

Abstract
The presence of epidermal growth factor receptor (EGFR) somatic mutations in non‐small‐cell lung cancer patients is associated with response to treatment with EGFR‐tyrosine kinase inhibitors, such as gefitinib and erlotinib. More than 100 mutations in the kinase domain of EGFR have been identified. In particular there are many variations of deletion mutations in exon 19. In this study, using yellow fluorescent protein‐tagged fragments of the EGFR intracellular domain, we examined the differences in sensitivity to gefitinib, erlotinib and afatinib between several exon 19 mutants and other common EGFR mutations. We also used serum of patients undergoing treatment with EGFR‐tyrosine kinase inhibitors in this system. In addition, we examined the relative kinase activity of these mutants by measuring relative fluorescent intensity after immunofluorescence staining. We found that both sensitivity to EGFR‐tyrosine kinase inhibitors and relative kinase activity differed among several EGFR mutations found in the same region of the kinase domain. This study underscores the importance of reporting the clinical outcome of treatment in relation to different EGFR mutations.
About half of lung adenocarcinoma patients in Japan have somatic mutations in the kinase domain of epidermal growth factor receptor (EGFR),1, 2 and the presence of these mutations is known to be associated with increased response to treatment with EGFR‐tyrosine kinase inhibitors (EFGR‐TKI).3, 4, 5 To date, more than 100 EGFR somatic mutations have been identified in lung cancer patients, as detailed in the COSMIC database (www.sanger.ac.uk/genetics/CGP/cosmic/). About 90% of EGFR mutations consist of either short deletion mutations in exon 19 or a point mutation in exon 21 (L858R). Mitsudomi et al.6 reported the response rate to gefitinib was higher in patients with exon 19 deletion mutations (81%) than in those with an L858R mutation (71%) in exon 21. The L861Q point mutation in exon 21 accounts for 1–2% of EGFR mutations,2, 7 and the response rate of affected patients to EGFR‐TKI is reportedly 60%.8 G719X is a point mutation in exon 18 of EGFR, in which the glycine at codon 719 is substituted with cysteine, alanine or serine; it comprises <5% of all EGFR mutations. Patients with these mutations are reported to be less sensitive to EGFR‐TKI, and the response rate is around 56%.6 Mutations in exon 20, such as the T790M second mutation, are known to be resistant to EGFR‐TKI. Thus, there is variability in sensitivity of different EGFR somatic mutations to these drugs.9 Furthermore limited data exist regarding the more uncommon EGFR mutations, including the less frequent variants in exon 19. In this study, using yellow fluorescent protein (YFP)‐tagged fragments of the EGFR intracellular domain (YFP‐EGFR‐ICD), we examined the sensitivity of various exon 19 mutations and other common EGFR mutations to EGFR‐TKI.10, 11 We then compared the autophosphorylation levels of the kinase domains of the different mutants by measuring fluorescent intensity.
Materials and Methods
Plasmid construction and site‐directed mutagenesis
The mutant YFP‐EGFR‐ICD constructs were generated as described previously.10, 11 A KOD ‐Plus‐ Mutagenesis kit (TOYOBO, Tokyo, Japan) was used to construct EGFR mutants according to the manufacturer's protocol with WT YFP‐EGFR‐ICD as a template. Primers for each mutation are described in Data S1.
Cell culture, transfections and drug treatments
The human breast cancer cell line Michigan Cancer Foundation (MCF)‐7 was grown in DMEM supplemented with 10% FBS, 100‐U/mL penicillin and 100‐μg/mL streptomycin (Gibco, Carlsbad, CA, USA). Cells were seeded onto sterile glass coverslips in six‐well plates and transfected with 1‐μg plasmid DNA using the X‐tremeGENE 9 Transfection Reagent (Roche, Indianapolis, IN, USA) according to the manufacturer's protocol. Gefitinib, erlotinib and afatinib were added at the indicated concentrations at 24 h after transfection, and the cells were incubated for 12 h before they were processed for immunofluorescence analyses. Drug treatments were always performed in standard culture medium containing 10% FBS. Gefitinib and erlotinib were purchased from Cayman Chemical (Ann Arbor, MI, USA), and afatinib was purchased from Selleck Chemicals (Houston, TX, USA).
