

Abstract
Stearoyl‐CoA desaturase‐1 (SCD1) is an endoplasmic reticulum anchored enzyme catalyzing the synthesis of monounsaturated fatty acids, mainly palmytoleyl‐CoA and oleyl‐CoA. Recent studies have revealed a function for SCD1 in the modulation of signaling processes related to cell proliferation, survival and transformation to cancer. We used MCF7 and MDA‐MB‐231 cells to analyze the role of SCD1 in the metastatic acquisition of breast cancer cells. Silencing SCD1 expression in breast cancer cells has no effect on cell viability but the levels of cell proliferation, cell cycle genes' expressions and the phosphorylation state of ERK1/2 MAPK are significantly reduced. Decreasing SCD1 expression also reduces the level of GSK3 phosphorylation, indicating higher activity of the kinase. Using cells fractionation, immunofluorescence and a β‐catenin/TCF‐responsive reporter construct, we demonstrate that lowering SCD1 expression leads to a decrease of β‐catenin amounts within the nucleus and to inhibition of its transactivation capacity. Moreover, MDA‐MB‐231 cells transfected with the SCD1 siRNA show a lower invasive potential than the control cells. Taken together, our data demonstrate that low SCD1 expression is associated with a decrease in the proliferation rate of breast cancer cells associated with a decrease in ERK1/2 activation. SCD1 silencing also inhibits GSK3 phosphorylation, lowering β‐catenin translocation to the nucleus, and, subsequently, its transactivation capacity and the expression of its target genes. Finally, we show that silencing SCD1 impairs the epithelial to mesenchymal transition‐like behavior of the cells, a characteristic of metastatic breast cancer. (Cancer Sci 2013; 104: 36–42)
The process of cell proliferation and survival in mammalian organisms requires the production of new membranes, involving the production of new lipids with the appropriate composition. Saturated fatty acids (SFA) and mono‐unsaturated fatty acids (MUFA) are the major components of the cellular membrane phospholipids. Balance alterations among the different lipid species may modify the appropriate cell functions, such as survival and proliferation.
Several studies reveal a relationship between obesity and breast cancer.1 We and others have demonstrated that high levels of stearoyl‐CoA desaturase 1 (SCD1) expression in human adipose tissues are associated with obesity.2, 3 SCD1 is a key regulator of fatty acid composition in the membranes of mammalian cells. This enzyme catalyzes the introduction of a double bound in the Δ‐9 position of palmitoyl‐CoA and stearoyl‐CoA to form palmitoleoyl‐CoA and oleoyl‐CoA, respectively.4
In cancer cells and tissues, increased content of MUFA has been observed, suggesting a role of SCD1 in the transformation process.5, 6, 7 The level of SCD1 expression and activity are also increased in lung and breast cancer cells.8, 9. In addition, high SCD1 gene expression is observed in cancer cells and tissues, such as breast, bladder, colon and lungs, and in prostate tumors (www.oncomine.org).10 Furthermore, fatty acid profiles and, in particular, the balance between SFA and MUFA can be used as predictors for breast cancer in women.11, 12 A positive correlation is observed between unbalanced levels of SFA and MUFA and higher risk for breast cancer.13 In addition, the content of MUFA in cholesterol esters has been associated with a higher rate of death in patients.14 In women diagnosed with breast cancer, an elevated level of oleic acid has been observed in adipose tissue and lymph node metastasis, suggesting increased activity of SCD1,15 whereas low levels of stearic acid (a SCD1 substrate) in phosphatidylcholine have been measured in breast cancer tumors associated with subsequent metastasis and poor prognostic.6 Another study observed a positive correlation between MUFA concentration and the apparition of metastasis in patients.16 It has also been shown that alterations of the SFA to MUFA ratio in breast cancer tumors reflect the change in fatty acid metabolism in cells.17 Together, these studies strongly point towards a central role of MUFA in metastatic breast cancer.
The loss of epithelial phenotype and acquisition of mesenchymal characteristics is a key feature of cells that become metastatic. This epithelial to mesenchymal transition (EMT) is normally activated during embryonic programming . In abnormal situations , EMT is reactivated, especially during tumor invasion and dissemination of cancer cells.18 One of the first events occurring in EMT is the loss of E‐cadherin expression.19 E‐cadherin is a cell–cell adhesion molecule interacting with elements of adherent junctions, such as β‐catenin.20 If β‐catenin is liberated in the cytosol, it is rapidly degraded by the proteasome system.21 This degradation is GSK3β‐dependent.22 Upon loss of E‐cadherin, GSK3β is inhibited by phosphorylation23 and, consequently, β‐catenin is no longer degraded. It can translocate into the nucleus, activating transcription of genes involved in the invasion process.24
In the present study, we evaluate the role of SCD1 in breast cancer and in EMT. We show that a decrease of SCD1 expression is associated with a lower proliferating rate of breast cancer cells and with a switch to a more epithelial phenotype, suggesting that SCD1 accounts for the apparition of metastasis in breast cancer cells.
