

Abstract
As CD20 has become an established target for treating B‐cell malignancies, there is interest in developing anti‐CD20 antibodies with different functional activity from rituximab that might translate into improved efficacy. Obinutuzumab (GA101) is a glycoengineered, humanized type II anti‐CD20 monoclonal antibody that has demonstrated superior activity to type I antibodies in preclinical studies and is currently being investigated in phase III trials. In this phase I dose‐escalating study in Japanese patients with relapsed/refractory B‐cell non‐Hodgkin lymphoma, the primary endpoint was to characterize the safety of GA101; secondary endpoints were efficacy, pharmacokinetics and pharmacodynamics. Patients received up to nine doses of GA101 with up to 52 weeks' follow up. Most adverse events were grade 1 or 2 infusion‐related reactions, and 10 grade 3/4 adverse events occurred. No dose‐limiting toxicities were observed and the maximum tolerated dose was not identified. Out of 12 patients, 7 responded (end‐of‐treatment response rate 58%), with 2 complete responses and 5 partial responses. Responses were observed from low to high doses, and no dose‐efficacy relationship was observed. B‐cell depletion occurred in all patients after the first infusion and was maintained for the duration of treatment. Serum levels of GA101 increased in a dose‐dependent fashion, although there was inter‐patient variability. This phase I study demonstrated that GA101 has an acceptable safety profile and offers encouraging activity to Japanese patients with relapsed/refractory B‐cell non‐Hodgkin lymphoma. (Cancer Sci 2013; 104: 105–110)
The advent of the type I chimeric anti‐CD20 antibody rituximab in 1997 led to significant improvements in outcomes for patients with CD20‐positive B‐cell hematologic malignancies. Rituximab in combination with chemotherapy (R‐chemo) is now the standard of care worldwide in B‐cell non‐Hodgkin lymphoma (B‐NHL) and chronic lymphocytic leukemia (CLL), and maintenance therapy with rituximab is the standard of care in follicular lymphoma (FL) in many countries, based on a series of randomized phase III trials.1, 2, 3, 4, 5, 6, 7
Despite the major advances in treatment achieved with rituximab, a significant number of patients relapse and some may be refractory to treatment. There remains a clinical need for improved treatment options for these patients. Because CD20 has become established as an important immunotherapeutic target for B‐NHL, there is interest in developing anti‐CD20 antibodies with different functional activity from rituximab that might translate into improved efficacy. Antibodies that target CD20 can be classified as one of two types based on their mode of CD20 binding and mechanism of action.8, 9, 10, 11 Type I antibodies, such as rituximab and ofatumumab, exhibit strong complement‐dependent cytotoxicity (CDC) and antibody‐dependent cell‐mediated cytotoxicity (ADCC). Type II antibodies, such as obinutuzumab (GA101), in contrast, show effective ADCC but only weakly induce CDC. Type II antibodies, unlike type I antibodies, are effective in inducing non‐apoptotic direct cell death of CD20‐expressing cells via an actin‐dependent, lysosome‐mediated mechanism.9, 12, 13 In addition, GA101 has been glycoengineered by afucosylation of the Fc region, resulting in an improved capacity for GA101 to recruit and activate immune effector cells and mediate ADCC.14 GA101 has demonstrated superior activity compared with rituximab in vitro and in vivo, including increased induction of direct cell death of lymphoma cells, increased ADCC induction, enhanced B‐cell depletion from whole human blood,14, 15 increased antitumor activity in human xenograft models,14, 16 and superior B‐cell depletion from lymphoid tissues of non‐human primates.14
Based on these encouraging data, GA101 is the first type II anti‐CD20 glycoengineered antibody to enter clinical development. Data from completed and ongoing phase I and II trials suggest that GA101 has an acceptable safety profile and has demonstrated promising efficacy as monotherapy in patients with relapsed/refractory non‐Hodgkin lymphoma (NHL) and CLL of B‐cell phenotype, as well as in combination with chemotherapy in patients with relapsed/refractory FL in the EU and North American countries.17, 18, 19, 20, 21, 22 GA101 plus chemotherapy is currently being investigated in a series of phase III trials in patients with rituximab‐refractory indolent NHL (iNHL), first‐line iNHL and first‐line diffuse large B‐cell lymphoma. We report here the safety, pharmacokinetics, pharmacodynamics and efficacy data from a phase I dose‐escalating study of GA101 monotherapy in Japanese patients with relapsed/refractory B‐NHL, who had previously received rituximab.