Immunofluorescence and microscopy analysis
To evaluate EGFR autophosphorylation, a rabbit anti‐phosphorylated EGFR‐Y1068 antibody (#3777S, diluted 1:200, Cell Signaling Technology, Danvers, CO, USA) was used. The immunostaining procedure was used as previously described.12 Briefly, cells were fixed using 4% formaldehyde in PBS for 20 min and permeabilized with 0.2% Triton X‐100 in PBS for 15 min. After a blocking step with Blocking One Histo (Nakarai, Kyoto, Japan) for 30 min, primary antibody diluted in PBS with 0.1% Tween20 was applied for 1 h. After being washed with PBS, samples were incubated with Alexa Fluor 594 (AF‐594)‐conjugated anti‐rabbit secondary antibody (diluted 1:400, Life Technologies, Carlsbad, CA, USA) for 1 h. Finally, the coverslips were mounted onto microscopic slides with VECTASHIELD Mounting Medium with DAPI (Vector Laboratories Inc., Burlingame, CA, USA).
Slides were examined with a Keyence BZ‐8100 fluorescence microscope (Osaka, Japan). BZ Analyzer software (Keyence) was used to collect images, and exposure time was kept constant to allow for comparison of the signal intensities among different samples.
Semi‐quantitative comparison of YFP‐EGFR‐ICD autophosphorylation levels was performed with computer‐assisted image analysis. With a ×20 objective lens, images of several transfected cells were taken, and the fluorescent intensity in the green and red channels was measured within a cytoplasmic area (YFP signal and AF‐594 signal) and within an area outside the cells (background). The intensity of the YFP and AF‐594 signals for each cell was plotted on a scatter plot and an approximation straight lines was obtained. Then, the angles of inclination were compared on a bar graph. All plotted signals in each group were subjected to the analysis of covariance. P‐values less than 0.05 were considered to be statistically significant. Excel 2008 (Microsoft Corporation, Redmond, WA, USA) was used for these analyses.
Results
Sensitivity to EGFR‐TKI
Michigan Cancer Foundation‐7 cells were transfected with plasmids encoding YFP‐EGFR‐ICD for WT EGFR or mutant variants of EGFR. Twenty four hours post‐transfection gefitinib or erlotinib at a final concentration ranging from 10 nM to 10 μM and afatinib at a final concentration ranging from 10 to 500 nM were added to the culture medium. As reported previously, the YFP‐tagged EGFR fragments used in this study lacked the extracellular and juxtamembrane domains of the receptor.10, 11 Thus, we could reduce interference from the experimental context and introduce mutagenesis more efficiently to shorter EGFR fragments. When treated with EGFR‐TKI, these YFP‐EGFR‐ICD fusion proteins relocate to fibril‐like formation. Although the mechanism of this relocation is unclear, it parallels the sensitivity to EGFR‐TKI, and correlates with decreasing downstream phosphor‐PKB signal.10, 11 In this study, when 70% of cells changed the fusion protein location, we determined there was sensitivity to the EGFR‐TKI. Gefitinib had no effect on WT YFP‐EGFR‐ICD and del746–750/T790M YFP‐EGFR‐ICD double mutant‐transfected cells (Fig. 1a). However, low concentrations of gefitinib (20–100 nM) induced relocation of exon 19 deletion mutant YFP‐EGFR‐ICD and L858R YFP‐EGFR‐ICD (Figs 1a, 2). Among various mutations in exon 19, there was a small difference in sensitivity ranging from 20‐ to 50‐nM gefitinib. All exon 19 deletion mutants and/or insertion mutants were more sensitive to EGFR‐TKI than to L858R (Table 1). However, an exon 19 insertion variant (745–746 ins VPVAIK [insertion mutation valine‐proline‐valine‐alanine‐isoleucine‐lysine]) was less sensitive to gefitinib (500 nM) than other exon 19 deletion mutants (Table 1, Fig. 2).