Materials and Methods
Materials
MCF7 and MDA‐MB‐231 cells were a gift from Dr J. J. Lebrun (McGill University, Montreal PQ, Canada). The siRNA, the DharmaFECT and the DharmaFECT Duo transfection reagents were obtained from Ambion (Burlington, ON, Canada). The α‐tubulin, phospho‐ERK1/2 (Thr202/Tyr204) and phospho‐GSK3α/β (Ser21/9) antibodies were from Cell Signaling Technology (Danvers, MA, USA). The β‐catenin antibody was purchased from Santa‐Cruz (Santa‐Cruz, CA, USA). The SCD1 antibody was obtained from Abcam (Cambridge, MA, USA). The Lap‐2 antibody was purchased from Sigma‐Aldrich (Oakville, Ontario, Canada). The DMEM was from Wisent Bioproducts (St‐Bruno, PQ, Canada). TOP and FOP flash vectors were a gift from Dr N. Rivard (Sherbrooke University, PQ, Canada). Most other reagents were obtained from Sigma‐Aldrich.
Cell culture and transfection procedure
MCF7 and MDA‐MB‐231 were grown in DMEM supplemented with 10% FBS, 100 μg/mL streptomycin and 100 U/mL penicillin. Cells were plated and transfected the day after with either a control siRNA (siRNA‐CTL) or a siRNA targeting SCD1 (siRNA‐SCD1) using the DharmaFECT transfection reagent. For the experiments implicating the use of TOP and FOP flash vector, cells were transfected using the DharmaFECT Duo transfection reagent.
Semi‐quantitative RT‐PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and subjected to reverse transcription using the Qiagen Omniscript RT kit (Montreal, PQ, Canada) and Oligo‐dT (Roche Diagnostics, Laval, PQ, Canada), and then to semi‐quantitative PCR analysis. The primers' sequences are indicated in Table 1.
Table 1.
Sequences of oligonucletotides used in RT‐PCR analyses
| Gene | Sens | Antisense |
|---|---|---|
| Human SCD1 | CTCCACTGCTGGACATGAGA | AATGAGTGAAGGGGCACAAC |
| Human CdK4 | TGGACAAGGCACCCCCACCA | GGCCAGGCCAAAGTCAGCCA |
| Human Cyclin D1 | CCGCACGATTTCATTGAAC | CACAGAGGGCAACGAAGGTC |
| Human c‐Myc | CGTCTCCACACATCAGCACAA | TCTTGGCAGCAGGATAGTCCTT |
| Human E‐Cadherin | GAGTGCCAACTGGACCATTC | CCCACCTCTAAGGCCATCT |
| Human Vimentin | AGATGGCCCTTGACATTGAG | CCAGAGGGAGTGAATCCAGA |
| Human HPRT | ACCAGTCAACAGGGGACATAA | AAGCTTGCGACCTTGACC |
| Human β‐actin | CGTGACATTAAGGAGAAGCTGT | CTCAGGAGAGCAATGATCTTGAT |
Cell viability assay
Cell viability was measured using the CellTiter 96 Aqueous One Solution Assay (Promega, Madison, WI, USA). Cells were seeded at a density of 10 000 (MCF7) or 5000 cells (MDA‐MB‐231) per well in a 96‐well plate. The day after, cells were transfected with different concentrations of siRNA. Methanethiosulfonate and phenazinemethosulfate reagents (MTS/PMS) were added 48 h post‐transfection. One hour later, the production of formazan was measured at 490 nm.
Cell cycle analysis
Cells were trypsinized and fixed with 70% ethanol. Cells were resuspended in propidium iodide solution (40 μg propidium iodide and 100 μg RNaseA in 1 mL PBS) for 30 min at 37°C. Cell cycle distribution was determined using a FACScan flow cytometer (FACScan [Becton Dickinson, Franklin Lakes, NJ, USA]).
Western blot analysis
Nuclear and cytoplasmic proteins were extracted using the Cytoplasmic and Nuclear Proteins Extraction Kit (ZmTech Scientifique, Montreal, PQ. Canada), and 20 to 50 μg of proteins were transferred to Immobilon‐P membranes. Membranes were incubated for 1 h in blocking buffer (1X TBS, 0,1% Tween‐20: TBST, 5% milk) and then overnight in TBST + 5% BSA and antibodies for SCD1 (1/1000), phospho‐ERK1/2 (Thr202/Tyr204) (1/1000), phospho‐GSK3α/β (Ser21/9) (1/1000), β‐catenin (1/1000), GAPDH (1/2500), α‐tubulin (1/5000) and Lap 2 (1/1000). Anti‐rabbit (or anti‐mouse) IgG bound to the HRP (1/10 000) was used as a secondary antibody. Signals were revealed using the ECL plus western blotting detection reagent (Roche, Laval, PQ, Canada).
Immunofluorescence assay
Cells were plated on glass coverslips and transfected with siRNA‐CTL or siRNA‐SCD1. Cells were fixed in 3.7% paraformaldehyde 72 h after transfection. Cells were then permeabilized and blocked for 1 h with 0.1% Triton X‐100, 10% goat serum and 10% BSA. Coverslips were then incubated with the first antibodies (SCD1 [1/100] and β‐catenin [1/100]) for 2 h at room temperature. After three successive washes in PBS, cells were incubated with the Alexa Fluor‐466 and 568 antibodies (1/1000) (Invitrogen). After washes in PBS, coverslips were incubated for 5 min with DAPI (1/200 000). Coverslips were mounted onto slides using Prolong Gold antifade reagent (Invitrogen) and observed by immunofluorescence microscopy (Eclipse Ti fluorescent microscope, Nikon, Mississauga, ON, Canada).
