deaminase gene, and enhanced cytosolic 5′‐nucleotidase II (cN‐II) activity have been reported to be associated with ara‐C resistance in vitro.30, 31, 32 Clinically, low hENT1 transcript level, increased cN‐II expression, decreased dCK expression, and increased cN‐II/dCK expression ratio were associated with poor therapeutic outcomes of ara‐C‐based chemotherapy.34, 35, 36 Moreover, resistance to ara‐C might develop due to the antiapoptotic nature of cancer cells including the X‐linked inhibitor of apoptosis protein (XIAP) and Bcl‐2.37, 38 Thus, the development of cellular resistance to ara‐C is mainly a result of decreased capability to produce intracellular ara‐CTP and inhibition of apoptosis.
Here, we have demonstrated a reduced capability to yield ara‐CTP (Fig. 1) and resistance to the induction of apoptosis (Fig. 3) in a newly established ara‐C‐resistant leukemic cell line (HL‐60/ara‐C60). In this resistant cell line, reduced ara‐CTP production was attributable to a reduction in dCK protein (Fig. 1c) while its antiapoptotic nature was due to overexpression of Bcl‐2 and Bcl‐XL (Fig. 3). The addition of the purine nucleoside analog ara‐G to ara‐C augmented the dCK protein level (Fig. 1c), thereby increasing ara‐CTP production (Fig. 2a,b) and cytotoxicity (Fig. 2d). The combination of the Bcl‐2 antagonist YC137 with ara‐C provided a greater amount of apoptosis than ara‐C alone (Fig. 4). The contribution of Bcl‐2 overexpression was also noted in the exertion of cytotoxic effects of dFdC and 2CdA (Fig. 5). The effect of three drugs combination was the most in ara‐C resistance cells line.
Ara‐G is a guanosine nucleoside analog that exerts specific cytotoxicity in T‐lymphoblasts compared with myeloblasts and B‐lymphoblasts.20, 21 No cytotoxic effect was shown in HL‐60 and HL‐60/ara‐C60 cell lines (Table 1). Ara‐GTP was produced in these cells (Fig. 2c), although ara‐GTP production was a small scale level. This is reported to be a result of reduced ara‐GTP half‐life in these cells when compared with T‐cells.39, 40, 41 Like ara‐C, ara‐G must be phosphorylated intracellularly to ara‐G triphosphate for its cytotoxic effect as well as the augmentation of ara‐CTP production. However, the activation pathway of ara‐G is not identical to that of ara‐C.19, 20, 21, 22 Ara‐G is transported into the cell via both nitrobenzylthioinosine‐sensitive and ‐insensitive equilibrative nucleoside transporters including hENT1 and concentrative nucleoside transporter 3.19 Inside the cell, ara‐G is phosphorylated to its monophosphate form by not only dCK but also the mitochondrial enzyme deoxyguanosine kinase. Therefore, ara‐GTP has been suggested to be an active enhancer of ara‐C cytotoxicity even in ara‐C‐resistant leukemic cells that possessed decreased dCK activity (Fig. 2c). We have demonstrated that the addition of ara‐G to ara‐C increased dCK expression, ara‐CTP production and subsequent cytotoxicity in HL‐60/ara‐C60 cells (Table 1, Figs 1c and 2b). To the contrary, F‐ara‐A, which uses the activation pathway in common with ara‐C, was less effective for enhancing ara‐CTP production in HL‐60/ara‐C60 cells (Fig. 2b). Thus, these results suggest that ara‐G has potential to overcome cellular resistance to ara‐C.
Apoptosis is important for normal development, host defense, and suppression of oncogenesis, and faulty regulation of apoptosis has been implicated in cancer. The Bcl‐2 family of proteins contains key regulators of the mitochondrial pathway of apoptosis and includes both antiapoptotic molecules such as Bcl‐2 and Bcl‐XL, and proapoptotic molecules such as Bax, Bak, Bid, and Bad.23 Many types of cancer overexpress antiapoptotic Bcl‐2 family members.24 Here, Bcl‐2 protein was overexpressed in HL‐60/ara‐C60 cells (Fig. 3), suggesting that the altered apoptotic function would contribute to the development of resistance to ara‐C. Addition of the Bcl‐2 inhibitor YC137 to ara‐C induced more apoptosis in HL‐60/ara‐C60 cells than ara‐C alone (Fig. 4b,e). Apoptosis was also enhanced when YC137 was combined with other nucleoside analogs (dFdC, 2CdA), to which HL‐60/ara‐C60 cells showed cross resistance (Fig. 5). These results suggest that the antiapoptotic nature mediated by Bcl‐2 overexpression plays a critical role in ara‐C resistance in leukemic cells.