Patients and Methods
Study design and objectives
This open‐label, multicenter, dose‐escalating phase I study was designed to evaluate the safety, tolerability, pharmacokinetics and efficacy of GA101 in Japanese patients with relapsed/refractory CD20‐positive B‐NHL for whom no treatment of higher priority was available. The primary endpoint was safety of GA101, including frequency of dose‐limiting toxicities (DLT), maximum tolerated dose (MTD), and type, grade and frequency of adverse events (AE). Secondary endpoints included pharmacokinetics, pharmacodynamics and preliminary efficacy of GA101. The study was conducted between 2008 and 2011 at the Nagoya Daini Red Cross Hospital, Nagoya, Japan, the National Cancer Center Hospital, Tokyo, Japan, and the Cancer Institute Hospital, Tokyo, Japan. The study was reviewed and approved by the Ethics Review Boards of the relevant institutions and was conducted in accordance with the Declaration of Helsinki.
Eligible patients were 20–75 years of age with at least one bi‐dimensionally measurable lesion (>1.5 cm). Patients with a lymphocyte count >25 000/mm3 were excluded. All patients had a life expectancy of ≥12 weeks and an Eastern Cooperative Oncology Group performance status of 0–1. Patients were required to have appropriate hematologic parameters and adequate renal and liver function. All patients enrolled in the study provided written informed consent.
Prior use of any investigational monoclonal antibody (24 weeks), rituximab (8 weeks), or chemotherapy, radiotherapy or any investigational product (4 weeks) before enrollment was not permitted. Patients with previous cancers were required to have had a disease‐free period of at least 5 years, and patients who were positive for hepatitis B (viral DNA, surface or core antigen), hepatitis C (antibody) or human immunodeficiency virus (antigen or antibody), and those with serious heart disease, serious bronchospasms or symptoms of central nervous system invasion by underlying disease were excluded.
Treatment
Four treatment cohorts of 3 patients each were planned; cohort 1 received 200 mg at first dose and 400 mg at the second and subsequent doses (200/400 mg), cohort 2 received 400/800 mg, cohort 3 received 800/1200 mg and cohort 4 received 1200/2000 mg. The dose of the first infusion was decreased in each cohort due to initial concerns about severe infusion‐related reaction (IRR). If there were no DLT during the first infusion, the full assigned doses were given for the remaining eight infusions. GA101 was administered intravenously on days 1 and 8 of cycle 1, and on day 1 of cycles 2–8, in 3‐weekly cycles for a total of 9 doses. The infusion rate for the first dose was 50 mg/h for the first 30 min, and was then raised by 50 mg/h every 30 min to an upper limit of 400 mg/h if no problems were observed. For all subsequent doses, the initial infusion rate was 100 mg/h (if no problems were observed with the first dose) and in the absence of any observed problems this was increased by 100 mg/h every 30 min to the upper limit of 400 mg/h.
Dose escalation
In the present study 3 patients were treated in each cohort. Dose escalation to the next dose level was based on DLT observed at the previous dose. Two categories of DLT were defined. Any grade (National Cancer Institute – Common Toxicity Criteria for Adverse Events version 3.0: CTCAE v3.0) 4 IRR, or grade 3 IRR failing to respond to corticosteroid medication, occurring during or within 24 h of the first infusion, was defined as DLT1. All other grade 3 or 4 AE, regardless of which infusion they were associated with, were defined as DLT2, with the exception of:
Lymphopenia
Neutropenia that did not result in febrile neutropenia and resolved from grade 4 to grade 3 within 1 week and from grade 3 to grade 2 within 1 week
Thrombocytopenia that did not result in bleeding and resolved from grade 4 to grade 3 within 1 week and from grade 3 to grade 2 within 1 week.
If no DLT were observed then dose escalation proceeded to the next cohort. If 1 DLT1 or DLT2 was observed in a cohort then additional subjects were recruited in order to provide 6 cases for assessment of the particular DLT. If 2 or more patients in a single cohort experienced DLT1 then MTD1 was established and the first dose administered to the next cohort was given at MTD1 followed by dose‐escalation as planned for the second dose onwards. If 2 or more cases of DLT2 were observed with subsequent doses then the MTD2 was established from the previous cohort, and the dose was not escalated to the next level.