Cell invasion assay
MDA‐MB‐231 cells were transfected with siRNA‐CTL or siRNA‐SCD1. After 48 h, 1 × 105 cells were resuspended in medium containing 0.1% BSA and seeded in the upper chamber of matrigel‐coated (10 mg/mL) transwell inserts (8‐μm pore size; Costar, Lowell, MA, USA ). The lower chamber was filled with medium containing 10% FBS. Cells were allowed to migrate for 48 h at 37°C. Following incubation, non‐migrated cells were scraped off from the upper surface of the membranes. Cells that invaded the lower chamber were fixed for 1 h in 3.7% formaldehyde and stained for 30 min with 20% methanol and 0.25% crystal violet. Cells were counted and analyzed using the ImageJ software.
Statistical analysis
Data are given as a mean with ± standard deviation (SD) of the mean. Densitometry analyses were performed using the ImageJ software. Statistical analyses were performed by two‐tailed Student's test for multiple comparisons. P < 0.05 defined the significance of the test.
Results
Inhibition of stearoyl‐CoA desaturase‐1 expression does not affect cells viability
Using an RNAi approach, we showed that transfection of 10 nM of siRNA‐SCD1 was sufficient to greatly reduce SCD1 mRNA expression in MCF7 and MDA‐MB‐231 cells (Fig. 1a and b). An inhibition was also observed at the protein level (40% of inhibition in MCF7 cells and 50% in MDA‐MB‐231). Compared to non‐transfected cells, the viability of MCF7 cells transfected with siRNA‐CTL or with siRNA‐SCD1 (from 5 to 20 nM) was comparable between the two conditions and reached 80% (Fig. 1c). Similar results were obtained with 50 nM of siRNA (data not shown). The viability of siRNA‐transfected MDA‐MB‐231 cells was even slightly higher than that of the non‐transfected cells (Fig. 1d).
Figure 1.
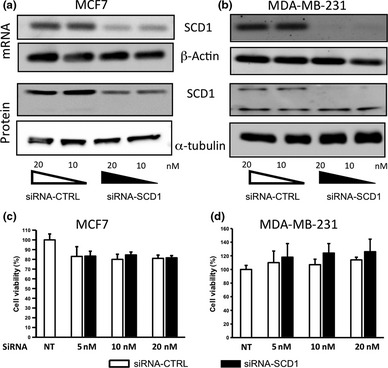
Inhibition of SCD1 expression does not affect cell viability. MCF7 (a) and MDA‐MB‐231 (b) cells were transfected with 10 or 20 nM of siRNA‐SCD1 or siRNA‐CTRL. The SCD1 silencing was confirmed at the mRNA (top panels) and protein levels (bottom panels). β‐actin and α‐tubulin were both used as loading controls. The figure shows representative results of at least three independent experiments. (c and d) Cell viability was evaluated using the MTS/PMS technique 48 h after siRNA transfection. Results are expressed as a percentage of viable cells in the non‐transfected control. n = 3 for (c) and n = 5 for (d).
Inhibition of stearoyl‐CoA desaturase‐1 expression affects cell cycle and ERK1/2 phosphorylation
Decreasing SCD1 expression significantly reduced the percentage of cells in S phase compared to cells transfected with the control siRNA (25 vs 31%, respectively; P = 0.003) (Fig. 2a). A similar observation was made on the percentage of cells in G2/M (2.5% in cells transfected with siRNA‐SCD1 vs 7.5% in control cells). Consequently, the percentage of cells in the resting phase (G0–G1) was greatly increased in the siRNA‐SCD1‐transfected cells compared to the control cells (72.5 vs 61.5%, respectively, P = 0.03). In both cell types, SCD1 silencing reduced the mRNA expression levels of cell cycle genes, such as Cdk4, CyclinD1 and c‐Myc (Fig. 2b and c). These results suggest that low SCD1 expression is associated with a lower proliferation rate of breast cancer cells. Regulators of the cell cycle progression, such as cyclin D1, are regulated by numerous kinases, including ERK1/2.25, 26 In agreement with a reduction of cell proliferation, we observed that inhibition of SCD1 expression leads to a significant decrease in ERK1/2 MAPK phosphorylation in both cell types (Fig. 2d and e).
Figure 2.