Synergistic effects were observed for the combination of ara‐G with ara‐C and YC137 with ara‐C in both HL‐60/ara‐C60 cells and HL‐60/AD cells (Tables 2 and 3, Fig. 4b,c,e,f). Moreover, the three‐drug combination provided greater cytotoxicity than ara‐C+ara‐G (Tables 2 and 3, Fig. 4b,c,e,f). These results suggest that the addition of two drugs of different nature could dramatically augment ara‐C cytotoxicity in ara‐C resistant‐leukemic cells.
In conclusion, the development of drug resistance in cancer cells is usually multifactorial. Our findings suggest that reduced ara‐CTP production and refractoriness of the induction of apoptosis may be major mechanisms of resistance to ara‐C in leukemic cells. Moreover, optimal combination strategies should be developed based on the understanding of the mechanisms of drug resistance in chemotherapy for leukemia. The mechanisms of ara‐C resistance mainly involved two factors; the reduced intracellular ara‐CTP production due to decreased dCK and hENT1 and the enhanced antiapoptosis due to overexpression of antiapoptotic Bcl‐2. Therefore, these results suggested that augmentation of ara‐CTP production by ara‐G and reversal of high Bcl‐2‐mediated antiapoptosis by YC137 might collaborate to sensitize HL‐60/ara‐C60 cells to ara‐C.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (23501307), and a Grant from the Japan Gout Research Foundation (2008, 2009, 2010).
(Cancer Sci 2013; 104: 502–507)
References
- 1. Garcia‐Carbonero R, Ryan DP, Chabner BA. Cytidine analogs In: Chabner BA, Longo DL, eds. Cancer Chemotherapy and Biotherapy. Philadelphia: Lippincott‐Raven Publishers, 1996; 265–94. [Google Scholar]
- 2. Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia 2001; 15: 875–90. [DOI] [PubMed] [Google Scholar]
- 3. Ferrara F. Unanswered questions in acute myeloid leukemia. Lancet Oncol 2004; 5: 443–50. [DOI] [PubMed] [Google Scholar]
- 4. Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood 2005; 106: 1154–63. [DOI] [PubMed] [Google Scholar]
- 5. Jabbour EJ, Estey E, Kantarjian HM. Adult acute myeloid leukemia. Mayo Clin Proc 2006; 81: 247–60. [DOI] [PubMed] [Google Scholar]
- 6. Fernandez HF, Rowe JM. Induction therapy in acute myeloid leukemia: intensifying and targeting the approach. Curr Opin Hematol 2010; 17: 79–84. [DOI] [PubMed] [Google Scholar]
- 7. Wiley JS, Jones SP, Sawyer WH. Cytosine arabinoside influx and nucleoside transport sites in acute leukemia. J Clin Invest 1982; 69: 479–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Inagaki A, Nakamura T, Wakisaka G. Studies on the mechanism of action of 1‐beta‐d‐arabinofuranosylcytosine as an inhibitor of DNA synthesis in human leukemic leukocytes. Cancer Res 1969; 29: 2169–76. [PubMed] [Google Scholar]
- 9. Graham FL, Whitmore GF. Studies in mouse L‐cells on the incorporation of 1‐β‐d‐arabinofuranosylcytosine into DNA and on inhibition of DNA polymerase by 1‐β‐d‐arabinofuranosylcytosine 5'‐triphosphate. Cancer Res 1970; 30: 2636–44. [PubMed] [Google Scholar]
- 10. Graham FL, Whitmore GF. The effect of 1‐beta‐d‐arabinofuranosylcytosine on growth, viability, and DNA synthesis of mouse L‐cells. Cancer Res 1970; 30: 2627–35. [PubMed] [Google Scholar]
- 11. Kufe DW, Major PP, Egan EM et al Correlation of cytotoxicity with incorporation of ara‐C into DNA. J Biol Chem 1980; 255: 8997–9000. [PubMed] [Google Scholar]
- 12. Yamauchi T, Ueda T, Nakamura T. A new sensitive method for determination of intracellular 1‐β‐d‐arabinofuranosylcytosine 5'‐triphosphate content in human materials in vivo . Cancer Res 1996; 56: 1800–4. [PubMed] [Google Scholar]