Figure 1.
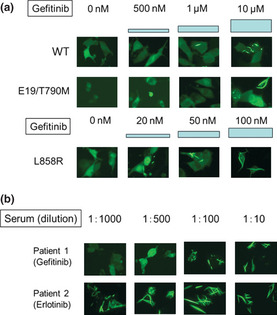
Evaluation of the sensitivity of EGFR mutations to gefitinib. YFP‐EGFR‐ICD‐transfected cells were treated with gefitinib at the indicated concentrations for 12 h, and cells were then subjected to image analysis by fluorescence microscopy. (a) The WT and del746–750/T790M YFP‐EGFR‐ICD‐transfected cells showed only partial relocation of the YFP signal at 10‐μM gefitinib. (b) Serum from lung cancer patients treated with gefitinib or erlotinib was diluted at the indicated ratios and then added to del746–750 YFP‐EGFR‐ICD‐transfected cells. Del, deletion mutation; E19, exon 19; EGFR, epidermal growth factor receptor; YFP, yellow fluorescent protein; YFP‐EGFR‐ICD, YFP‐tagged fragments of the EGFR intracellular domain.
Figure 2.
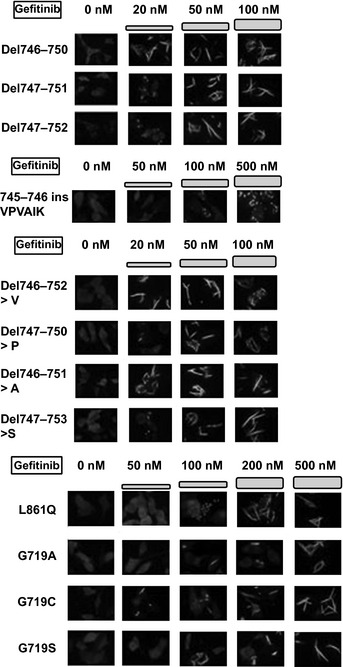
Sensitivity of various mutations in exon 19 and common EGFR mutations to gefitinib. The various YFP‐EGFR‐ICD exon 19 mutant‐transfected cells showed YFP signal relocation at lower concentrations of gefitinib than WT YFP‐EGFR‐ICD. Sensitivity to gefitinib was different among these mutations. The L861Q and G719X YFP‐EGFR‐ICD‐transfected cells showed YFP signal relocation at 200–500‐nM gefitinib. A, alanine; Del, deletion mutation; EGFR, epidermal growth factor receptor; ins, insertion mutation; P, proline; S, serine; V, valine; VPVAIK, valine‐proline‐valine‐alanine‐isoleucine‐lysine; YFP, yellow fluorescent protein; YFP‐EGFR‐ICD, YFP‐tagged fragments of the EGFR intracellular domain.
Table 1.
Sensitivity of EGFR mutations to EGFR‐TKI
| Gefitinib | Erlotinib | Afatinib | |
|---|---|---|---|
| Del746–750 | 20 nM | 20 nM | NP |
| Del747–751 | 50 nM | 20 nM | NP |
| Del747–752 | 50 nM | 50 nM | NP |
| Del746–751>A | 20 nM | 20 nM | NP |
| Del747–750>P | 50 nM | 50 nM | NP |
| Del747–753>S | 50 nM | 50 nM | NP |
| Del746–752>V | 20 nM | 20 nM | NP |
| E19 ins VPVAIK | 500 nM | 500 nM | 20 nM |
| L858R | 100 nM | 50 nM | NP |
| L861Q | 200 nM | 200 nM | 50 nM |
| G719C | 500 nM | 500 nM | 50 nM |
| G719S | 500 nM | 500 nM | 50 nM |
| G719A | 500 nM | 500 nM | 50 nM |
| Del746–750/T790M | >10 μM | >10 μM | 200 nM |
| WT | >10 μM | >10 μM | 200 nM |
EGFR‐TKI were added to transfected cells at the iNPicated concentrations.
