

Abstract
The incidence of colorectal cancer has been increasing and is associated with obesity and diabetes. We have found that type 2 diabetes model KK‐A y/TaJcl (KK‐A y) mice develop tumors within a short period after treatment with azoxymethane (AOM). However, factors that contribute to the promotion of carcinogenesis have not been clarified. Therefore, we looked at the genetic background of KK‐Ay, including two genetic characteristics of KK/TaJcl (KK) mice and C57BL/6J‐Ham‐Ay/+ (Ay) mice, compared with other non‐obese and non‐diabetic mouse strains C57BL/6J and ICR, and induced colorectal premalignant lesions, aberrant crypt foci (ACF), and tumors using AOM (150 μg/mouse/week for 4 weeks and 200 μg/mouse/week for 6 weeks, respectively). The mice with a diabetes feature, KK‐Ay and KK, developed significantly more ACF, 67 and 61 per mouse, respectively, whereas ICR,Ay, and C57BL/6J mice developed 42, 24, and 18 ACF/mouse, respectively, at 17 weeks of age. Serum insulin and triglyceride levels in KK ‐ Ay and KK mice were quite high compared with other non‐diabetic mouse strains. Interestingly, KK‐Ay mice developed more colorectal tumors (2.7 ± 2.3 tumor/mouse) than KK mice (1.2 ± 1.1 tumor/mouse) at 25 weeks of age, in spite of similar diabetic conditions. The colon cancers that developed in both KK‐Ay and KK mice showed similar activation of β‐catenin signaling. However, mRNA levels of inflammatory factors related to the activation of macrophages were significantly higher in colorectal cancer of KK‐Ay mice than in KK. These data indicate that factors such as insulin resistance and dyslipidemia observed in obese and diabetic patients could be involved in susceptibility to colorectal carcinogenesis. In addition, increase of tumor‐associated macrophages may play important roles in the stages of promotion of colorectal cancer.
Excessive accumulation of visceral adipose tissue can induce many disorders, such as type 2 diabetes mellitus (elevated fasting glucose, insulin, and insulin‐like growth factor levels), and dyslipidemia (elevated triglyceride or low high‐density lipoprotein cholesterol levels). Obesity is common in Western countries, and is currently increasing almost ubiquitously across the globe. Recently, obesity has attracted much interest as a risk factor for colorectal cancer. The World Cancer Research Fund and American Institute for Cancer Research have evaluated causal relationships between accumulation of visceral adipose tissue and cancer, and concluded “confident evidence” for colorectal cancers.1 In males in Japan, an overweight condition and obesity (body mass index ≥25) are reported to be associated with colorectal cancer.2, 3
We have reported that obese KK‐A y mice are highly susceptible to induction of colorectal premalignant lesions, ACF, and to development of colorectal cancers by AOM treatment.4 The KK‐A y mice feature severe hyperinsulinemia, severe hypertriglyceridemia, excessive abdominal obesity, and resultant elevation of serum adipocytokines, such as IL‐6, leptin, and Pai‐1 compared with values for lean C57BL/6J mice. Such a consequent abnormality is suggested to be involved in the promotion of colorectal carcinogenesis, and is partly considered as a factor for high cancer susceptibility.
The KK‐A y mice were established by cross‐mating KK mice, a type 2 diabetes mellitus model, with A y mice,5, 6 which carry the Agouti yellow (A y) gene and feature severe hyperphagia, hyperinsulinemia, and dyslipidemia. Non‐diabetic or non‐obese mice could be set as control, e.g., young A y mice or C57BL/6J‐Ham‐+/+ (+/+) mice. In addition to this, the body weight of ICR mice is almost the same as KK‐A y mice at a young age, and they are also susceptible to induction of colorectal carcinogenesis by AOM treatment. The strains and features that could be the most susceptible to induction of colorectal carcinogenesis by AOM treatment have not been clarified. Thus, we aimed to investigate and find the features and molecules involved in obesity‐associated cancer by comparing mouse strains KK‐A y, KK, A y, +/+, C57BL, and ICR. In the present study, we showed colorectal ACF development was strongly affected by diabetic conditions, and additional features of inflammation derived from agouti gene overexpression may lead to further promotion of cancer development.
