

Abstract
Barasertib, an aurora B inhibitor, terminates cell division, introduces polyploidy, and consequently causes apoptosis. In the present study, we evaluated the effect of the combination of barasertib and cytarabine (ara‐C), a key agent for leukemia chemotherapy, on leukemic cells in vitro. Human leukemia HL‐60 cells and HL‐60/ara‐C20 cells, a 20‐fold ara‐C‐resistant variant, were used. The 50% growth inhibitory concentrations of an active metabolite of barasertib, barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA), and ara‐C were 51 nM and 300 nM for HL‐60 cells and 70 nM and 5300 nM for HL‐60/ara‐C20 cells, respectively. Barasertib‐HQPA induced polyploidy with a subsequent induction of sub‐G1 phase apoptosis, indicating the M‐phase specific cytotoxicity. Cells treated with the S‐phase specific ara‐C accumulated in S phase and subsequently died through apoptosis. When HL‐60 cells were treated with barasertib‐HQPA and ara‐C in combination, a greater‐than‐additive apoptosis was induced. This enhancement was obtained when the cells were treated with barasertib‐HQPA prior to ara‐C (37.9% sub‐G1) or with both concurrently (31.2% sub‐G1), but not with ara‐C prior to barasertib‐HQPA (17.8% sub‐G1). The combination effects were similarly obtained in HL‐60/ara‐C20 cells with 19.7% sub‐G1 for barasertib‐HQPA→ara‐C, 18.4% sub‐G1 for both concurrently, and 13.8% sub‐G1 for ara‐C→barasertib‐HQPA, and another leukemic U937 cells with 25.4% sub‐G1 for barasertib‐HQPA→ara‐C, 28.2% sub‐G1 for both concurrently, and 16.0% sub‐G1 for ara‐C→barasertib‐HQPA. Barasertib‐HQPA inhibited aurora B autophosphorylation and histone H3 phosphorylation in all the cell lines. Barasertib‐HQPA did not inhibit DNA synthesis, allowing ara‐C incorporation into DNA for its cytotoxicity. Thus, barasertib‐HQPA and ara‐C provided a greater‐than‐additive cytotoxicity in leukemic cells in vitro.
In mammalian cells, the aurora family of serine/threonine protein kinases is composed of three paralogous proteins (aurora A, B, and C), among which aurora A and aurora B are essential regulators of mitotic entry and progression.1, 2, 3 Aurora B is the catalytic component of the chromosomal passenger complex. This complex regulates the spindle checkpoint and ensures the accurate segregation of chromatids, and correct microtubule/kinetochore attachment during the mitotic phase of the cell cycle. Therefore, aurora kinases are indispensable for accurate mitosis and cell division,1, 2, 3, 4, 5, 6, 7, 8 and are attractive targets for anticancer therapy.5, 6, 7, 8
Barasertib (formally AZD1152) is a highly potent and selective aurora B kinase inhibitor. After administration, barasertib is converted into the active moiety, barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA), which is a reversible ATP‐competitive inhibitor of aurora B. Barasertib inhibits the activity of aurora B, thereby inducing chromosome misalignment, and terminating cell division with the introduction of polyploidy. This agent consequently reduces cell viability and induces apoptosis in cancer cells.9, 10, 11, 12, 13 The antitumor action of barasertib is therefore M‐phase specific. A phase I study was conducted in Japan to assess the safety, pharmacokinetics and efficacy of barasertib in patients with refractory or relapsed acute myeloid leukemia (AML).14 A promising overall hematologic response rate of 19% was achieved, which warrants further investigation in leukemic patients.
A key agent used for the treatment of AML is the pyrimidine nucleoside analog cytarabine (ara‐C).15, 16, 17 However, long‐term survivors accounted for only 40% of the young adult AML patients who were treated with ara‐C‐based chemotherapy, suggesting a need for improvement of the treatment modality.18, 19 If barasertib is to be combined with ara‐C regimens, one should consider the difference in the mechanism of action of these two agents. Ara‐C is intracellularly activated to ara‐C triphosphate (ara‐CTP), which then inhibits DNA synthesis and induces cytotoxicity.20, 21 This cytotoxicity is S‐phase‐specific.20, 21 When the M‐phase specific baraserib is combined with ara‐C, barasertib induces polyploidy and inhibits cell division, but DNA synthesis still occurs; thus, ara‐C may still exert its cytotoxicity. Alternatively, ara‐C terminates the cell cycle at S‐phase and prevents tumor cells from entering M‐phase, which may then abrogate the cytotoxic effect of barasertib. Therefore, the incorporation of barasertib into AML chemotherapy should be carefully planned based on the pharmacological interaction of these two agents.
In the present study, we investigated the combination effects of barasertib and ara‐C in cultured leukemia cells in vitro. First, the cytotoxicity of barasertib was evaluated in relationship to the cell cycle. Next, the combination effects of barasertib and ara‐C on the induction of apoptosis and the cell growth inhibition were determined. The cytotoxicity achieved from different combination schedules was compared. Finally, the ara‐C‐resistant leukemic cell line was similarly evaluated to discover whether barasertib could be cytotoxic in the context of ara‐C resistance.
Materials and Methods
Chemicals and reagents
The active compound of barasertib, barasertib‐HQPA, was kindly supplied by AstraZeneca (Macclesfield, UK) and dissolved in dimethylsulfoxide to a stock concentration of 1 mM. Ara‐C, vincristine, paclitaxel, and docetaxel were purchased from Sigma (St. Louis, MO, USA).