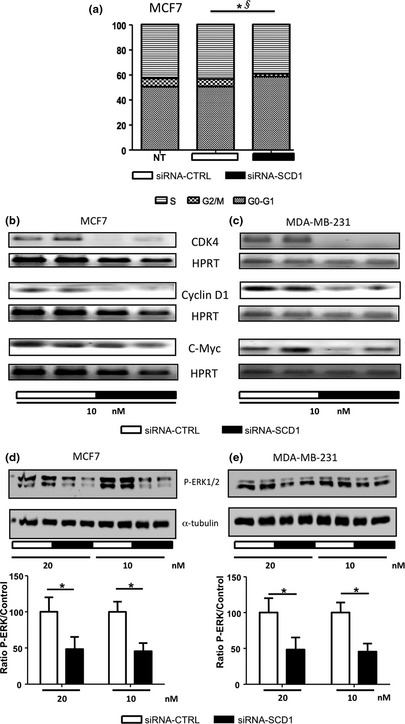
Inhibition of SCD1 expression affects cell cycle and ERK1/2 phosphorylation. (a) MCF7 cells were transfected with 10 nM of siRNA‐CTL or siRNA‐SCD1. Cell cycle distribution was determined 72 h post‐transfection. The graph represents the percentage of cells present in each phase of the cell cycle. n = 3. *P = 0.03 comparing percentage of cells in the G0–G1 phases between siRNA‐CTL and siRNA‐SCD1 cells. §P = 0.003 comparing percentage of cells in the S phase between siRNA‐CTL and siRNA‐SCD1 cells. (b) MCF7 and (c) MDA‐MB‐231 cells were transfected with 10 nM of siRNA‐CTL or siRNA‐SCD1. 48 h after transfection, total RNA was extracted and Cdk4, cyclin D1 and c‐Myc mRNA levels were evaluated by RT‐PCR. Hypoxanthine‐guanine phosphoribosyltransferase was used as a control. (d) MCF7 and (e) MDA‐MB‐231 cells were transfected with 10 or 20 nM of siRNA‐CTL or siRNA‐SCD1. 48 h after transfection, the level of ERK1/2 phosphorylation was measured by western blot analysis. α‐Tubulin was used as a control. The figure shows a representative western blot of at least three independent experiments. Bottom graphs represent densitometry quantification of the blots. *P < 0.05.
Inhibition of stearoyl‐CoA desaturase‐1 decreases GSK3 phosphorylation
Inhibition of GSK3, through increased phosphorylation, is also involved in the activation of the G1/S cell cycle progression.27, 28 This kinase is implicated in EMT through its effect on the degradation of β‐catenin.22, 24 In lung cancer cells, SCD1 inhibition decreases Akt and GSK3 phosphorylation.29 We show here that reducing SCD1 expression in breast cancer cells significantly impairs the phosphorylation of both GSK3α and GSK3β (Fig. 3a and b), indicating that GSK3 activity is enhanced when SCD1 expression is decreased.
Figure 3.
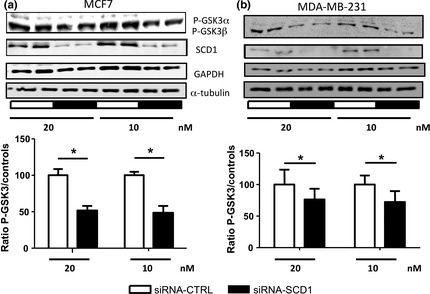
Inhibition of SCD1 expression impairs GSK3 phosphorylation. MCF7 (a) and MDA‐MB‐231 (b) were transfected with 10 or 20 nM of siRNA‐CTL or siRNA‐SCD1. The level of GSK3 phosphorylation was analyzed using a phospho‐specific antibody 48 h after transfection. GAPDH and α‐tubulin were used as loading control. The figures show representative western blots of at least three independent experiments. Bottom graphs represent densitometry quantification of the p‐GSK3 blots. *P < 0.05.
Inhibition of stearoyl‐CoA desaturase‐1 decreases the nuclear amount and the transactivation potential of β‐catenin impairing epithelial to mesenchymal transition‐like behavior of the cells
Epithelial to mesenchymal transition, which is reactivated during tumor invasion and dissemination of cancer cells,18 is characterized by an increase of GSK3 phosphorylation inhibiting the kinase activity.23 A known target of GSK3 is β‐catenin. When phosphorylated by GSK3, β‐catenin is ubiquitinated and degraded and, therefore, no longer targeted to the nucleus where it can activate its target genes. Many of these genes, such as vimentin,30 contain β‐catenin (TCF/LEF) response elements in their promoters and are involved in EMT.31, 32 Because GSK3 phosphorylation was decreased in cells transfected with siRNA‐SCD1 compared to control cells, we suspected that SCD1 inhibition would result in a reduction of β‐catenin in the nucleus. Indeed, we show here that SCD1 silencing decreases the amount of β‐catenin in the cytosol and even more significantly in the nucleus (Fig. 4a). A similar observation was made in MDA‐MB‐231 but the decrease of cytosolic β‐catenin was less pronounced (Fig. 4b). By immunofluorescence, we observed that decreasing SCD1 expression clearly reduced the amount of β‐catenin expressed in MCF7 cells (Fig. 4c). Moreover, in cells transfected with siRNA‐SCD1, β‐catenin is present at the cell periphery. A colocalization with a DAPI staining clearly demonstrated that silencing SCD1 decreased the amount of β‐catenin in the nucleus (Fig. 4c, right panels). Given the fact that less β‐catenin is available in the nucleus, we tested the possibility that its transactivation potential might also be decreased. We used a β‐catenin/TCF‐responsive reporter construct: the TOP flash vector and showed that the transactivation activity of β‐catenin was reduced by 40% (Fig. 4d). This reduction was associated with a decrease in the expression of the mesenchymal marker vimentin. In accordance with an acquisition of a more epithelial phenotype, the cells transfected with the siRNA‐SCD1 showed a higher level of expression of the epithelial marker E‐Cadherin compared to the control cells (Fig. 4e and f). SCD1 silencing also resulted in the reduction of MCF7 cells spreading and in the establishment of tighter cell–cell junctions (Fig. 4g). Furthermore, MDA‐MB‐231 cells, when transfected with siRNA‐SCD1, displayed a less fibroblastic appearance (Fig. 4h) and became significantly less invasive than the control cells (Fig. 4i).