Statistical analysis
The sample size of this study was based on a three‐case cohort design, with 12 subjects being the minimum number needed to confirm the initial safety and tolerability of GA101 in Japanese patients. Efficacy (proportion of response and complete response) was determined by point estimates and using the Clopper–Pearson interval with a 95% degree of confidence. Serum GA101 levels were measured by ELISA. Pharmacokinetics analyses, concentration and pharmacokinetic parameters were derived using non‐compartmental model analysis. Pharmacokinetic parameters, including AUClast, Cmax, t1/2, CL and Vd, were estimated by non‐compartmental pharmacokinetic analysis using Phoenix WinNonlin 6.1 (Tripos, St Louis, MO, USA). The relationships between pharmacokinetic parameters (CL and Vd) and body size (body weight and surface area) were assessed by exploratory analysis using TIBCO Spotfire S+8.2 (TIBOC Software, Palo Alto, CA, USA).
The frequency and incidence of DLT (as defined above in the subsection “Dose escalation”) was summarized in each cohort and for all subjects. In the case of DLT events, these were to be used to form the basis for estimating MTD. The type, frequency and incidence of other AE were also summarized in each cohort and for all subjects. The severity of AE was graded according to the CTCAE v3.0.
Efficacy assessments were based on point estimates of the response rate and complete remission rate, for each of which 95% confidence intervals were estimated. Tumour response was evaluated according to the International Workshop Criteria for NHL.23
This trial is registered with www.clinicaltrials.jp (number JapicCTI‐121787).
Results
Patient demographics
A total of 12 patients, all of whom had previously been treated with rituximab or rituximab‐containing chemotherapy, were enrolled in the four dose cohorts (Table 1). Lymphoma subtypes were follicular (n = 8), small lymphocytic (n = 2), nodal marginal zone (n = 1) and mucosa‐associated lymphoid tissue (n = 1). At the time of initial presentation, 9 patients had clinical stage (Ann Arbor classification) III or IV disease, and 9 patients had extranodal involvement.
Table 1.
Patient baseline characteristics
| Characteristic | Cohort 1 200/400 mg n = 3 | Cohort 2 400/800 mg n = 3 | Cohort 3 800/1200 mg n = 3 | Cohort 4 1200/2000 mg n = 3 | Total N = 12 |
|---|---|---|---|---|---|
| Male sex, n (%) | 3 (100) | 2 (67) | 1 (33) | 2 (67) | 8 (67) |
| Median age, years | 57 | 48 | 59 | 58 | 58 |
| Lymphoma subtype, n (%) | |||||
| Follicular NHL | 3 (100) | 2 (67) | 1 (33) | 2 (67) | 8 (67) |
| Other indolent B‐NHL | – | 1 (33) | 2 (67) | 1 (33) | 4 (33) |
| Clinical stage (Ann Arbor classification), n (%) | |||||
| I/II | 1 (33) | – | – | 2 (67) | 3 (25) |
| III/IV | 2 (67) | 3 (100) | 3 (100) | 1 (33) | 9 (75) |
| Prior rituximab, n (%) | 3 (100) | 3 (100) | 3 (100) | 3 (100) | 12 (100) |
B‐NHL, B cell non‐Hodgkin lymphoma (NHL).
Safety and tolerability
All 12 patients recruited to the study received at least one dose of GA101 and were included in the safety analysis. All patients experienced at least 1 AE during the course of the study (Table 2). There were a total of 107 AE (all grades) recorded and these were distributed evenly across all dose groups with no obvious evidence for a dose‐dependent effect. The majority of adverse events were grade 1 or 2 (97 of 107 total AE, 91%) and resolved with appropriate treatment, with the most commonly reported AE being grade 1 or 2 IRR. Other common AE included leukopenia (67%), nasopharyngitis (58%), thrombocytopenia (58%), neutropenia (58%), anemia (33%) and elevation of liver enzymes (alanine transaminase or aspartate transaminase) (33%). Given the small number of patients in this trial, an association between dose and safety and tolerability cannot be confirmed.
Table 2.