Cell culture
The human leukemia cell lines HL‐60 and 20‐fold more ara‐C‐resistant HL‐60 variant, HL‐60/ara‐C20, were used.22 HL‐60/ara‐C20 cells exhibited reduced expression of human Equilliblative Nucleoside Transporter 1 and deoxycitidine kinase.22 HL‐60/ara‐C20 cells were maintained in the medium without ara‐C. Nevertheless, the cells kept their ara‐C‐resistant nature. The human monocytic leukemia cell line U937 was also used for comparison. These cell lines were cultured in RPMI1640 media supplemented with 10% FCS in a 5% CO2‐humidified atmosphere at 37°C.
Proliferation assay
To evaluate the growth inhibition effects, the sodium 3′‐(1‐[(phenylamino)‐carbonyl‐3,4‐tetrazolium])‐bis(4‐methoxy‐6‐nitro) benzene sulfonic acid hydrate (XTT) assay was performed according to the manufacturer's instructions (Roche, Indianapolis, IN, USA) with slight modifications.22
Cell cycle distribution
Flow cytometric analysis was performed to evaluate the cell cycle distribution. The treated samples were fixed in 80% ethanol, stained with 20 μg/mL propidium iodide (Beckman Coulter, Fullerton, CA, USA), and analyzed using FACSCanto II (BD Bioscience, Franklin Lakes, NJ, USA).
Morphological evaluation
The cells having been treated with barasertib‐HQPA were collected on a microscope slide by centrifugation (500g, 10 min) using the Shandon Cytospin III (Thermo Fisher Scientific, Waltham, MA, USA). The samples were then stained with May‐Grünwald and Giemsa dye, and evaluated by light microscopy.
Quantitation of apoptotic cell death
The population of cells in the sub‐G1 phase was determined using flow cytometry to assess apoptotic cell death.
Western blot analysis
The levels of aurora B, phospho‐aurora B, histone H3, and phospho‐histone H3 proteins were determined by standard Western blotting.22 Rabbit polyclonal anti‐aurora B (Abcam, Cambridge, CB4 0FL, UK), rabbit polyclonal anti‐phospho‐aurora B (Enzo Life Sciences, Farmingdale, NY, USA), rabbit polyclonal anti‐histone H3 (Cell Signaling Technology, Beverly, MA, USA), rabbit polyclonal anti‐phospho‐histone H3 (Cell Signaling Technology), and anti‐actin (Sigma) antibodies were used as primary antibodies.
Determination of intracellular ara‐CTP concentrations
The intracellular active metabolite of ara‐C, ara‐CTP was determined using HPLC as described previously.21 Briefly, the ara‐C‐treated sample was centrifuged to collect the cell pellet (400 g, 10 min, 4°C). The acid‐soluble fraction, the nucleotide pool, was extracted from the sample, and subjected to HPLC using a TSK gel DEAE‐2 SW column (length, 250 mm; internal diameter, 4.6 mm, TOSOH, Tokyo, Japan) and 0.06 M Na2HPO4 (pH 6.9) −20% acetonitrile buffer. The ara‐CTP peak was identified by its retention time and quantitated from its peak area at an absorbance of 269 nm.
Evaluation of DNA synthesis
DNA synthesis was determined as the incorporation of tritiated thymidine into DNA.23 In brief, the cells were preincubated with or without barasertib‐HQPA for 24 h, and then treated with tritiated thymidine for 6 h. The sample was then added to perchloric acid, and the DNA‐containing acid‐insoluble fraction was obtained. This fraction was dissolved in NaOH overnight and then subjected to scintillation counting.23
Ara‐C incorporation into DNA
The cells were preincubated with or without barasertib‐HQPA for 24 h, and were then treated with 10 μM tritiated ara‐C for 6 h. DNA‐containing acid‐insoluble fraction was subjected to scintillation counting.23
Calculation of the combination index
Combination index (CI) analysis provides quantitative information on the nature of drug interaction. The CI method was based on that described by Chou and Talalay,24 and the values were determined by using the computer software CalcuSyn (version 2.0; Biosoft, Great Shelford, Cambridge, UK). The CI values of less than, equal to, and more than 1 indicate synergy, additivity, and antagonism, respectively.24
Statistical analyses
All of the statistical analyses were performed using Microsoft Excel 2007 software (Microsoft, Redmond, WA, USA). All graphs were generated using GraphPad Prism software (version 5.0; GraphPad Software, San Diego, CA, USA). Values of P ≤ 0.05 were considered statistically significant.
Results
Cytotoxicity of barasertib‐HQPA in ara‐C‐sensitive and ara‐C‐resistant cell lines
The antileukemic effect of barasertib was evaluated in HL‐60 cells and HL‐60/ara‐C20 cells. HL‐60/ara‐C20 cells were 20‐fold more ara‐C‐resistant than were HL‐60 cells (Table 1). Both of the cell types were incubated with an active compound of barasertib, barasertib‐HQPA, for 72 h, followed by the evaluation of the growth inhibition using the XTT assay. Barasertib‐HQPA equally inhibited the growth of HL‐60 cells and HL‐60/ara‐C20 cells (Table 1). Both of the cell lines were also sensitive to several antimitotic agents (Table 2). The results thus indicated the efficacy of barasetib against leukemic cells regardless of the cellular sensitivity to ara‐C.
Table 1.