- 13. Takemura H, Urasaki Y, Yoshida A et al Simultaneous treatment with 1‐beta‐d‐arabinofuranosylcytosine and daunorubicin induces cross‐resistance to both drugs due to a combination‐specific mechanism in HL60 cells. Cancer Res 2001; 61: 172–7. [PubMed] [Google Scholar]
- 14. Yamauchi T, Kawai Y, Ueda T. 1‐β‐d‐Arabinofuranosylcytosine is cytotoxic in quiescent normal lymphocytes undergoing DNA excision repair. Cancer Sci (Jpn J Cancer Res) 2002; 93: 1334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamauchi T, Ueda T. A sensitive new method for clinically monitoring cytarabine concentrations at the DNA level in leukemic cells. Biochem Pharmacol 2005; 69: 1795–803. [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto S, Yamauchi T, Kawai Y et al Fludarabine‐mediated circumvention of cytarabine resistance is associated with fludarabine triphosphate accumulation in cytarabine‐resistant leukemic cells. Int J Hematol 2007; 85: 108–15. [DOI] [PubMed] [Google Scholar]
- 17. Yamauchi T, Negoro E, Kishi S et al Intracellular cytarabine triphosphate production correlates to deoxycytidine kinase / cytosolic 5'‐nucleotidase II expression ratio in primary acute myeloid leukemia cells. Biochem Pharmacol 2009; 77: 1780–6. [DOI] [PubMed] [Google Scholar]
- 18. Negoro E, Yamauchi T, Urasaki Y et al Characterization of cytarabine‐resistant leukemic cell lines established from five different blood cell lineages using gene expression and proteomic analyses. Int J Oncol 2011; 38: 911–9. [DOI] [PubMed] [Google Scholar]
- 19. Prus KL, Averett DR, Zimmerman TP. Transport and metabolism of 9‐beta‐d‐arabinofuranosylguanine in a human T‐lymphoblastoid cell line: nitrobenzylthioinosine‐sensitive and ‐insensitive influx. Cancer Res 1990; 50: 1817–21. [PubMed] [Google Scholar]
- 20. Rodriguez CO Jr, Gandhi V. Arabinosylguanine‐induced apoptosis of T‐lymphoblastic cells: incorporation into DNA is a necessary step. Cancer Res 1999; 59: 4937–43. [PubMed] [Google Scholar]
- 21. Rodriguez CO Jr, Mitchell BS, Ayres M et al Arabinosylguanine is phosphorylated by both cytoplasmic deoxycytidine kinase and mitochondrial deoxyguanosine kinase. Cancer Res 2002; 62: 3100–5. [PubMed] [Google Scholar]
- 22. Lotfi K, Månsson E, Peterson C et al Low level of mitochondrial deoxyguanosine kinase is the dominant factor in acquired resistance to 9‐beta‐d‐arabinofuranosylguanine cytotoxicity. Biochem Biophys Res Commun 2002; 293: 1489–96. [DOI] [PubMed] [Google Scholar]
- 23. Enyedy IJ, Ling Y, Nacro K et al Discovery of small‐molecule inhibitors of Bcl‐2 through structure‐based computer screening. J Med Chem 2001; 44: 4313–24. [DOI] [PubMed] [Google Scholar]
- 24. Real PJ, Cao Y, Wang R et al Breast cancer cells can evade apoptosis‐mediated selective killing by a novel small molecule inhibitor of Bcl‐2. Cancer Res 2004; 64: 7947–53. [DOI] [PubMed] [Google Scholar]
- 25. Yamauchi T, Nishi R, Kitazumi K et al A new high‐performance liquid chromatography method determines low production of 9‐beta‐d‐arabinofuranosylguanine triphosphate, an active metabolite of nelarabine, in adult T‐cell leukemia cells. Oncol Rep 2010; 23: 499–504. [PubMed] [Google Scholar]
- 26. Yoshio N, Kawai Y, Hori H et al Resistance to 9‐beta‐d‐arabinofuranosyl‐2‐fluoroadenine due to reduced incorporation into DNA from competition by excess deoxyadenosine triphosphate: implications for different sensitivities to nucleoside analogues. Int J Hematol 2005; 81: 405–12. [DOI] [PubMed] [Google Scholar]