Adverse events (all grades) experienced by 2 or more patients and grade 3 or 4 events
| AE | All grades | Grade 3 or 4a | ||
|---|---|---|---|---|
| N | % | N | % | |
| Total patients with at least 1 AE | 12 | 100 | 5 | 42 |
| Total number of AE | 107 | – | 10 | – |
| Hematologic events, n (%) | ||||
| Leukopenia | 8 | 67 | 2 | 17 |
| Thrombocytopenia | 7 | 58 | 1 | 8 |
| Neutropenia | 7 | 58 | 2 | 17 |
| Anemia | 4 | 33 | – | – |
| Non‐hematologic events, n (%) | ||||
| Infusion‐related reaction | 12 | 100 | 2 | 17 |
| Nasopharyngitis | 7 | 58 | – | – |
| AST increase | 4 | 33 | – | – |
| ALT increase | 4 | 33 | – | – |
| Hypophosphatemia | 3 | 25 | 1 | 8 |
| Blood lactate dehydrogenase increase | 3 | 25 | – | – |
| Constipation | 3 | 25 | – | – |
| Insomnia | 3 | 25 | – | – |
| Headache | 3 | 25 | – | – |
| Hyperbilirubinemia | 2 | 17 | – | – |
| Malaise | 2 | 17 | – | – |
| Diarrhea | 2 | 17 | – | – |
| Back pain | 2 | 17 | – | – |
Other Grade 3 or 4 events not listed include anorexia (1) and herpes zoster infection (1). Multiple occurrences of the same adverse events in 1 individual counted only once. AE, adverse event; ALT, alanine transaminase; AST, aspartate transaminase.
In the present study, 5 patients experienced a total of 10 grade 3 or 4 events. Of these, 2 patients, both in cohort 3 (800/1200 mg), had a grade 3 IRR; however, these did not meet the definition of a DLT. Grade 3 or 4 neutropenia with grade 3 leukopenia was observed in 2 patients: 1 grade 4 neutropenia with grade 3 leukopenia in cohort 2 (400/800 mg) and 1 grade 3 neutropenia with grade 3 leukopenia in cohort 4 (1200/2000 mg). These events were considered to be related to the study drug and both patients were withdrawn at cycle 3. One patient in cohort 3 (800/1200 mg) experienced transient grade 3 thrombocytopenia, and this was considered to be related to the study drug. The patient recovered without medication and continued in the study. One patient in cohort 4 experienced grade 3 herpes zoster infection approximately 2 months after the treatment period; this event was considered an SAE and related to the study drug. No deaths were reported during the course of the study.
No DLT1 or DLT2 events were observed, so no MTD was identified.
Pharmacokinetics of GA101
The pharmacokinetics of GA101 following the first, second and final doses are summarized in Table 3. GA101 displayed linear pharmacokinetics; generally exposure (AUClast and Cmax) to GA101 increased with increasing dose, whereas t1/2, CL and Vd were comparable across the different dose levels. GA101 serum concentrations reached a peak by the end of each infusion before slowly decreasing over time (Fig. 1). Serum GA101 levels generally increased in a dose‐dependent manner over the investigated range (doses for the first/subsequent infusion ranged from 100/200 mg to 1200/2000 mg). No clear and significant relationship between pharmacokinetic parameters (CL and Vd) at cycle 8 and body size (body weight and body surface area) was observed (Fig. 2).
Table 3.