Drug sensitivity in HL‐60, HL‐60/ara‐C20, and U937
| Drugs | IC50 (nM) | ||
|---|---|---|---|
| HL‐60 | HL‐60/ara‐C20 | U937 | |
| Barasertib‐HQPA | 51 | 70 | 29 |
| Ara‐C | 300 | 5300 | 71 |
Cells were incubated with various concentrations of barasertib‐HQPA or ara‐C for 72 h. The IC50 was then determined by using the XTT assay. Barasertib‐HQPA, Barasertib ‐hydroxyquinazoline‐pyrazol‐aniline; Ara‐C, cytarabine.
Table 2.
Sensitivity to antimitotic agents in HL‐60 and HL‐60/ara‐C20
| Drugs | IC50 (nM) | |
|---|---|---|
| HL‐60 | HL‐60/ara‐C20 | |
| PTX | 4.0 | 4.2 |
| DTX | 4.2 | 3.9 |
| VCR | 4.3 | 3.7 |
Cells were incubated with various concentrations of a given anticancer agent for 72 h. The IC50 was then determined by using the XTT assay. DTX, docetaxel; PTX, paclitaxel; VCR, vincristine.
Cell cycle analysis after treatment with baraseritb‐HQPA
The cell cycle distribution of HL‐60 cells treated with barasertib‐HQPA for different length of time was evaluated using flow cytometry. The cells exhibited increased DNA contents of 4N and 8N, indicative of polyploidy, within 24–48 h of treatment (Fig. 1). After 48–72 h, barasertib‐HQPA induced apoptotic cell death, as detected by an increased sub‐G1 population compared for that of untreated cells (Fig. 1). Morphological changes were observed using May‐Giemsa stain at each time point (Fig. 2). The induction of polyploidy was obvious at 24–48 h, and thereafter, the nuclei showed morphology typical of apoptosis, such as nuclear fragmentation and condensation (Fig. 2). These observations were in accordance with the findings of the flow cytometric analysis (Fig. 1). When HL‐60 cells were treated with ara‐C, the G2M population was decreased within 24–48 h, reflecting ara‐C‐mediated inhibition of DNA synthesis in S phase (Fig. 1). Subsequently, the sub‐G1 population was increased at 48–72 h, suggestive of the induction of apoptosis (Fig. 1). These results thus suggested that barasertib‐HQPA and ara‐C modulated the cell cycle distribution differently, which might be attributable to the difference in the mechanisms of action of barasertib, which is M‐phase specific, and ara‐C, which is S‐phase specific.
Figure 1.
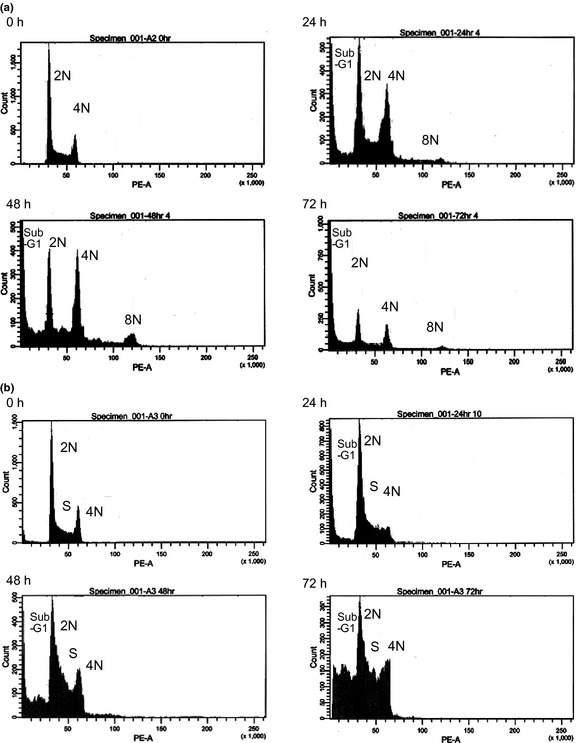
Flow cytometric analysis. (a, b) HL‐60 cells were treated with 25 nM barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA) (a) or 150 nM ara‐C (b) for the indicated time periods (0 h, 24 h, 48 h, and 72 h), and cell cycle analysis was performed. The flow cytometry results shown are representative of three independent experiments.
Figure 2.
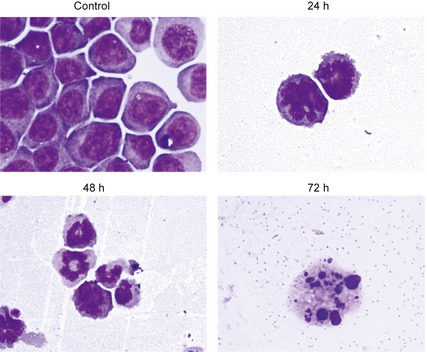
Morphological changes after barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA) treatment. HL‐60 cells having been treated with 25 nM barasertib‐HQPA for the indicated time periods (0 h, 24 h, 48 h, and 72 h) were collected and stained with May‐Giemsa dye (×400).
Inhibition of aurora B kinase activity
The expression levels of both aurora B and the autophosphorylation of aurora B were evaluated. When HL‐60 cells were treated with barasertib‐HQPA for 48 h, the expression of aurora B was unchanged, but the autophosphorylation was apparently inhibited (Fig. 3). The phosphorylation of histone H3, a representative substrate of aurora B, was also inhibited by barasertib‐HQPA, whereas the expression level of histone H3 was not altered. The inhibition of the autophosphorylation of aurora B and the phosphorylation of histone H3 by barasertib‐HQPA was also observed in ara‐C‐resistant HL‐60/ara‐C20 cells and other leukemic U937 cells (Fig. 3). Thus, barasertib‐HQPA effectively inhibited the activity of aurora B kinase in leukemic cells.