Figure 4.
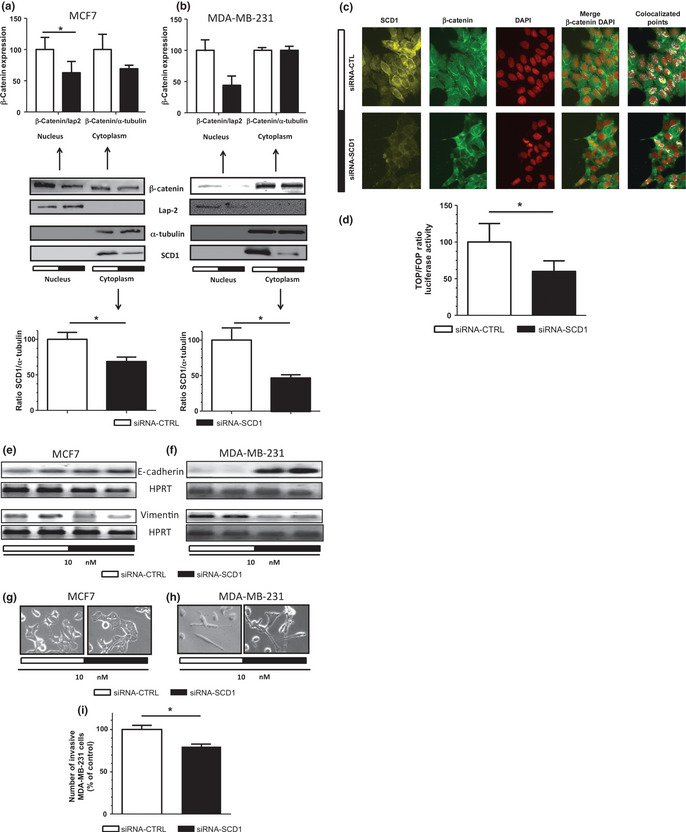
Inhibition of SCD1 expression decreases the nuclear localization and the transactivation potential of β‐catenin and impairs EMT‐like behavior. MCF7 and MDA‐MB‐231 were transfected with 10 nM of siRNA‐CTL or siRNA‐SCD1. After 48 h, the nuclear localization of β‐catenin was evaluated using cell fractionation and western blot analysis in MCF7 (a) and MDA‐MB‐231 (b) cells. Lap2 was used as a nuclear marker while α‐tubulin was used as a cytosolic marker. Upper graphs represent densitometry analysis of β‐catenin blots using Lap‐2 and α‐tubulin as nuclear and cytosolic loading controls, respectively. The bottom graphs represent densitometry quantification of SCD1 blots using α‐tubulin as a loading control. *P < 0.05. (c) The effect of SCD1 silencing was evaluated on the nuclear localization of β‐catenin. MCF7 cells were labeled with DAPI to stain the nuclei while β‐catenin and SCD1 were detected by immunofluorescence using specific antibodies. (d) MCF7 cells were co‐transfected with 10 nM of siRNA‐CTL (white bar) or siRNA‐SCD1 (black bars) mRNA and the TOP flash or the FOP flash (negative control) reporter constructs. After 48 h, cells were lysed and luciferase activity was determined.47 The results are expressed as a percentage of the ratio of TOP flash/FOP flash activity corrected by β‐galactosidase activity measured in cells transfected with the siRNA‐CTL. Results are representative of three independent experiments. *P < 0.05. Total RNA was extracted from MCF7 (e) and MDA‐MB‐231 cells (f). E‐cadherin and vimentin mRNA levels were evaluated by RT‐PCR. Hypoxanthine‐guanine phosphoribosyltransferase was used as a loading control. MCF7 (g) and MDA‐MB‐231 (h) cells were visualized and photographed under bright field illumination at ×20 magnification. MDA‐MB‐231 cells (i) were transfected and 48 h later cell invasion assays were performed. The graph compares the number of invasive siRNA‐CTL (white bar) and siRNA‐SCD1 (black bar) transfected cells. Results, representative of the three experiments, are expressed as a percentage of cells transfected with siRNA‐CTL. *P < 0.05.
Our data suggest that reducing SCD1 expression in breast cancer cells lowers their proliferation rate because of the lower expression of cell cycle genes and activation of the ERK1/2. We also showed that silencing SCD1 is associated with a decrease of the characteristic factors of the mesenchymal phenotype, such as increased GSK3 activity, reduction of β‐catenin nuclear localization and transactivation activity. SCD1 silencing also modifies the shape of the cells and their invasive potential, suggesting that high SCD1 expression is associated with increased transformation of cancer cells, which is a characteristic of the acquisition of a mesenchymal phenotype.