Summary of pharmacokinetic parameters following dose 1 and 2, and the last dose of GA101
| Parameter | Cohort 1 n = 3 | Cohort 2 n = 3 | Cohort 3 n = 3 | Cohort 4 n = 3 |
|---|---|---|---|---|
| Cycle 1 day 1 (mean ± SD) | ||||
| Dose (mg) | 200 | 400 | 800 | 1200 |
| AUClast (μg × day/mL) | 305 ± 39.5 | 768 ± 109 | 1470 ± 297 | 1970 ± 624 |
| Cmax (μg/mL) | 79.0 ± 17.2 | 157 ± 24.4 | 358 ± 37.2 | 415 ± 81.7 |
| CL (mL/day) | 257 ± 12.6 | 220 ± 49.0 | 347 ± 82.4 | 248 ± 155 |
| t 1/2 (day) | 9.65 ± 2.00 | 10.9 ± 1.80 | 6.11 ± 0.617 | 14.5 ± 8.87 |
| Cycle 1 day 8 (mean ± SD) | ||||
| Dose (mg) | 400 | 800 | 1200 | 2000 |
| AUClast (μg × day/mL) | 1630 ± 283 | 4190 ± 1190 | 6540 ± 1070 | 9340 ± 2060 |
| Cmax (μg/mL) | 201 ± 40.5 | 421 ± 56.4 | 669 ± 124 | 833 ± 186 |
| CL (mL/day) | 115 ± 43.9 | 80.2 ± 43.4 | 108 ± 16.9 | 69.1 ± 11.2 |
| t 1/2 (day) | 17.1 ± 4.68 | 28.1 ± 19.7 | 14.2 ± 1.44 | 30.3 ± 7.88 |
| Cycle 8 (mean ± SD) | ||||
| Dose (mg) | 400 | 800 | 1200 | 2000 |
| AUClast (μg × day/mL) | 3470 ± 1820 | 11 900 ± 7130 | 10 800 ± 722 | 25 100 ± 4970 |
| Cmax (μg/mL) | 359 ± 118 | 1200 ± 698 | 1040 ± 146 | 1910 ± 156 |
| CL (mL/day) | 38.8 ± 31.5 | 29.3 ± 2.61 | 44.1 ± 9.29 | NC |
| t 1/2 (day) | 37.0 ± 19.4 | 25.2 ± 15.0 | 27.2 ± 6.23 | NC |
AUClast, area under curve from time zero to last quantifiable result; CL, clearance; Cmax, maximum concentration; NC, not calculated; t1/2, half‐life.
Figure 1.
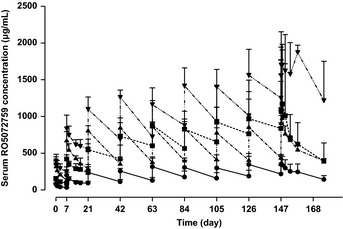
Mean serum GA101 profiles following multiple ascending doses. Three patients per cohort; error bars show standard deviation. Closed circle, cohort 1; closed square, cohort 2; closed triangle, cohort 3; closed point‐down triangle, cohort 4.
Figure 2.
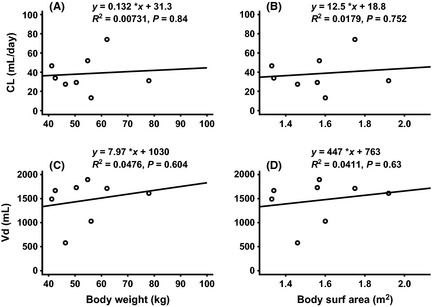
Relationship between pharmacokinetic parameters (CL and Vd) and body size (body weight and body surface area): (A) CL versus body weight at cycle 8, (B) CL versus body surface area at cycle 8, (C) Vd versus body weight at cycle 8 and (D) Vd versus body surface area at cycle 8.
Pharmacodynamics of GA101
All patients had previously received rituximab, and 11 of the 12 patients had an absolute B‐cell count in the range 5–240 cells/μL at baseline. One patient had a baseline peripheral blood B‐cell count of 2930 cells/μL. The observed number of B‐cells rapidly decreased following the first infusion, and the nadir was achieved in most patients shortly after infusion on day 1. For all patients (n = 12), B‐cell depletion (<80 cells/μL) was observed at the end of the treatment period (Fig. 3). Following treatment, 4 patients (2 in cohort 1 [200/400 mg] and 2 in cohort 3 [800/1200 mg]) displayed B‐cell recovery (over baseline or 80 cells/μL) within 52 weeks of follow‐up, and 3 patients displayed persistent B‐cell depletion during the follow‐up period. The remaining 5 patients could not be followed up for B‐cell recovery due to receiving alternative lymphoma treatment. Human antichimeric antibodies (HACA), presumably from previous rituximab treatment, were detected in serum samples of 2 patients at the time of enrollment. Human anti‐human antibodies (HAHA) were not detected in any patient during the treatment period; however, 1 patient developed HAHA during the post‐treatment 52‐week follow‐up period. There is no further data of HAHA after 52‐week follow‐up.
Figure 3.
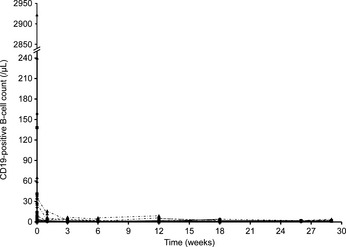
Individual peripheral B‐cell (CD19‐positive) level at each dose level. Two patients withdrew from the study at cycle 3. Closed circle, cohort 1; closed square, cohort 2; closed triangle, cohort 3; closed point‐down triangle, cohort 4.