Materials and Methods
Animals
Female 5‐week‐old KK‐A y, KK, C57BL, and ICR mice were purchased from CLEA Japan (Tokyo, Japan), and A y and +/+ mice were purchased from SLC Japan (Shizuoka, Japan). All animals were acclimated to laboratory conditions for 1 week. Three to four mice were housed per plastic cage with sterilized softwood chips as bedding in a barrier‐sustained animal room at 22 ± 1°C and 55% humidity on a 12:12 h light:dark cycle, and fed AIN‐76A powdered basal diet (CLEA Japan). Food and water were available ad libitum. The animals were observed daily for clinical signs, including anal bleeding and mortality. Body weights and food and water consumption were measured weekly. The experiments were carried out according to the Guidelines for Animal Experiments in the National Cancer Center (National Cancer Center, Tokyo, Japan) and were approved by the Institutional Ethics Review Committee for Animal Experimentation in the National Cancer Center.
Azoxymethane‐induced colorectal ACF development
For the induction of ACF by AOM (Nard Institute, Amagasaki, Japan), 6‐week‐old female KK‐A y (n = 13), KK (n = 13), A y (n = 12), +/+ (n = 12), C57BL/6J (n = 13), and ICR (n = 13) mice were given i.p. injections of AOM (150 μg/mouse) weekly for 4 weeks. Six mice were injected with saline as a control group. At the end of the experimental period, the colorectum was removed, opened longitudinally, and fixed flat between sheets of filter paper in 10% buffered formalin for more than 24 h. They were divided into the proximal segment, rectum (15% in length), then the proximal (middle) and distal halves of the remainder. These were stained with 0.2% methylene blue (Merck, Darmstadt, Germany) and the mucosal surface was assessed for ACF with a stereoscopic microscope, as previously reported.7
Azoxymethane‐induced colorectal tumor development
Six‐week‐old female KK‐A y (n = 15), KK (n = 15), ICR (n = 15), A y (n = 18), and +/+ (n = 17) mice were given i.p. injections of AOM (200 μg/mouse) weekly for 6 weeks for induction of colorectal tumors. Nine mice were injected with saline as a control group. The KK‐A y, KK, and ICR mice were anesthetized with ether and killed at the age of 25 weeks. Because there were no symptoms of tumor development, A y and +/+ mice were killed at 55 weeks. The colorectum was opened longitudinally and colorectal tumors were noted for their location, number, and size. Colorectal tumors along with non‐tumorous parts were fixed in 10% buffered formalin and embedded in paraffin blocks for histopathological evaluation. Diagnosis of colorectal tumors using H&E stained sections was carried out according to the classification of Pozharisski.8 The organs, including heart, liver, lung, spleen, and kidney were also excised and observed macroscopically, and blood samples from the abdominal aorta were collected. Abnormal findings were further histopathologically examined.
Determination of serum parameter levels for diabetes
Serum samples from 17‐week‐old female KK‐A y, KK, A y, +/+, C57BL/6J, and ICR mice and from 25‐week‐old female KK‐A y, KK, and ICR mice were measured for serum concentrations of insulin (n = 6 for saline‐treated group, n = 6–9 for AOM‐treated group; Millipore, Billerica, MA, USA) and leptin (n = 6 for saline‐treated group, n = 9 for AOM‐treated group; BioVendor, Brno, Czech Republic), respectively, by an enzyme‐linked immunoassay according to the manufacturer's protocol. The level of serum triglycerides was analyzed using the Fuji Dri‐Chem system (n = 6 for saline‐treated group, n = 15 for AOM‐treated group; Fujifilm, Tokyo, Japan). Blood glucose was measured using a GR‐102 blood glucose monitor (n = 6 for saline‐treated group, n = 15 for AOM‐treated group; Terumo, Tokyo, Japan).
Immunohistochemical staining
Paraffin‐embedded tissue sections of colorectal tumors were used for further immunohistochemical examination with the avidin–biotin complex immunoperoxidase technique after heating with 10 mM citrate buffer (pH 6.0). As the primary antibodies, monoclonal mouse anti‐β‐catenin Ab (BD Transduction Laboratories, Franklin Lakes, NJ, USA), polyclonal rabbit anti‐macrophage colony‐stimulating factor Ab (Abbiotec, San Diego, CA, USA) and polyclonal rabbit anti‐F4/80 Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 100×, 200×, and 100× dilution, respectively, were used. The secondary Ab, biotinylated horse anti‐mouse IgG or biotinylated goat anti‐rabbit IgG was used at 200× dilution (Vector Laboratories, Burlingame, CA, USA). Staining was carried out using avidin–biotin reagents (Vectastain ABC reagents; Vector Laboratories), 3,3′‐diaminobenzidine, and hydrogen peroxide, and the sections were counterstained with hematoxylin to facilitate orientation. As a negative control, consecutive sections were immunostained without exposure to the primary Ab.