Discussion
The activation of de novo lipogenesis has been associated with aggressive cancer.33 Among the enzymes involved in this pathway, SCD1 catalyzes the production of MUFA from SFA precursors. These fatty acids are the most abundant in plasma membrane and the ratio of SFA to MUFA can affect membrane fluidity. Evidence has now emerged showing an implication of SCD1 in human lung, colon, prostate and breast cancer cells survival.8, 9, 10, 29, 34, 35, 36, 37, 38 In two breast cancer cell models, MCF7 and MDA‐MB‐231, decreasing SCD1 expression does not affect cell viability 48 h post‐transfection (Fig. 1c and d). However, in lung cancer cells, inhibition of SCD1 affects cell viability.39 This effect is observed in cells cultured with 2% serum but not with 10% serum. The difference in cell sensitivity to SCD1 inhibition could be attributed to the presence of a higher concentration of MUFA in the media containing 10% serum compared to 2% serum 39. Pharmaceutical SCD1 inhibition on MCF7 and MDA‐MB‐231 also affects the viability of cells cultured with 10% serum.37. Our experiments were performed in serum‐free medium. Therefore, it seems that serum concentration might not modify the effect of SCD1 on MCF7 and MDA‐MB‐231 survival. In addition, SCD1 depletion does not universally affect cell survival because the HCT116 colon cancer cells are more affected by SCD1 inhibition than the SKOV3 ovarian cancer cells.35 Therefore, the cancer cell type and serum concentrations might influence the effect of SCD1 inhibition on cell viability.
Upon silencing of SCD1, we also observed a significant diminution of ERK1/2 phosphorylation associated with a modification of cell cycle distribution and a decrease in cell cycle gene expression. Similar observations have been made in prostate cancer cells treated with a pharmaceutical SCD1 inhibitor.38 Although ERK1/2 phosphorylation decreased, the percentage of cells in G0–G1 increased by 11% and the proportion of cells in S and G2‐M phases decreased by 6 and 5%, respectively (Fig. 2). In lung cancer cells, the treatment of cells with a specific SCD1 pharmaceutical inhibitor increases the number of cells in the G0–G1 phase and decreases that in the S phase.40 Therefore, a lower SCD1 expression level is probably associated with an increased number of cells in the resting state.
Epithelial to mesenchymal transition represents one of the key events of cancer progression leading to cell dissemination and metastasis. The activation of the Wnt/β‐catenin signaling pathway is associated with the induction of EMT41, 42 and markers of its activation, such as GSK3 inhibition and nuclear β‐catenin, are positively correlated in breast cancer patients with poor prognosis.43 We observed a diminution of GSK3β phosphorylation on serine 9 in siRNA‐SCD1 cells compared to the control, suggesting an augmentation of GSK3 activity.23 Fritz and collaborators also observed the same phenomenon in prostate cancer cell lines transfected with a SCD1 siRNA or treated with a specific SCD1 inhibitor.38 As GSK3 is part of the complex that drives β‐catenin for proteasomal degradation,22 we analyzed the effect of SCD1 inhibition on the sub‐cellular localization of β‐catenin, a marker of the acquisition of the mesenchymal stage. The cytosolic amount and the nuclear localization of β‐catenin in MCF7 and MDA‐MB‐231 cells were modified upon SCD1 inhibition (Fig. 4a–c). The decrease of β‐catenin amounts within the nucleus was also reflected by its lower capacity to transactivate TCF target genes, such as cyclin D1 and vimentin (Fig. 2b–4f). These latter genes are mainly implicated in cell cycle progression and in the acquisition of a mesenchymal phenotype.44 An impairment of β‐catenin nuclear translocation has also been observed in prostate cancer cells,38 suggesting that decreasing SCD1 expression might have a positive effect on other types of cancers.
To our knowledge, the present study constitutes the first report showing an impairment of the Wnt/β‐catenin signaling pathway in SCD1 inhibited‐breast cancer cells. In cancer cells, secreted factors, such as the wingless proteins (Wnt‐1 and Wnt‐3a), are implicated in the initiation of Wnt/β‐catenin pathway activation.45 These proteins also create a positive feedback loop, contributing to sustain cancer cell proliferation. Of interest, Wnt‐3a has been shown to be acylated by one of the main products of SCD1: palmitoleic acid. Moreover, this acylation is a prerequisite for Wnt‐3a secretion.46 Hence, SCD1 inhibition might perturb Wnt‐3a acylation and secretion. Consequently, it modifies the Wnt/β‐catenin pathway activation, leading to an impairment of cell proliferation and mesenchymal behavior acquisition.
Another potential consequence of the effect of SCD1 inhibition could be the activation of the PI3K/Akt pathway, which is implicated in cancer cell survival. Indeed, inhibition of SCD1 in lung and prostate cancer cells also impairs Akt activation and, as a result, its inhibitory action on its target GSK3β.29, 38 However, the mechanism of Akt activation remains to be determined in breast cancer cells.