Efficacy
Response assessments at the end of treatment were available for all 12 patients in the study (Table 4). In total, 7 patients (58%) responded at the end of treatment. Of these, 2 (17%) achieved a complete response (CR), 5 (42%) achieved a partial response (PR) and the remaining 5 (42%) had stable disease. For the 8 patients with FL, 6 responded (75%), of whom 2 achieved a CR. Furthermore, 1 of the 2 rituximab‐refractory patients achieved a response. Responses were observed from low to high doses, with no obvious dose response observed. Responses were observed in patients with both high‐affinity and low‐affinity polymorphisms at residue 158 of the Fcγ receptor; 1 CR and 3 PR were observed for patients with the F/F genotype (n = 7) and 1 CR and 2 PR for patients with the F/V genotype (n = 5). None of patients in the study had a V/V genotype. No patients progressed or relapsed by the end of treatment. Of the 12 patients, 10 displayed a reduction in tumor mass at the end of the treatment. Reduction in mass was observed across the low to high dose cohorts and for both Fcγ genotypes. Both rituximab‐refractory patients experienced a reduction in tumor mass.
Table 4.
Summary of overall response at end of administration
| Response, n | Cohort 1 200/400 mg n = 3 | Cohort 2 400/800 mg n = 3 | Cohort 3 800/1200 mg n = 3 | Cohort 4 1200/2000 mg n = 3 | Total (%) N = 12 |
|---|---|---|---|---|---|
| Overall response | 2 | 2 | – | 3 | 7 (58) |
| Complete response | – | 2 | – | – | 2 (17) |
| Partial response | 2 | – | – | 3 | 5 (42) |
| Stable disease | 1 | 1 | 3 | – | 5 (42) |
Discussion
This dose‐escalating, 3 + 3, phase I study was designed primarily to assess the safety of up to nine doses of GA101 (200 to 2000 mg) over eight 3‐weekly cycles in pretreated Japanese patients with relapsed/refractory B‐NHL. The data for the 12 patients were sufficient to confirm the initial safety and tolerability of GA101 in this group and to allow assessment of possible DLT. Most AE were grade 1 or 2 infusion‐related reactions, and 10 grade 3/4 AE occurred. No dose‐limiting toxicities were observed and the maximum tolerated dose was not identified. Furthermore, our results suggest that GA101 has encouraging activity in Japanese patients with relapsed/refractory B‐NHL, the end‐of‐treatment response rate was 58% (7 of 12 patients).
Our results demonstrate that GA101 has an acceptable safety profile in pretreated Japanese patients. The observed safety and tolerability of GA101 is comparable to that seen for rituximab.1, 2, 3, 4, 5, 6, 7 GA101 was well tolerated at all doses: there was no evidence for increased toxicity with increasing dose, and no maximum tolerated dose of GA101 was identified. IRR were the most frequently observed adverse events and were commonly associated with the first infusion and resolved with appropriate management. A total of 12 IRR were observed and included pyrexia, chills, hypotension, nausea and vomiting. The majority of IRR were grade 1 or 2, and 2 were grade 3. The rate, type and grade of IRR observed in this study are broadly similar to those reported for trials of GA101 monotherapy or rituximab therapy in non‐Japanese patients.1, 2, 3, 4, 5, 6, 7, 21
While exposure (AUClast and Cmax) to GA101 was dose‐dependent, there was a high degree of inter‐individual variability in the observed pharmacokinetic parameters for GA101; this is to be expected for a study in patients as opposed to healthy volunteers. This variability might be explained by the small number of patients included in each cohort and their heterogeneity in histology and tumor load. Treatment with GA101 resulted in rapid and sustained peripheral B‐cell depletion in all patients, with 4 patients experiencing B‐cell recovery in the 12‐month follow‐up period. No significant relationship was observed between pharmacokinetic parameters (CL and Vd) and body size (body weight and body surface area). While the number of patients is small, this supports the use of a fixed dose of GA101, as used in this study, rather than adjusting the dose by body weight or surface area.