Real‐time PCR analysis
Tissue samples from the non‐cancerous and cancerous parts of the colorectum of KK‐A y, KK, and ICR mice were rapidly deep‐frozen in liquid nitrogen and stored at −80°C. Total RNA was isolated using Isogen (Nippon Gene, Tokyo, Japan) treated with DNase (Invitrogen, Carlsbad, CA, USA), and 2‐μg aliquots in a final volume of 10 μL were used for synthesis of cDNA using an iScript cDNA Synthesis kit (Bio‐Rad Laboratories, Hercules, CA, USA). Real‐time PCR was carried out using a CFX96/384 system (n = 3 for saline‐treated group, n = 2–4 for colon non‐cancerous parts in AOM‐treated group, n = 5–6 for colon cancerous parts in AOM‐treated group; Bio‐Rad Laboratories, Tokyo, Japan) with SsoAdvanced SYBR Green Supermix (Bio‐Rad Laboratories, USA) according to the manufacturer's instructions. Primers for mouse actB (5′‐ACGAGGCCCAGAGCAAGAGA‐3′, 5′‐TGGCTGGGGTGTTGAAGGTC‐3′), COX‐2 (5′‐AATGAGTACCGCAAACGCTT‐3′, 5′‐GAGAGACTGAATTGAGGCAG‐3′), Pai‐1 (5′‐CTTCATGCCCCACTTCTTCAA‐3′, 5′‐TCTGAGCCATCATGGGCAC‐3′), cyclin D1 (5′‐CCATGGAACACCAGCTCCTG‐3′, 5′‐CGGTCCAGGTAGTTCATGGC‐3′), F4/80 (5′‐CCTGGACGAATCCTGTGAAG‐3′, 5′‐GGTGGGACCACAGAGAGTTG‐3′), MCP1 (5′‐TGTTGGCTCAGCCAGATGCA‐3′, 5′‐CTTCTTGGGGTCAGCACAGA‐3′), CSF1 (5′‐GCATCATCCTAGTCTTGCTG‐3′, 5′‐ACTGGCCAGTTCCACCTGTCT‐3′), IL‐1β (5′‐CTTCATCTTTGAAGAAGAGC‐3′, 5′‐TGTACAAAGCTCATGGAGAA‐3′), and OPN (5′‐CTTGCGCCACAGAATGCTG‐3′, 5′‐TGACCTCAG TCCATAAGCCA‐3′) were used. To assess the specificity of each primer set, amplicons generated from the PCR reaction were analyzed for melting curves.
Statistical analysis
The significance of difference in the incidence of AOM‐induced mouse tumors was analyzed using the χ2‐test, and other statistical analyses were carried out with Student's t‐test. Differences were considered to be statistically significant at P < 0.05.
Results
Obese status observed in KK‐A y, KK, and ICR mice
To clarify the causative factors susceptible to colorectal carcinogenesis, strain differences in the line of KK‐A y strains were investigated. At 6 weeks of age, mean body weights of KK‐A y and ICR mice were almost the same at approximately 27 g, but body weight gains were marked in KK‐A y mice compared with those in ICR mice (Fig. 1a). Mean body weights of KK mice were slightly less at 6 weeks of age, but became greater than that of ICR mice during the experiment. Compared to these mouse strains, the body size of mouse strains with the C57BL/6J background was smaller. Mean body weights of A y mice of C57BL/6J background were similar to the control +/+ mice and C57BL/6J mice at 6 weeks of age (18–16 g), but markedly higher than those mice at the end of the experiment. In the ACF experiment with 4‐times AOM treatment (ACF experiment), the final mean body weights at 17 weeks of age of KK‐A y, KK, ICR, A y, +/+, and C57BL/6J mice with AOM treatment were 48.3 ± 4.7 g (mean ± SD), 44.8 ± 2.4, 40.6 ± 5.9, 30.8 ± 4.5, 20.4 ± 1.4, and 22.1 ± 1.3 g, respectively, and mean daily food intakes were 4.2 ± 1.1, 3.8 ± 0.3, 3.8 ± 0.3, 3.3 ± 0.6, 2.9 ± 0.1 and 2.9 ± 0.1 g/mouse/day, respectively. Thus, KK‐A y and A y mice ate more than the respective control mouse strains. In the colorectal cancer experiment with 6‐times AOM treatment (colorectal cancer experiment), the final mean body weights at 25 weeks of age of KK‐A y and KK mice with AOM treatment were significantly higher than those of ICR mice, being 54.2 ± 4.8, 46.9 ± 4.8, and 41.1 ± 5.9 g, respectively; furthermore, KK‐A y mice were significantly heavier than KK mice. Of note, weights of visceral fat in KK‐A y and KK mice were also significantly higher than those of ICR mice (Fig. 1b).