Figure 3.
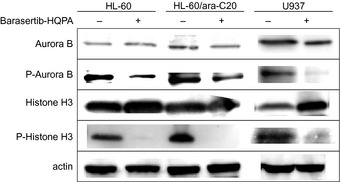
Inhibition of aurora B kinase by barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA). HL‐60 cells and HL‐60/ara‐C20 cells were incubated with 25 nM barasertib‐HQPA for 48 h, while U937 cells were incubated with 15 nM barasertib‐HQPA for 48 h. The protein levels of aurora B, phospho‐aurora B, histone H3, and phospho‐histone H3 were determined by Western blotting.
Increase in ara‐C‐induced cytotoxicity by the addition of barasertib‐HQPA
To evaluate the cytotoxicity of the combination of ara‐C with barasertib‐HQPA, HL‐60 cells and HL‐60/ara‐C20 cells were treated for 48 h with ara‐C, barasertib‐HQPA, or these two agents in combination. Flow cytometric analysis was performed to determine apoptotic cell death thereafter. Comparison of the resultant sub‐G1 populations revealed that the combination treatment of ara‐C and barasertib‐HQPA produced more‐than‐additive apoptotic cell death compared with that from either agent individually or that from the sum of the two agents (Fig. 4). The combination effect occurred when the cells were treated with both agents simultaneously (31.2% sub‐G1 for HL‐60, 18.4% sub‐G1 for HL‐60/ara‐C20) or when the cells were treated with barasertib‐HQPA 24 h prior to ara‐C administration (37.9% sub‐G1 for HL‐60, 19.7% sub‐G1 for HL‐60/ara‐C20). However, the cytotoxic effect was reduced, when ara‐C was administered 24 h prior to the addition of barasertib‐HQPA (17.8% sub‐G1 for HL‐60, 13.8% sub‐G1 for HL‐60/ara‐C20). The advantage of concurrent administration and barasertib‐HQPA→ ara‐C over ara‐C→barasertib‐HQPA was not statistically significant in HL‐60/ara‐C20 cells. This schedule‐dependency was similarly observed in another leukemic U937 cells with 25.4% sub‐G1 for barasertib‐HQPA→ara‐C, 28.2% sub‐G1 for both concurrently, and 16.0% sub‐G1 for ara‐C→barasertib‐HQPA (Fig. 4). Importantly, the dose and the incubation period (25 nM, 48 h) of barasertib‐HQPA that were required for the combination effect with ara‐C (Fig. 4) in HL‐60 cells and HL‐60/ara‐C20 cells inhibited autophosphorylation of aurora B and phosphorylation of histone H3 in these cell lines (Fig. 3). Moreover, the incubation with 15 nM barasertib for 48 h, which exerted the combination effect with ara‐C in U937 cells (Fig. 4), also inhibited autophosphorylation of aurora B and phosphorylation of histone H3 in U937 cells (Fig. 3). Apart from this short incubation experiment to determine apoptosis, when the cells were incubated for 72 h with both agents concurrently, the combination effects on the cell growth inhibition were determined by using the CI value. The CI values were 0.973 for HL‐60 cells, 0.733 for HL‐60/ara‐C20 cells, and 0.970 for U937 cells, respectively (Fig. 5), suggesting that the combination effects were at least additive in all of these cell lines. Thus, ara‐C combined with barasertib‐HQPA produced enhanced cytotoxicity.
Figure 4.
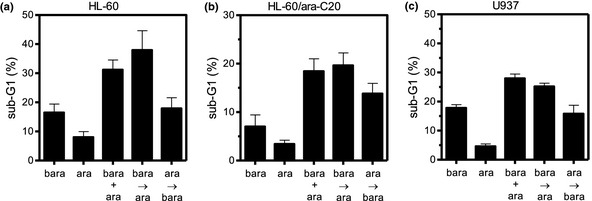
Schedule dependency of the cytotoxic effects of ara‐C and barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA) in combination. HL‐60 cells (a) and HL‐60/ara‐C20 cells (b) were treated for 48 h with 25 nM barasertib‐HQPA or a minimally toxic concentration of ara‐C (100 nM for HL‐60, 2 μM for HL‐60/ara‐C20) or both in combination, followed by the evaluation of apoptotic cell death using flow cytometry. Another leukemic cell line U937 was also evaluated in the parallel experiment using 15 nM barasertib‐HQPA and 80 nM ara‐C (c). Bara, barasertib‐HQPA alone; ara, ara‐C alone; bara + ara, concurrent administration of barasertib‐HQPA + ara‐C; bara→ara, barasertib‐HQPA administered 24 h prior to the addition of ara‐C; ara→bara, ara‐C administered 24 h prior to the addition of barasertib‐HQPA. Bara + ara versus bara→ara, P = 0.394; bara + ara versus ara→bara, P = 0.027; and bara→ara versus ara→bara, P = 0.022 in HL‐60 cells. Bara + ara versus bara→ara, P = 0.764; bara + ara versus ara→bara, P = 0.193; and bara→ara versus ara→bara, P = 0.123 in HL‐60/ara‐C20 cells. Bara + ara versus bara→ara P = 0.16; bara + ara versus ara→bara, P = 0.003; and bara→ara versus ara→bara, P = 0.01 in U937 cells. The values are the mean ± SD of at least three independent experiments.