Sensitivities to EGFR‐TKI were determined by observation of YFP signal relocation.
A, alanine; Del, deletion mutation; E19, exon 19; EGFR, epidermal growth factor receptor; ins, insertion mutation; NP, not performed; P, proline; S, serine; TKI, tyrosine kinase inhibitor; V, valine; VPVAIK, valine‐proline‐valine‐alanine‐isoleucine‐lysine; YFP, yellow fluorescent protein.
Serum obtained from patients who underwent treatment with gefitinib or erlotinib for at least 1 month were diluted and added to del746–750 YFP‐EGFR‐ICD‐transfected cells. The serum from patient 1, who had received gefitinib treatment, induced YFP signal relocation at a dilution ratio below 1:100. The serum from patient 2, who had received erlotinib treatment, induced relocation at a 1:1000 dilution (Fig. 1b). Erlotinib has a higher potency than gefitinib and is commonly administered at the maximum tolerated dose of 150 mg/day. In contrast, gefitinib is commonly used at a dose (250 mg/day) that is less than half of the maximum tolerated dose. The trough serum concentrations of 250‐mg/day gefitinib and 150‐mg/day erlotinib are reported to be approximately 400 nM and 1.5–3.0 μM, respectively.13, 14 We examined the serum of 34 additional patients; all except one sample was of sufficiently high concentration to induce relocation at 1:10–1:1000 dilution (Table S1).
L861Q and G719X mutations are known to be moderately sensitive to EGFR‐TKI.15, 16 Gefitinib at 200 nM induced relocation in L861Q YFP‐EGFR‐ICD‐transfected cells (Fig. 2). A relatively high gefitinib concentration (500 nM) was needed to induce relocation of G719X (Fig. 2). We also evaluated the effect of erlotinib or afatinib (BIBW 2992) treatment (Table 1). Erlotinib treatment showed similar results to gefitinib. Afatinib is an irreversible EGFR and human epidermal growth factor receptor type 2 (HER2) inhibitor, predicted to overcome the acquired resistance caused by T790M that covalently binds in the catalytic pocket of EGFR.17 In a phase I clinical trial, the trough serum concentration of afatinib at a dose of 40–50 mg/day was under 100 nM.18 The del746–750/T790M YFP‐EGFR‐ICD‐transfected cells showed fibril‐like formation of YFP signals at an afatinib concentration of 100 nM or more. Cells transfected with intermediately‐sensitive mutants (L861Q, G719X) all presented a similar response to afatinib at 50 nM.
Relative kinase activity of various EGFR mutations
We transfected WT YFP‐EGFR‐ICD and a different YFP‐EGFR‐ICD mutation into MCF‐7 cells. Twenty four hours post‐transfection, immunofluorescence staining was performed using a specific primary antibody to detect phosphorylation of EGFR at Y1092. Secondary antibody conjugated to AF‐594 was used, and cells were examined with fluorescence microscopy. A YFP signal is a marker of transfection, and non‐transfected cells did not contain a detectable AF‐594 signal. We used computer‐assisted analysis to compare the AF‐594 and YFP signal intensity of individual cells. We then evaluated the relative kinase activities (i.e. the levels of autophosphorylated Y1092 at the same YFP level) (Fig. 3a). The phosphorylation status of WT EGFR was very low. Among the exon 19 mutations, all had similar or significantly higher levels of autophosphorylation than WT EGFR, with the exception of del747–752, which had a much lower autophosphorylation level than the other mutations (Fig. 3b).