Figure 1.

Body weight changes during the experiment (a) and weight of visceral fat tissue at the end of the experiment (17 weeks of age) (b) in KK‐Ay/TaJcl (KK‐Ay), KK/TaJcl (KK), C57BL/6J‐Ham‐Ay/+ (Ay), C57BL/6J‐Ham‐+/+ (+/+), C57BL/6J, and ICR mice treated with or without azoxymethane (AOM) four times. (a) A solid line indicates mice treated with AOM, and a dotted line indicates mice treated without AOM. ●, KK‐Ay; ○, KK; ▲, A y; ∆, +/+; ■, ICR; □, C57BL/6J. (b) The data of the indicated mice treated with AOM are shown in an open box, and without AOM are shown in a closed box. Data are mean ± SD. *P < 0.05, **P < 0.01 vs +/+ mice.
Diabetes status observed in KK‐A y and KK mice
Serum concentrations of glucose, insulin, leptin, and triglycerides at age 17 weeks were measured to evaluate characteristics of glucose metabolism between the strains (Fig. 2). Among the strains, KK‐A y and KK mice showed high serum glucose, insulin, and triglyceride levels, a feature of diabetes, in non‐AOM‐treated mice. Serum insulin and triglyceride levels in non‐diabetic mice (A y, +/+, C57BL/6J mice, and ICR) were all less than 15 ng/dL and 250 mg/dL, respectively. However, serum insulin levels in KK‐A y and KK mice were 478.3 ± 201.5 (mean ± SD) (P < 0.01 vs A y) and 299.4 ± 201.6 ng/dL (P < 0.01 vs A y), respectively. Serum triglyceride levels in KK‐A y and KK mice were 1158 ± 748 (mean ± SD) (P < 0.01 vs A y) and 910 ± 191.0 mg/dL (P < 0.01 vs A y), respectively. Serum leptin levels in A y mice were also elevated compared to those of +/+ mice, but lower than those of KK‐A y and KK mice. Similar results were obtained in the mice treated with AOM.
Figure 2.
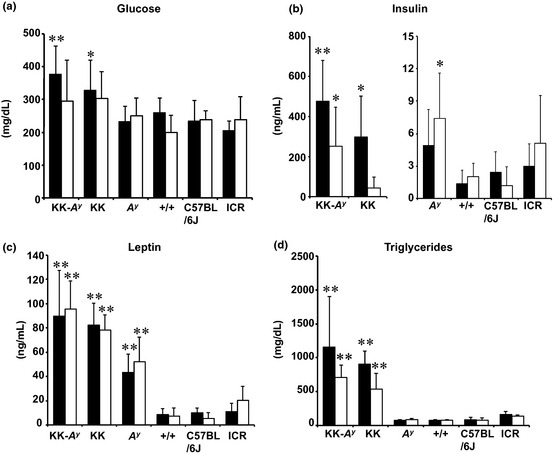
Parameters for diabetes in KK‐Ay/TaJcl (KK‐Ay), KK/TaJcl (KK), and ICR mice treated with or without azoxymethane (AOM). The data of the group treated with AOM six times are shown in an open box, and without AOM are shown in a closed box. Parameters for diabetes, such as blood glucose (a), serum insulin levels (b), serum leptin levels (c), and serum triglyceride levels (d) are shown in the indicated mice strain. Data are mean ± SD. *P < 0.05, **P < 0.01 vs ICR mice. +/+, C57BL/6J‐Ham‐+/+; Ay, C57BL/6J‐Ham‐Ay/+.