- 27. Yamauchi T, Nowak BJ, Keating MJ et al DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4‐hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin Cancer Res 2001; 7: 3580–9. [PubMed] [Google Scholar]
- 28. Yamauchi T, Kawai Y, Ueda T. Inhibition of nucleotide excision repair by fludarabine in normal lymphocytes in vitro, measured by the alkaline single cell gel electrophoresis (Comet) assay. Cancer Sci (Jpn J Cancer Res) 2002; 93: 567–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kasimir‐Bauer S, Beelen D, Flasshove M et al Impact of the expression of P glycoprotein, the multidrug resistance‐related protein, bcl‐2, mutant p53, and heat shock protein 27 on response to induction therapy and long‐term survival in patients with de novo acute myeloid leukemia. Exp Hematol 2002; 30: 1302–8. [DOI] [PubMed] [Google Scholar]
- 30. Certo M, Moore VDG, Nishino M et al Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL‐2 family members. Cancer Cell 2006; 9: 351–65. [DOI] [PubMed] [Google Scholar]
- 31. Bhalla K, Nayak R, Grant S. Isolation and characterization of a deoxycytidine kinase‐deficient human promyelocytic leukemic cell line highly resistant to 1‐beta‐d‐arabinofuranosylcytosine. Cancer Res 1984; 44: 5029–37. [PubMed] [Google Scholar]
- 32. Higashigawa M, Ido M, Nagao Y et al Decreased DNA polymerase sensitivity to 1‐beta‐d‐arabinofuranosylcytosine 5'‐triphosphate in P388 murine leukemic cells resistant to vincristine. Leuk Res 1991; 15: 675–81. [DOI] [PubMed] [Google Scholar]
- 33. Schröder JK, Kirch C, Flasshove M et al Constitutive overexpression of the cytidine deaminase gene confers resistance to cytosine arabinoside in vitro . Leukemia 1996; 10: 1919–24. [PubMed] [Google Scholar]
- 34. Hubeek I, Stam RW, Peters GJ et al The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br J Cancer 2005; 93: 1388–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kawasaki H, Carrera CJ, Piro LD et al Relationship of deoxycytidine kinase and cytoplasmic 5'‐nucleotidase to the chemotherapeutic efficacy of 2‐chlorodeoxyadenosine. Blood 1993; 81: 597–601. [PubMed] [Google Scholar]
- 36. Galmarini CM, Graham K, Thomas X et al Expression of high Km 5'‐nucleotidase in leukemic blasts is an independent prognostic factor in adults with acute myeloid leukemia. Blood 2001; 98: 1922–6. [DOI] [PubMed] [Google Scholar]
- 37. Wang S, Vrana JA, Bartimole TM et al Agents that down‐regulate or inhibit protein kinase C circumvent resistance to 1‐beta‐d‐arabinofuranosylcytosine‐induced apoptosis in human leukemia cells that overexpress Bcl‐2. Mol Pharmacol 1997; 52: 1000–9. [DOI] [PubMed] [Google Scholar]
- 38. Schimmer AD, Estey EH, Borthakur G et al Phase I/II trial of AEG35156 X‐linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J Clin Oncol 2009; 27: 4741–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roecker AM, Stockert A, Kisor DF. Nelarabine in the treatment of refractory T‐cell malignancies. Clin Med Insights Oncol 2010; 4: 133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rodriguez CO, Stellrecht CM, Gandhi V. Mechanisms for T‐cell selective cytotoxicity of arabinosylguanine. Blood 2003; 102: 1842–8. [DOI] [PubMed] [Google Scholar]
- 41. Kisor DF, Plunkett W, Kurtzberg J et al Pharmacokinetics of nelarabine and 9‐beta‐d‐arabinofuranosyl guanine in pediatric and adult patients during a phase I study of nelarabine for the treatment of refractory hematologic malignancies. J Clin Oncol 2000; 18: 995–1003. [DOI] [PubMed] [Google Scholar]