Figure 3.
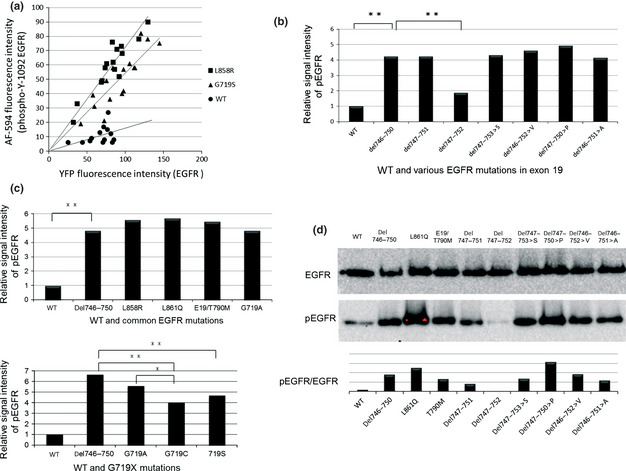
Relative kinase activity of EGFR exon 19 mutations. (a) Semi‐quantitative comparison of YFP‐EGFR‐ICD autophosphorylation levels based on computer‐assisted image analyses. The intensity of YFP and AF‐594 for each cell line was plotted on a scatter plot and an approximation straight lines as obtained. The angles of inclination were compared on a bar graph for each mutation. (b) Comparison of phosphorylation levels of WT EGFR and EGFR exon 19 mutants. (c) Comparison of phosphorylation levels of WT and common EGFR mutations. The y‐axis indicates the relative phosphorylation levels of EGFR. The phosphorylation of WT EGFR is shown as 1. Statistical analysis was performed as described in the Materials and Methods. (d) EGFR and pEGFR levels of several EGFR mutations were quantified by Western blotting. *P < 0.01. **P < 0.05. A, alanine; AF‐594, Alexa Fluor 594; Del, deletion mutation; E19, exon 19; EGFR, epidermal growth factor receptor; P, proline; pEGFR, phosphorylated epidermal growth factor receptor; S, serine; V, valine; YFP, yellow fluorescent protein; YFP‐EGFR‐ICD, YFP‐tagged fragments of the EGFR intracellular domain.
With regard to other common EGFR mutations, G719X showed weaker phosphorylation levels than the exon 19 deletion mutants (Fig. 3c). Among G719X mutations, G719C and G719S mutations showed lower levels of autophosphorylation than G719A (P < 0.01). The other common mutations (L861Q, L858R and exon 19 del/T790M) showed similarly enhanced phosphorylation to the exon 19 deletion mutations. The results of fluorescent microscopy‐based EGFR phosphorylation analysis were confirmed by Western blot analysis (Fig. 3d).