Overall, the exposure level of GA101 in this population of Japanese patients was broadly comparable to those observed in a phase I study of non‐Japanese patients.24 Various dose schedules were investigated in two overseas phase I/II studies. Based upon all available clinical data and PK modelling studies, a flat dose of 1000 mg 3‐weekly cycles was selected for Phase III studies, with GA101 administered on days 1, 8 and 15 of the first cycle to rapidly achieve and maintain adequate exposure levels. This identified dose and schedule of GA101 is expected to achieve serum GA101 concentrations sufficient to saturate the target site and provide clinical efficacy for the majority of patients (data available on request from authors).21, 22
The end‐of‐treatment response in these 12 patients was 58% (7 patients), with 2 patients (17%) achieving a CR. While these data are preliminary, response rates in this Japanese population were higher than observed in a recently reported phase I study of GA101 monotherapy in non‐Japanese patients, where the end of treatment response was 33% (4 CR or CR unconfirmed and 3 PR).21 However, this difference might, in part, be attributed to the small sample size, and the less heavily pre‐treated population reported here that comprised patients solely of indolent histology.
HACA for rituximab were detected in serum samples of 2 patients at the time of enrollment. The PK data and safety observations for these 2 patients were generally similar to those of other patients in the study. One patient developed HAHA for GA101 at week 52 of the follow‐up period. The serum GA101 concentration of this patient was not different from that of other patients and was undetectable at week 28 of follow‐up (data not shown).
In conclusion, these preliminary data show that the type II glycoengineered humanized antibody GA101 is well tolerated and has encouraging activity in this group of Japanese patients with indolent B‐NHL previously treated with rituximab, with an end of treatment response of 58%. Most studies of rituximab report no ethnic differences in response or safety in B‐NHL. As previously reported,24 our results suggest that there are no apparent differences in the safety or efficacy of GA101 monotherapy in Japanese and non‐Japanese indolent B‐NHL patients. The safety profile of GA101 was broadly similar for the two populations; no dose‐limiting toxicities and no dose reductions were observed in either study. While efficacy was higher in the Japanese patients, this can potentially be explained by differences in demographics of the two populations. However, analyses of larger databases are required to confirm these initial results. These data support the ongoing clinical investigation of GA101 in Japanese patients with B‐NHL.
Disclosure Statement
Authors received honoraria, research grants and/or consultancy fees as follows: Michinori Ogura from Chugai; Kensei Tobinai from Eisai and Kyowa Hakko Kirin; Kiyohiko Hatake from Ono Pharmaceutical, Chugai, Eisai, J&J K.K., F. Hoffman‐La Roche and Takeda Pharmaceutical; Masahiro Yokoyama from Chugai; and Tomomitsu Hotta from Chugai. The other authors have no conflicts of interest to report.
Acknowledgments
The authors thank the patients, doctors, nurses and staff members who participated in this trial. Chugai Pharmaceutical (Chugai) sponsored this study, provided the study design and performed statistical analyses in collaboration with F. Hoffmann‐La Roche. Support for third‐party writing assistance for this manuscript was provided by Chugai. This trial is registered with www.clinicaltrials.jp, number JapicCTI‐121787.
(Cancer Sci, doi: 10.1111/cas.12040, 2012)
References
- 1. Coiffier B, Lepage E, Brière J et al CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large‐B‐cell lymphoma. N Engl J Med 2002; 346: 235–42. [DOI] [PubMed] [Google Scholar]
- 2. Hiddemann W, Kneba M, Dreyling M et al Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced‐stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low‐Grade Lymphoma Study Group. Blood 2005; 106: 3725–32. [DOI] [PubMed] [Google Scholar]
- 3. Pfreundschuh M, Trümper L, Osterborg A et al CHOP‐like chemotherapy plus rituximab versus CHOP‐like chemotherapy alone in young patients with good‐prognosis diffuse large‐B‐cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 2006; 7: 379–91. [DOI] [PubMed] [Google Scholar]
- 4. Marcus R, Imrie K, Solal‐Céligny P et al Phase III study of R‐CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol 2008; 26: 4579–86. [DOI] [PubMed] [Google Scholar]