Figure 5.
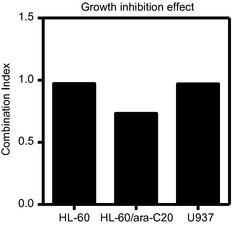
The Combination Index (CI) in each cell line (HL‐60, HL60/ara‐C20, U937) for the combination treatment using barasertib‐HQPA and ara‐C. When the cells were incubated for 72 h with both agents concurrently, the combination effects on the cell growth were determined as the CI by using CalcuSyn software. The CI values of less than, equal to, and more than 1 indicated synergy, additivity, and antagonism, respectively.
Alteration of the cell cycle by the treatment with ara‐C and barasertib‐HQPA in combination
To further investigate the schedule dependency of the combination effect, the cell cycle distribution was evaluated in HL‐60 cells that had been treated with ara‐C and barasertib‐HQPA in combination (Figs 6, 7). Polyploidy was induced, followed by an increase in the proportion of cells in the sub‐G1 phase, when the cells were treated with ara‐C and barasertib‐HQPA simultaneously or with barasertib‐HQPA→ara‐C (Figs 6, 7). Conversely, when ara‐C preceded barasertib‐HQPA, the cells accumulated in S phase due to the action of ara‐C, resulting in a reduction of the G2‐M population and the formation of polyploidy cells (Figs 6, 7). This was accompanied by the lower amount of sub‐G1 induction (Figs 6, 7). The results thus suggested that the inhibition of the cell cycle progression from S phase to G2M phase by ara‐C might attenuate the cytotoxicity of barasertib‐HQPA.
Figure 6.
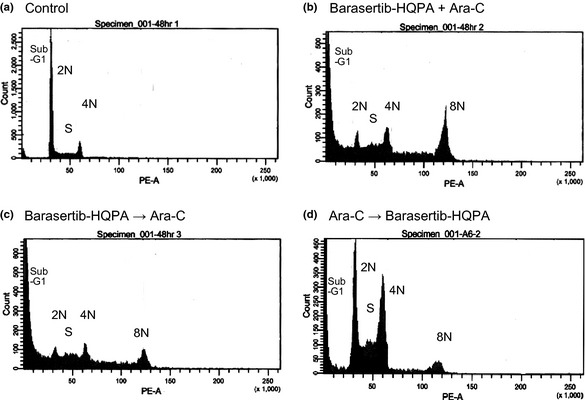
Flow cytometric analysis of the combination treatment with ara‐C and barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA). HL‐60 cells were treated with ara‐C plus barasertib‐HQPA for 48 h, followed by the cell cycle analysis. (a) Untreated control cells. (b) Cells treated with ara‐C and barasertib‐HQPA simultaneously. (c) Cells treated with barasertib‐HQPA for 24 h prior to the addition of ara‐C, (d) Cells treated with ara‐C for 24 h prior to the addition of barasertib‐HQPA. The images are representative of three independent experiments.
Figure 7.
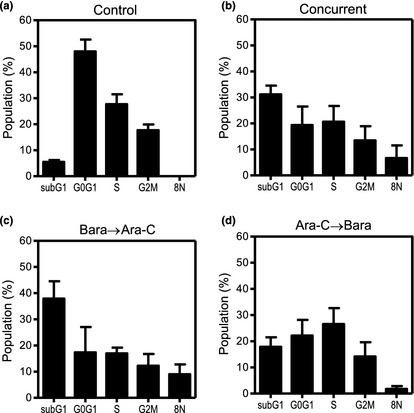
Alteration of the cell cycle by the combination treatment with ara‐C and barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA). HL‐60 cells were treated with ara‐C and barasertib‐HQPA for 48 h, followed by the cell cycle analysis using flow cytometry. (a) Untreated control cells. (b) Cells concurrently treated with ara‐C and barasertib‐HQPA. (c) Cells treated with barasertib‐HQPA for 24 h prior to the addition of ara‐C, or (d) Cells treated with ara‐C for 24 h prior to the addition of barasertib‐HQPA. The values are the mean ± SD of at least three independent experiments.
Cellular DNA synthesis in the presence or absence of barasertib‐HQPA
Tritiated thymidine and tritiated ara‐C were used to determine whether cellular DNA synthesis was affected by barasertib‐HQPA. The cells were incubated for 24 h with or without barasertib‐HQPA, followed by the incubation with tritiated thymidine or tritiated ara‐C. The incorporation of thymidine, an indicative of ongoing DNA synthesis, was not altered by preincubation with barasertib‐HQPA (Fig. 8a). Moreover, ara‐C incorporation into DNA, a critical event for the exertion of its cytotoxicity, was not reduced in the presence of barasertib‐HQPA (Fig. 8b). Ara‐C must be activated intracellularly to ara‐CTP before being incorporated into DNA. The production of ara‐CTP was unchanged by the preincubation with barasertib‐HQPA (Fig. 8c). Equivalent findings were obtained with HL‐60 cells and HL‐60/ara‐C20 cells. Therefore, these results suggested that a short incubation with barasertib‐HQPA did not alter cellular DNA synthesis, which would preserve the cytotoxicity of ara‐C.
Figure 8.