Discussion
Analysis of the COSMIC database revealed that inframe deletion mutations and deletion/insertion mutations comprise 76% and 21% of EGFR mutations in exon 19, respectively. The most frequent mutation is del746–750, which comprises 64% of exon 19 mutations, followed by del747–753>S (6%), del747–751 (4.2%), del747–750>P (4%), del746–752>V (2.4%), del747–752 (2.1%) and del746–751>A (1%), respectively. The frequency is similar to that reported in the Somatic Mutations in EGFR Database. As for insertion mutations in exon 19, this type of mutation is described as less sensitive to EGFR‐TKI than other exon 19 deletion mutations.19 These insertion mutations comprise approximately 1% of all EGFR mutations identified by DNA sequencing of lung tumor specimens in the USA. Although many types of EGFR exon 19 mutations have been reported, little is known about their characteristics. Therefore, we studied several EGFR exon 19 variants using the YFP‐EGFR‐ICD assay. Previous reports demonstrated that the assay is useful for evaluating the sensitivity of EGFR mutations to EGFR‐TKI. We also used HEK/293 and BEAS‐2B cell lines in addition to MCF‐7 cells, and the results were almost identical (data not shown). We used mainly MCF‐7 cells because an irrelevant cell line seemed to be a better method to examine the effect of exogenous EGFR without unknown factors influencing EGFR signaling. Sensitivity to EGFR‐TKI was determined by YFP‐EGFR fusion protein relocation, not by cell proliferation or apoptosis. The relocation and signaling of YFP‐EGFR may be independent of cell addiction to endogenous EGFR signaling. In addition, in our study the assay was used to evaluate the concentration of EGFR‐TKI in serum from non‐small‐cell lung cancer patients undergoing EGFR‐TKI treatment. As del746–750 YFP‐EGFR‐ICD‐transfected cells showed YFP relocation at 20‐nM gefitinib, we could estimate the approximate serum concentration of gefitinib by the addition of diluted serum. The concentration in serum from patients administered gefitinib treatment was approximately 5–10 times lower than that of patients who received erlotinib treatment (Table S1). The results seemed to confirm the previously reported pharmacokinetics of EGFR‐TKI. Therefore, this assay might be useful as an alternative method for monitoring serum concentrations.
Several reports described the sensitivity of exon 19 mutation variants.15, 20 According to the previous data, del746–750 and del747–753>S were a little more sensitive (IC50 < 10 nM) than other variants. In one report, del746–752>V‐transfected cells were less sensitive (IC50 = 306 nM) to gefitinib than other variants (IC50 < 100 nM). In our study, del746–750, del746–751>A and del746–752>V were more sensitive (<20 nM) than other variants (<50 nM). Although there is a little discrepancy in results between our data and previous studies, in part because of different methods of detection, the most common mutation, del746–750, generally seems to be the most sensitive. As the amino acid residues at position 747–750 in human EGFR protein (leucine, arginine, glutamic acid and alanine) are highly conserved, it is presumed that single amino acid changes in exon 19 variants may influence sensitivity to EGFR‐TKI and then partly contribute to the individual differences in clinical benefit from EGFR‐TKI treatment. As variants of exon 19 deletions have not been distinguished in clinical trials, the clinical relevance of these variants remains unclear.
A small difference in sensitivity to EGFR‐TKI was also found between del746–750 and L858R. In several clinical trials of gefitinib or erlotinib treatment, lung cancer patients with exon 19 deletion mutations had superior response rates, progression free survival and overall survival rates than patients with L858R.6, 21, 22 The differences in clinical outcome might partially reflect the differences in sensitivity.
YFP‐tagged fragments of the EGFR intracellular domain L861Q‐or G719X‐transfected cells showed intermediate sensitivity to gefitinib and erlotinib, and L861Q was more sensitive than G719X. No difference in sensitivity between these mutations was found for afatinib. Kancha et al.23 noted that because L861Q retained high binding affinity for ATP, irreversible inhibitors of EGFR might be more beneficial than reversible inhibitors to patients with L861Q EGFR mutation. In our study, the L861Q mutation was relatively sensitive to reversible and irreversible tyrosine kinase inhibitors. Erlotinib may be more effective than gefitinib in patients with G719X that need 500 nM of either drug, because trough levels of gefitinib are lower than effective concentrations when the drug is given at standard dose.
In approximately half of patients with acquired resistance after treatment with first generation EGFR‐TKI, T790M secondary mutation will occur.24, 25, 26, 27 In the present study, afatinib needed to be administered at a concentration of at least 100 nM to alter the location of YFP‐EGFR‐ICD fusion protein with the del746–750/T790M mutation, whereas afatinib had a median trough concentration of 30–60 nM in a clinical study.18 In addition, a recent in vitro study showed that T790M secondary mutation was also involved in acquired resistance after treatment with afatinib.28 Clinical studies targeting T790M mutations are necessary. Higher intermittent doses of afatinib may be more effective than standard continuous dosing in this patient population.