In summary, we have shown that low SCD1 expression is linked to a decrease in the proliferation rate of breast cancer cells. This was accompanied by an increase in GSK3 activity. Consequently, the nuclear translocation of β‐catenin was decreased, and its transactivation capacity. This strongly suggests a role of SCD1 in EMT and cancer progression. Therefore, SCD1 could be an efficient new therapeutic target for the treatment of metastatic breast cancer.
Disclosure Statement
The authors have no conflicts of interest to declare.
Acknowledgments
We thank Dr N. Rivard (Sherbrooke University) for the TOP and FOP flash vectors. We thank D. Flipo (University of Quebec in Montreal [UQÀM]) for the confocal microscopy analysis. This work was supported by Fondation UQÀM to DM and CC.
(Cancer Sci, doi: 10.1111/cas.12032, 2012)
References
- 1. Stephenson GD, Rose DP. Breast cancer and obesity: an update. Nutr Cancer 2003; 45: 1–16. [DOI] [PubMed] [Google Scholar]
- 2. Roberts R, Hodson L, Dennis AL et al Markers of de novo lipogenesis in adipose tissue: associations with small adipocytes and insulin sensitivity in humans. Diabetologia 2009; 52: 882–90. [DOI] [PubMed] [Google Scholar]
- 3. Caron‐Jobin M, Mauvoisin D, Michaud A et al Stearic acid content of abdominal adipose tissues in obese women. Nutrition and Diabetes 2012; 2: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Enoch HG, Catala A, Strittmatter P. Mechanism of rat liver microsomal stearyl‐CoA desaturase Studies of the substrate specificity, enzyme‐substrate interactions, and the function of lipid. J Biol Chem 1976; 251: 5095–103. [PubMed] [Google Scholar]
- 5. Ruggieri S, Roblin R, Black PH. Lipids of whole cells and plasma membrane fractions from Balb/c3T3, SV3T3, and concanavalin A‐selected revertant cells. J Lipid Res 1979; 20: 760–71. [PubMed] [Google Scholar]
- 6. Bougnoux P, Chajes V, Lanson M et al Prognostic significance of tumor phosphatidylcholine stearic acid level in breast carcinoma. Breast Cancer Res Treat 1992; 20: 185–94. [DOI] [PubMed] [Google Scholar]
- 7. Williams CM, Maunder K. Fatty acid compositions of inositol and choline phospholipids of breast tumours and normal breast tissue. Eur J Clin Nutr 1993; 47: 260–7. [PubMed] [Google Scholar]
- 8. Scaglia N, Caviglia JM, Igal RA. High stearoyl‐CoA desaturase protein and activity levels in simian virus 40 transformed‐human lung fibroblasts. Biochim Biophys Acta 2005; 1687: 141–51. [DOI] [PubMed] [Google Scholar]
- 9. Scaglia N, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase‐1 inactivates acetyl‐CoA carboxylase and impairs proliferation in cancer cells: role of AMPK. PLoS ONE 2009; 4: e6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Igal RA. Stearoyl‐CoA desaturase‐1: a novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010; 31: 1509–15. [DOI] [PubMed] [Google Scholar]
- 11. Chajes V, Hulten K, Van Kappel AL et al Fatty‐acid composition in serum phospholipids and risk of breast cancer: an incident case‐control study in Sweden. Int J Cancer 1999; 83: 585–90. [DOI] [PubMed] [Google Scholar]
- 12. Chajes V, Thiebaut AC, Rotival M et al Association between serum trans‐monounsaturated fatty acids and breast cancer risk in the E3N‐EPIC Study. Am J Epidemiol 2008; 167: 1312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pala V, Krogh V, Muti P et al Erythrocyte membrane fatty acids and subsequent breast cancer: a prospective Italian study. J Natl Cancer Inst 2001; 93: 1088–95. [DOI] [PubMed] [Google Scholar]
- 14. Zureik M, Ducimetiere P, Warnet JM, Orssaud G. Fatty acid proportions in cholesterol esters and risk of premature death from cancer in middle aged French men. BMJ 1995; 311: 1251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petrek JA, Hudgins LC, Ho M, Bajorunas DR, Hirsch J. Fatty acid composition of adipose tissue, an indication of dietary fatty acids, and breast cancer prognosis. J Clin Oncol 1997; 15: 1377–84. [DOI] [PubMed] [Google Scholar]
- 16. Zhu ZR, Agren J, Mannisto S et al Fatty acid composition of breast adipose tissue in breast cancer patients and in patients with benign breast disease. Nutr Cancer 1995; 24: 151–60. [DOI] [PubMed] [Google Scholar]
- 17. Simonsen NR, Fernandez‐Crehuet NJ, Martin‐Moreno JM et al Tissue stores of individual monounsaturated fatty acids and breast cancer: the EURAMIC study. European Community Multicenter Study on Antioxidants, Myocardial Infarction, and Breast Cancer. Am J Clin Nutr 1998; 68: 134–41. [DOI] [PubMed] [Google Scholar]
- 18. Yilmaz M, Christofori G, Lehembre F. Distinct mechanisms of tumor invasion and metastasis. Trends Mol Med 2007; 13: 535–41. [DOI] [PubMed] [Google Scholar]
- 19. Schmalhofer O, Brabletz S, Brabletz T. E‐cadherin, beta‐catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev 2009; 28: 151–66. [DOI] [PubMed] [Google Scholar]
- 20. Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol 2003; 19: 207–35. [DOI] [PubMed] [Google Scholar]