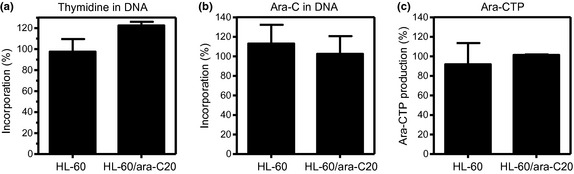
Barasertib‐hydroxyquinazoline‐pyrazol‐aniline (Barasertib‐HQPA) did not affect DNA synthesis. (a, b) HL‐60 cells and HL‐60/ara‐C20 cells were preincubated for 24 h with or without barasertib‐HQPA, followed by the additional incubation with tritiated thymidine (a) or tritiated ara‐C (b) for 6 h. The incorporation of each tritiated compound into DNA was determined by scintillation counting. The percentage of incorporation was determined as the ratio of the radioactivity counts of the cells preincubated with/without barasertib‐HQPA. (c) Barasertib‐HQPA did not affect intracellular ara‐CTP production levels. Intracellular ara‐CTP concentrations were measured using high performance liquid chromatography (HPLC) after the cells had been incubated with 10 μM ara‐C for 6 h. The percentage of ara‐CTP production was determined as the ratio of the ara‐CTP levels of the cells preincubated with/without barasertib‐HQPA. The values are the mean ± SD of at least three independent experiments.
Discussion
Among the aurora family members, aurora A and aurora B are overexpressed in many tumor types including leukemia,1, 2, 3, 25 suggesting aurora kinase as a therapeutic target. Barasertib has been shown to effectively inhibit the growth of many types of cancer cells, including leukemic cells,9, 10, 13, 26, 27, 28, 29, 30, 31, 32, 33 and as such, barasertib has been undergoing clinical evaluation.
The cytotoxicity of similar aurora kinase inhibitor SNS‐314 in combination with common chemotherapeutic agents was evaluated in cell culture and xenograft models.34 The colorectal carcinoma cell line HCT116 was treated with SNS‐314 and an anticancer agent from a panel composed of gemcitabine, 5‐fluorouracil, carboplatin, daunomycin, the active metabolite of irinotecan SN38, docetaxel, and vincristine. SNS‐314 had predominantly additive effects when administered concurrently with these agents.34 In another study, the combination effect of barasertib‐HQPA and SN38 was evaluated.35 When SN‐38 was given before or concomitantly with barasertib‐HQPA, SN‐38 blocked the effect of barasertib‐HQPA by arresting the cells in G2 and inhibiting them from undergoing polyploidy. With the reverse combination (barasertib‐HQPA followed by SN‐38), there was a significant induction of polyploidy and apoptosis.35 Moreover, the optimal sequence of barasertib‐HQPA and ionizing radiation combination therapy was investigated36 Enhanced cell killing was observed when cells were irradiated 24 h after exposure to barasertib‐HQPA, whereas the concomitant application of radiation and barasertib‐HQPA failed to yield an additive or synergistic effect.36 Thus, the administration schedule of combination chemotherapy using aurora kinase inhibitors should be carefully planned to obtain the maximal efficacy. Moreover, the effects of combining barasertib with ara‐C have not been previously evaluated.
The incorporation of barasertib into an ara‐C‐based regimen for treating AML should be carefully planned because the cytotoxicity of barasertib is M‐phase specific, but that of ara‐C is S‐phase specific. In the present study, barasertib‐HQPA was cytotoxic to HL‐60 cells and ara‐C‐resistant HL‐60/ara‐20 cells as a single agent (Table 1, Figs 1, 2). The combination of ara‐C with barasertib‐HQPA had a greater‐than‐additive cytotoxicity in HL‐60 cells compared to that of either agent individually or to that of the sum of the two agents; however, the effect was attained only by the concurrent use of ara‐C and barasertib‐HQPA or the sequential administration of barasertib‐HQPA→ara‐C (Fig. 4). The reverse administration schedule (ara‐C→barasertib‐HQPA) was less effective for inducing apoptosis (Fig. 4). The combination was similarly effective against ara‐C‐resistant HL‐60/araC20 cells and different cell line U937 cells (Fig. 4). Thus, the present study suggests that barasertib may be incorporated into an ara‐C‐based chemotherapy, but the efficacy will be attenuated if ara‐C is administered prior to barasertib.