Several lines of evidence indicated that EGFR mutants exhibit differentially enhanced kinase activities. In our study, exon 19 mutants, with the exception of del747–752, presented with almost similar autophosphorylation status. Reflecting previous data,29 del747–752, which accounts for <2% of mutations in exon 19, presented a low autophosphorylation status comparable to WT EGFR.2, 9 Pao et al. evaluated the phosphorylation of this mutation variant using a phospho‐Y1092‐specific antibody and an anti‐phosphotyrosine antibody; phosphorylation was at a low level compared to WT EGFR or L858R. Although it is unclear why only del747–752 is not as highly phosphorylated at Y1092, patients with del747–752 mutation benefit from EGFR‐TKI treatment.29, 30 A possible hypothesis is that this EGFR mutation may be activated by heterodimerization with HER2 or human epidermal growth factor receptor type 3 (HER3).
The autophosphorylation levels of G719X were less enhanced than other common mutations. Relatively low response rates in patients with EGFR G719X mutations to inhibitor treatment might indicate a lower dependence on EGFR signaling.
In conclusion, various EGFR exon 19 deletion mutants exhibited similar characteristics, with the exception of del747–752, in terms of phosphorylation of the EGFR tyrosine kinase domain. However, variants of EGFR exon 19 deletions showed differences in sensitivity to EGFR‐TKI. The difference of these characteristics of various EGFR mutations may be relevant to clinical outcome. Therefore, further studies to functionally characterize and determine the clinical relevance of EGFR mutations are warranted.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Doc. S1. List of the primers for site‐directed mutagenesis.
Table S1. Patients' characteristics and serum concentration of epidermal growth factor receptor‐tyrosine kinase inhibitors in this study.
Acknowledgments
We appreciate the technical support we received from the Research Support Center, Graduate School of Medical Sciences, Kyushu University (Fukuoka, Japan). This study was performed as part of the Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT), Ministry of Education, Culture, Sports, Science and Technology of Japan (Tokyo, Japan).
(Cancer Sci 2013; 104: 584–589)
References
- 1. Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer 2006; 118: 257–62. [DOI] [PubMed] [Google Scholar]
- 2. Shigematsu H, Lin L, Takahashi T et al Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97: 339–46. [DOI] [PubMed] [Google Scholar]
- 3. Mitsudomi T, Morita S, Yatabe Y et al Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harboring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomized phase 3 trial. Lancet Oncol 2010; 11: 121–8. [DOI] [PubMed] [Google Scholar]
- 4. Mok TS, Wu YL, Thongprasert S et al Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–57. [DOI] [PubMed] [Google Scholar]
- 5. Rosell R, Noran T, Queralt C et al Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009; 361: 958–67. [DOI] [PubMed] [Google Scholar]
- 6. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci 2007; 98: 1817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non‐small‐cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012; 13: e23–31. [DOI] [PubMed] [Google Scholar]
- 8. Wu J‐Y, Yu C‐J, Chang Y‐C, Yang C‐H, Shih J‐Y, Yang P‐C. Effectiveness of tyrosine kinase inhibitors on uncommon epidermal growth factor receptor mutations of unknown clinical significance in non‐small cell lung cancer. Clin Cancer Res 2011; 17: 3812–21. [DOI] [PubMed] [Google Scholar]
- 9. Murray S, Dahabreh IJ, Linardou H, Manoloukos M, Bafaloukos D, Kosmidis P. Somatic mutations of the tyrosine kinase domain of epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non‐small cell lung cancer: an analysis database. J Thorac Oncol 2008; 3: 832–9. [DOI] [PubMed] [Google Scholar]