- 5. Hallek M, Fischer K, Fingerle‐Rowson G et al Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open‐label, phase 3 trial. Lancet 2010; 376: 1164–74. [DOI] [PubMed] [Google Scholar]
- 6. van Oers MH, Van Glabbeke M, Giurgea L et al Rituximab maintenance treatment of relapsed/resistant follicular non‐Hodgkin's lymphoma: long‐term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol 2010; 28: 2853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salles G, Seymour JF, Offner F et al Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet 2011; 377: 42–51. [DOI] [PubMed] [Google Scholar]
- 8. Glennie MJ, French RR, Cragg MS et al Mechanisms of killing by anti‐CD20 monoclonal antibodies. Mol Immunol 2007; 44: 3823–37. [DOI] [PubMed] [Google Scholar]
- 9. Chan HT, Hughes D, French RR et al CD20‐induced lymphoma cell death is independent of both caspases and its redistribution into triton X‐100 insoluble membrane rafts. Cancer Res 2003; 63: 5480–9. [PubMed] [Google Scholar]
- 10. Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti‐CD20 reagents. Blood 2004; 103: 2738–43. [DOI] [PubMed] [Google Scholar]
- 11. Niederfellner G, Lammens A, Mundigl O et al Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 2011; 118: 358–67. [DOI] [PubMed] [Google Scholar]
- 12. Ivanov A, Beers SA, Walshe CA et al Monoclonal antibodies directed to CD20 and HLA‐DR can elicit homotypic adhesion followed by lysosome‐mediated cell death in human lymphoma and leukemia cells. J Clin Invest 2009; 119: 2143–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alduaij W, Ivanov A, Honeychurch J et al Novel type II anti‐CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin‐dependent, lysosome‐mediated cell death in B‐cell malignancies. Blood 2011; 117: 4519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mössner E, Brünker P, Moser S et al Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti‐CD20 antibody with enhanced direct and immune effector cell‐mediated B‐cell cytotoxicity. Blood 2010; 115: 4393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patz M, Isaeva P, Forcob N et al Comparison of the in vitro effects of the anti‐CD20 antibodies rituximab and GA101 on chronic lymphocytic leukaemia cells. Br J Haematol 2011; 152: 295–306. [DOI] [PubMed] [Google Scholar]
- 16. Dalle S, Reslan L, Besseyre de Horts T et al Preclinical studies on the mechanism of action and the anti‐lymphoma activity of the novel anti‐CD20 antibody GA101. Mol Cancer Ther 2011; 10: 178–85. [DOI] [PubMed] [Google Scholar]
- 17. Salles G, Morschhauser F, Lamy T et al Efficacy and safety of obinutuzumab (GA101) monotherapy in relapsed/refractory indolent non‐Hodgkin's lymphoma: results from a phase I/II study (BO20999). Blood 2011; 118: Abstract 268. [Google Scholar]
- 18. Sehn L, Goy A, Offner FC et al Randomized phase II trial comparing GA101 (obinutuzumab) with rituximab in patients with relapsed CD20+ indolent B‐cell non‐Hodgkin's lymphoma: preliminary analysis of the GAUSS study. Blood 2011; 118: Abstract 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Radford J, Davies A, Cartron G et al Obinutuzumab (GA101) in combination with FC or CHOP in patients with relapsed or refractory follicular lymphoma: final results of the Phase I GAUDI study (BO21000). Blood 2011; 118: Abstract 270. [Google Scholar]
- 20. Morschhauser F, Cartron G, Thieblemont C et al Encouraging activity of obinutuzumab (GA101) monotherapy in relapsed/refractory aggressive non‐Hodgkin's lymphoma: results from a phase II study (BO20999). Blood 2011; 118: Abstract 3655. [Google Scholar]
- 21. Salles G, Morschhauser F, Lamy T et al Phase 1 study results of the type II glycoengineered humanized anti‐CD20 monoclonal antibody obinutuzumab (GA101) in B‐cell lymphoma patients. Blood 2012; 119: 5126–32. [DOI] [PubMed] [Google Scholar]
- 22. Sehn L, Assouline SE, Stewart DA et al A phase I study of obinutuzumab induction followed by two years of maintenance in patients with relapsed CD20‐positive B‐cell malignancies. Blood 2012; 119: 5118–25. [DOI] [PubMed] [Google Scholar]
- 23. Cheson BD, Horning SJ, Coiffier B et al Report of an international workshop to standardize response criteria for non‐Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol 1999; 17: 1244. [DOI] [PubMed] [Google Scholar]
- 24. Ogura M, Morschhauser F, Tobinai K et al Comparison of single agent GA101 in Japanese and European patients with relapsed/refractory B‐cell non‐Hodgkin lymphoma (B‐NHL) – Results from two phase I studies. Blood 2010; 116: Abstract 4921. [Google Scholar]