- 21. Amit S, Hatzubai A, Birman Y et al Axin‐mediated CKI phosphorylation of beta‐catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 2002; 16: 1066–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Behrens J, Jerchow BA, Wurtele M et al Functional interaction of an axin homolog, conductin, with beta‐catenin, APC, and GSK3beta. Science 1998; 280: 596–9. [DOI] [PubMed] [Google Scholar]
- 23. Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL‐receptor‐related protein mediates Wnt signalling in mice. Nature 2000; 407: 535–8. [DOI] [PubMed] [Google Scholar]
- 24. Korinek V, Barker N, Morin PJ et al Constitutive transcriptional activation by a beta‐catenin‐Tcf complex in APC–/– colon carcinoma. Science 1997; 275: 1784–7. [DOI] [PubMed] [Google Scholar]
- 25. Albanese C, Johnson J, Watanabe G et al Transforming p21ras mutants and c‐Ets‐2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 1995; 270: 23589–97. [DOI] [PubMed] [Google Scholar]
- 26. Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 1996; 271: 20608–16. [DOI] [PubMed] [Google Scholar]
- 27. Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003; 2: 339–45. [PubMed] [Google Scholar]
- 28. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase‐3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12: 3499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scaglia N, Igal RA. Inhibition of Stearoyl‐CoA Desaturase 1 expression in human lung adenocarcinoma cells impairs tumorigenesis. Int J Oncol 2008; 33: 839–50. [PubMed] [Google Scholar]
- 30. Gilles C, Polette M, Mestdagt M et al Transactivation of vimentin by beta‐catenin in human breast cancer cells. Cancer Res 2003; 63: 2658–64. [PubMed] [Google Scholar]
- 31. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta‐catenin is a target for the ubiquitin‐proteasome pathway. EMBO J 1997; 16: 3797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan D, Avtanski D, Saxena NK, Sharma D. Leptin‐induced epithelial‐mesenchymal transition in breast cancer cells requires beta‐catenin activation via Akt/GSK3‐ and MTA1/Wnt1 protein‐dependent pathways. J Biol Chem 2012; 287: 8598–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamashita T, Honda M, Takatori H et al Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J Hepatol 2009; 50: 100–10. [DOI] [PubMed] [Google Scholar]
- 34. Morgan‐Lappe SE, Tucker LA, Huang X et al Identification of Ras‐related nuclear protein, targeting protein for xenopus kinesin‐like protein 2, and stearoyl‐CoA desaturase 1 as promising cancer targets from an RNAi‐based screen. Cancer Res 2007; 67: 4390–8. [DOI] [PubMed] [Google Scholar]
- 35. Mason P, Liang B, Li L et al SCD1 Inhibition causes cancer cell death by depleting mono‐unsaturated fatty acids. PLoS ONE 2012; 7: e33823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hess D, Igal RA. Genistein downregulates de novo lipid synthesis and impairs cell proliferation in human lung cancer cells. Exp Biol Med (Maywood) 2011; 236: 707–13. [DOI] [PubMed] [Google Scholar]
- 37. Paton CM, Ntambi JM. Loss of stearoyl‐CoA desaturase activity leads to free cholesterol synthesis through increased Xbp‐1 splicing. Am J Physiol Endocrinol Metab 2010; 299: E1066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fritz V, Benfodda Z, Rodier G et al Abrogation of de novo lipogenesis by stearoyl‐CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol Cancer Ther 2010; 9: 1740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roongta UV, Pabalan JG, Wang X et al Cancer cell dependence on unsaturated fatty acids implicates stearoyl‐CoA desaturase as a target for cancer therapy. Mol Cancer Res 2011; 9: 1551–61. [DOI] [PubMed] [Google Scholar]
- 40. Hess D, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS ONE 2010; 5: e11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prasad CP, Rath G, Mathur S, Bhatnagar D, Parshad R, Ralhan R. Expression analysis of E‐cadherin, Slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer 2009; 9: 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim K, Lu Z, Hay ED. Direct evidence for a role of beta‐catenin/LEF‐1 signaling pathway in induction of EMT. Cell Biol Int 2002; 26: 463–76. [DOI] [PubMed] [Google Scholar]
- 43. Logullo AF, Nonogaki S, Pasini FS, Osorio CA, Soares FA, Brentani MM. Concomitant expression of epithelial–mesenchymal transition biomarkers in breast ductal carcinoma: association with progression. Oncol Rep 2010; 23: 313–20. [PubMed] [Google Scholar]
- 44. Lin SY, Xia W, Wang JC et al Beta‐catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci USA 2000; 97: 4262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bjorklund P, Svedlund J, Olsson AK, Akerstrom G, Westin G. The internally truncated LRP5 receptor presents a therapeutic target in breast cancer. PLoS ONE 2009; 4: e4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takada R, Satomi Y, Kurata T et al Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell 2006; 11: 791–801. [DOI] [PubMed] [Google Scholar]
- 47. De Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol 1987; 7: 725–37. [DOI] [PMC free article] [PubMed] [Google Scholar]