The schedule dependency for the combination effect of ara‐C and barasetib‐HQPA appears to be mechanistically correlated to the cell cycle. The inhibition of aurora B inactivates the spindle checkpoint, resulting in aborted cell division without mitotic arrest.3, 7, 28 This inhibition results in an accumulation of the DNA content of the cell to 4N–8N, and continued suppression of the activity of aurora B leads to further rounds of genome replication without division. This process consequently results in tumor cell death by apoptosis.3, 7, 28 When HL‐60 cells were treated with barasertib‐HQPA alone, the treatment induced increase of the DNA content to 4N and 8N, indicative of polyploidy, and subsequently induced an increase in the sub‐G1 population, apoptosis (Figs 1, 2). This was compatible with those reported in previous studies,27, 32 and reflects the mechanism of action of the drug. Barasertib‐HQPA inhibited the autophosphorylation of aurora B in HL‐60 cells and ara‐C‐resistant HL‐60/ara‐C20 cells at 48 h (Fig. 3). In general, aurora B activation is triggered by autophosphorylation and is responsible for the mitotic phosphorylation of histone H3 that is necessary for chromosome condensation and entry into mitosis.1, 2, 24, 37 The inhibition of aurora B kinase (Fig. 3) was chronologically related to the induction of polyploidy, with both occurring by 48 h (Fig. 1). The results suggest a close association between cell cycle manipulation and the inhibition of aurora B kinase. Concurrent treatment with ara‐C and barasertib‐HQPA or sequential treatment with barasertib‐HQPA 24 h prior to the addition of ara‐C also caused the accumulation of DNA to 4N–8N, with a greater induction of apoptosis compared with barasertib‐HQPA alone (Figs 4, 5, 6, 7). The enhanced cytotoxicity attained using these combination schedules suggested that although the cells were under barasertib‐HQPA‐induced mitotic stress, DNA synthesis still occurred, providing a therapeutic window for ara‐C. In contrast, when ara‐C was administered before barasertib‐HQPA, the cells accumulated in S phase due to the inhibition of DNA synthesis by ara‐C (Figs 6, 7). This prevented cell cycle progression from the S phase to the G2M phase, in which barasertib‐HQPA could have exerted its cytotoxicity. The precise mechanism of leukemic cell killing induced by ara‐C and barasertib‐HQPA in combination remains to be clarified. However, the inhibition of cell division by barasertib‐HQPA combined with the termination of DNA synthesis by ara‐C may constitute a general strategy to promote mitotic catastrophe. This mechanism of action was also speculated for the effects of the combination of barasertib and ionizing radiation.36
As mentioned above, an important factor that might contribute to the combination effect of ara‐C and barasertib‐HQPA is that DNA replication was not affected by preincubation with barasertib‐HQPA (Fig. 8). This lack of an effect was apparent from the results demonstrating that thymidine uptake was unchanged in the presence of barasertib‐HQPA (Fig. 8a). This phenomenon enabled ara‐C to be incorporated into the DNA, which is indispensable to the cytotoxicity of ara‐C.21, 22 It was clearly demonstrated that intracellular ara‐CTP production and ara‐C incorporation into DNA were compatible in cells, regardless of whether they had been preincubated with barasertib‐HQPA (Fig. 8b,c). The results obtained with HL‐60 cells and HL‐60/ara‐C20 cells were similar. Thus, these results suggest that the presence of barasertib did not attenuate the cytotoxicity of ara‐C.
In conclusion, the combination of ara‐C with barasertib‐HQPA provided a greater‐than‐additive cytotoxicty in leukemic cells regardless of their ara‐C sensitivity. The concurrent or sequential administration of ara‐C with barasertib‐HQPA (barasertib‐HQPA→ara‐C) was more cytotoxic than the reverse regimen (ara‐C→barasertib‐HQPA). These preclinical data may facilitate optimization of the therapeutic potential of barasertib in combination with ara‐C for AML through scheduling.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (23501307, 2010) and by a Grant from the Gout Research Foundation (2010).
(Cancer Sci 2013; 104: 926–933)
References
- 1. Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol 2003; 4: 842–54. [DOI] [PubMed] [Google Scholar]
- 2. Ducat D, Zheng Y. Aurora kinases in spindle assembly and chromosome segregation. Exp Cell Res 2004; 301: 60–7. [DOI] [PubMed] [Google Scholar]
- 3. Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005; 8: 7–12. [DOI] [PubMed] [Google Scholar]
- 4. Keen N, Taylor S. Aurora‐kinase inhibitors as anticancer agents. Nat Rev Cancer 2004; 4: 927–36. [DOI] [PubMed] [Google Scholar]
- 5. Keen N, Taylor S. Mitotic drivers‐inhibitors of the aurora B kinase. Cancer Metastasis Rev 2009; 28: 185–95. [DOI] [PubMed] [Google Scholar]
- 6. Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res 2006; 12: 6869–75. [DOI] [PubMed] [Google Scholar]
- 7. Girdler F, Gascoigne KE, Eyers PA et al Validating aurora B as an anti‐cancer drug target. J Cell Sci 2006; 119: 3664–75. [DOI] [PubMed] [Google Scholar]
- 8. Andrews PD. Aurora kinases: shining lights on the therapeutic horizon? Oncogene 2005; 24: 5005–15. [DOI] [PubMed] [Google Scholar]
- 9. Keen N, Brown E, Crafter C et al Biological characterization of AZD1152, a highly potent and selective inhibitor of aurora kinase activity. Clin Cancer Res 2005; 11: B220. [Google Scholar]
- 10. Mortlock AA, Foote KM, Heron NM et al Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J Med Chem 2007; 50: 2213–24. [DOI] [PubMed] [Google Scholar]