- 10. De Gunst MM, Gallegos‐Ruiz MI, Giaccone G, Rodriguez JA. Functional analysis of cancer‐associated EGFR mutants using a cellular assay with YFP‐tagged EGFR intracellular domain. Mol Cancer 2007; 6: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harada T, Lopez‐Chavez A, Xi L, Raffeld M, Wang Y, Giaccone G. Characterization of epidermal growth factor receptor mutations in non‐small‐cell lung cancer patients of African–American ancestry. Oncogene 2011; 30: 1744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodriguez JA, Henderson BR. Identification of a functional nuclear export sequence in BRCA I. J Biol Chem 2000; 275: 38589–96. [DOI] [PubMed] [Google Scholar]
- 13. Baselga J, Rischin D, Ranson M et al Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J Clin Oncol 2002; 20: 4292–302. [DOI] [PubMed] [Google Scholar]
- 14. Hidalgo M, Siu LL, Nemunaitis J et al Phase I and pharmacologic study of OSI‐774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol 2001; 19: 3267–79. [DOI] [PubMed] [Google Scholar]
- 15. Yuza Y, Glatt KA, Jiang J et al Allele‐dependent variation in the relative cellular potency of distinct EGFR inhibitors. Cancer Biol Ther 2007; 6: 661–7. [DOI] [PubMed] [Google Scholar]
- 16. Kancha RK, von Bubnoff N, Peschel C, Duyster J. Functional analysis of epidermal growth factor receptor (EGFR) mutations and potential implications for EGFR targeted therapy. Clin Cancer Res 2009; 15: 460–7. [DOI] [PubMed] [Google Scholar]
- 17. Li D, Ambrogio L, Shimamura T et al BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008; 27: 4702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yap TA, Vidal L, Adam J et al Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol 2010; 28: 3965–72. [DOI] [PubMed] [Google Scholar]
- 19. He M, Capelletti M, Nafa K et al EGFR exon 19 insertions: a new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res 2012; 18: 1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engelman JA, Zejnullahu K, Gala C‐M et al PF00299804, an irreversible Pan‐ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007; 67: 11924–32. [DOI] [PubMed] [Google Scholar]
- 21. Jackman DM, Yeap BY, Sequist LV et al Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non‐small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res 2006; 12: 3908–14. [DOI] [PubMed] [Google Scholar]
- 22. Riely GJ, Pao W, Pham D et al Clinical course of patients with non‐small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res 2006; 12: 839–44. [DOI] [PubMed] [Google Scholar]
- 23. Kancha RK, Peschel C, Duyster J. The epidermal growth factor receptor‐L861Q mutation increases kinase activity without leading to enhanced sensitivity toward epidermal growth factor receptor kinase inhibitors. J Thorac Oncol 2011; 6: 387–92. [DOI] [PubMed] [Google Scholar]
- 24. Kobayashi S, Boggon TJ, Dayaram T et al EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 25. Pao W, Miller VA, Politi KA et al Acquired resistance of lung adenocarcinoma to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: 225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Balak MN, Gong Y, Riely GJ et al Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor‐mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006; 12: 6494–501. [DOI] [PubMed] [Google Scholar]
- 27. Kosaka T, Yatabe Y, Endoh H et al Analysis of epidermal growth factor receptor gene mutation in patients with non‐small‐cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res 2006; 12: 5764–9. [DOI] [PubMed] [Google Scholar]
- 28. Kim Y, Ko J, Cui Z et al The EGFR T790M mutation in acquired resistance to an irreversible second‐generation EGFR inhibitor. Mol Cancer Ther 2012; 11: 784–91. [DOI] [PubMed] [Google Scholar]
- 29. Pao W, Miller V, Zakowski M et al EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chung KP, Wu SG, Wu JY et al Clinical outcome in non‐small cell lung cancers harboring different exon 19 deletions in EGFR. Clin Cancer Res 2012; 18: 3470–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Doc. S1. List of the primers for site‐directed mutagenesis.
Table S1. Patients' characteristics and serum concentration of epidermal growth factor receptor‐tyrosine kinase inhibitors in this study.