- 11. Yang J, Ikezoe T, Nishioka C et al AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo . Blood 2007; 110: 2034–40. [DOI] [PubMed] [Google Scholar]
- 12. Evans RP, Naber C, Steffler T et al The selective Aurora B kinase inhibitor AZD1152 is a potential new treatment for multiple myeloma. Br J Haematol 2008; 140: 295–302. [DOI] [PubMed] [Google Scholar]
- 13. Oke A, Pearce D, Wilkinson RW et al AZD1152 rapidly and negatively affects the growth and survival of human acute myeloid leukemia cells in vitro and in vivo . Cancer Res 2009; 69: 4150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsuboi K, Yokozawa T, Sakura T et al A Phase I study to assess the safety, pharmacokinetics and efficacy of barasertib (AZD1152), an Aurora B kinase inhibitor, in Japanese patients with advanced acute myeloid leukemia. Leuk Res 2011; 35: 1384–9. [DOI] [PubMed] [Google Scholar]
- 15. Burnett AK, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol 2011; 29: 487–94. [DOI] [PubMed] [Google Scholar]
- 16. Fernandez HF. New trends in the standard of care for initial therapy of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program 2010; 2010: 56–61. [DOI] [PubMed] [Google Scholar]
- 17. Meyer RJ, Davis RB, Schiffer CA. Intensive postremission chemotherapy in adults with acute myeloid‐leukemia. N Engl J Med 1994; 331: 896–903. [DOI] [PubMed] [Google Scholar]
- 18. Appelbaum FR, Gundacker H, Head DR et al Age and acute myeloid leukemia. Blood 2006; 107: 3481–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blum W, Marcucci G. New approaches in acute myeloid leukemia. Best Pract Res Clin Haematol 2008; 21: 29–41. [DOI] [PubMed] [Google Scholar]
- 20. Garcia‐Carbonero R, Ryan DP, Chabner BA. Cytidine analogs In: Chabner BA, Longo DL, eds. Cancer Chemotherapy and Biotherapy. Philadelphia: Lippincott‐Raven Publishers, 1996; 265–94. [Google Scholar]
- 21. Yamauchi T, Ueda T, Nakamura T. A new sensitive method for determination of intracellular 1‐β‐D‐arabinofuranosylcytosine 5′‐triphosphate content in human materials in vivo . Cancer Res 1996; 56: 1800–4. [PubMed] [Google Scholar]
- 22. Negoro E, Yamauchi T, Urasaki Y et al Characterization of cytarabine‐resistant leukemic cell lines established from five different blood cell lineages using gene expression and proteomic analyses. Int J Oncol 2011; 38: 911–9. [DOI] [PubMed] [Google Scholar]
- 23. Yamauchi T, Nowak BJ, Keating MJ, Plunkett W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4‐hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin Cancer Res 2001; 7: 3580–9. [PubMed] [Google Scholar]
- 24. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 25. Ikezoe T, Yang J, Nishioka C et al A novel treatment strategy targeting Aurora kinases in acute myelogenous leukemia. Mol Cancer Ther 2007; 6: 1851–7. [DOI] [PubMed] [Google Scholar]
- 26. Wilkinson RW, Odedra R, Heaton SP et al AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res 2007; 13: 3682–8. [DOI] [PubMed] [Google Scholar]
- 27. Walsby E, Walsh V, Pepper C, Burnett A, Mills K. Effects of the aurora kinase inhibitors AZD1152‐HQPA and ZM447439 on growth arrest and polyploidy in acute myeloid leukemia cell lines and primary blasts. Haematologica 2008; 93: 662–9. [DOI] [PubMed] [Google Scholar]
- 28. Grundy M, Seedhouse C, Shang S, Richardson J, Russell N, Pallis M. The FLT3 internal tandem duplication mutation is a secondary target of the aurora B kinase inhibitor AZD1152‐HQPA in acute myelogenous leukemia cells. Mol Cancer Ther 2010; 9: 661–72. [DOI] [PubMed] [Google Scholar]
- 29. Payton M, Bush TL, Chung G et al Preclinical evaluation of AMG 900, a novel potent and highly selective pan‐aurora kinase inhibitor with activity in taxane‐resistant tumor cell lines. Cancer Res 2010; 70: 9846–54. [DOI] [PubMed] [Google Scholar]
- 30. Gully CP, Zhang F, Chen J et al Antineoplastic effects of an Aurora B kinase inhibitor in breast cancer. Mol Cancer 2010; 9: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mori N, Ishikawa C, Senba M, Kimura M, Okano Y. Effects of AZD1152, a selective Aurora B kinase inhibitor, on Burkitt's and Hodgkin's lymphomas. Biochem Pharmacol 2011; 81: 1106–15. [DOI] [PubMed] [Google Scholar]
- 32. Niermann KJ, Moretti L, Giacalone NJ et al Enhanced radio sensitivity of androgen‐resistant prostate cancer: AZD1152‐mediated Aurora kinase B inhibition. Radiat Res 2011; 175: 444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Libertini S, Abagnale A, Passaro C et al AZD1152 negatively affects the growth of anaplastic thyroid carcinoma cells and enhances the effects of oncolytic virus dl922–947. Endocr Relat Cancer 2011; 18: 129–41. [DOI] [PubMed] [Google Scholar]
- 34. VanderPorten EC, Taverna P, Hogan JN, Ballinger MD, Flanagan WM, Fucini RV. The Aurora kinase inhibitor SNS‐314 shows broad therapeutic potential with chemotherapeutics and synergy with microtubule‐targeted agents in a colon carcinoma model. Mol Cancer Ther 2009; 8: 930–9. [DOI] [PubMed] [Google Scholar]
- 35. Nair JS, de Stanchina E, Schwartz GK. The topoisomerase I poison CPT‐11 enhances the effect of the aurora B kinase inhibitor AZD1152 both in vitro and in vivo. Clin Cancer Res 2009; 15: 2022–30. [DOI] [PubMed] [Google Scholar]
- 36. Tao Y, Leteur C, Calderaro J et al The aurora B kinase inhibitor AZD1152 sensitizes cancer cells to fractionated irradiation and induces mitotic catastrophe. Cell Cycle 2009; 8: 3172–81. [DOI] [PubMed] [Google Scholar]
- 37. Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol 2001; 152: 669–82. [DOI] [PMC free article] [PubMed] [Google Scholar]