Increased number of colorectal aberrant crypt foci in KK‐A y and KK mice
All KK‐A y, KK, A y, +/+, C57BL/6J, and ICR mice developed ACF in the colon and rectum at 17 weeks with AOM treatment (Table 1). In spite of the treatment of all mice with AOM at the same dosage (150 μg/mouse [≈10 mg/kg], weekly for 4 weeks), induction of colorectal ACF was much greater in KK‐A y and KK mice compared to other non‐diabetic mice, that is, A y, +/+, C57BL/6J, and ICR mice. The numbers of total ACF per mouse in KK‐A y and KK mice were 66.7 ± 35.4 and 61.0 ± 17.4 (mean ± SD), which were almost three times higher than those in +/+ mice. Significantly higher numbers of colorectal ACF in KK‐A y and KK mice were observed in all portions of the colorectum compared with +/+ mice, but they were most abundant in the distal portion. The ICR mice were also susceptible to induction of ACF (41.9 ± 18.7/mouse), but the value was less than KK‐A y and KK mice. Saline‐treated mice did not develop colorectal ACF.
Table 1.
Development of colorectal aberrant crypt foci (ACF) in six strains of mice treated with azoxymethane
| Strain of mice | No. of mice with ACF | No. of ACF/colorectum | ||||
|---|---|---|---|---|---|---|
| Proximal | Middle | Distal | Rectum | Total | ||
| KK‐A y | 13/13 | 0.3 ± 0.5 | 16.7 ± 9.0** | 36.0 ± 24.0** | 13.7 ± 7.1* | 66.7 ± 35.4** |
| KK | 13/13 | 0.8 ± 1.0* | 14.2 ± 6.2** | 31.4 ± 8.2** | 14.6 ± 7.3** | 61.0 ± 17.4** |
| A y | 12/12 | 0.0 ± 0.0 | 1.9 ± 2.1 | 15.7 ± 6.7* | 6.1 ± 2.3 | 23.7 ± 8.6 |
| +/+ | 12/12 | 0.0 ± 0.0 | 3.4 ± 1.9 | 12.3 ± 4.6 | 6.6 ± 4.2 | 22.3 ± 6.9 |
| C57BL/6J | 13/13 | 0.1 ± 0.3 | 1.6 ± 2.3 | 9.3 ± 3.0 | 7.0 ± 2.2 | 18.0 ± 5.4 |
| ICR | 13/13 | 0.1 ± 0.3 | 7.7 ± 4.4* | 27.2 ± 12.0** | 6.9 ± 4.8 | 41.9 ± 18.7** |
Data are expressed as mean ± SD. *P < 0.05, **P < 0.01 versus C57BL/6J‐Ham‐+/+ (+/+). A y, C57BL/6J‐Ham‐A y/+; KK, KK/TaJcl; KK‐A y, KK‐A y/TaJcl.
Increased incidence, number, and size of colorectal tumors in KK‐A y mice
Serum concentrations of glucose, insulin, leptin, and triglycerides at the age of 25 weeks were similar to those at the age of 17 weeks. Serum insulin and triglyceride levels in KK‐A y, KK, and ICR mice at 25 weeks of age are shown in Figure 3.
Figure 3.
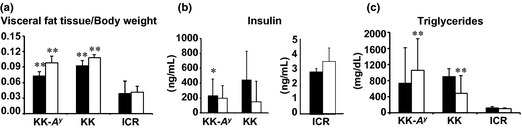
Parameters for diabetes in KK‐Ay/TaJcl (KK‐Ay), KK/TaJcl (KK), and ICR mice treated with or without azoxymethane (AOM). The data of the group treated with AOM six times are shown in an open box, and without AOM are shown in a closed box. Parameters for diabetes, such as visceral fat weight/body weight (a), serum insulin levels (b), and serum triglyceride levels (c) are shown in the indicated mice strain. Data are mean ± SD. *P < 0.05, ** P< 0.01 vs ICR mice.
As shown in Table 2, the incidence of colorectal tumors developed in the KK‐A y and KK mice by AOM treatment was 87% and 73%, respectively, the value in KK‐A y mice tending to be higher. No tumors were observed in ICR mice by 25 weeks of age. Incidences of colorectal tumors in A y and +/+ mice were both 0 out of 3 at 30 weeks of age (0%), and 0 out of 15 (0%) and 3 out of 14 (20%) at 55 weeks of age, respectively, showing that A y mice with a C57BL/6J background were not susceptible to colon carcinogenesis. Most colorectal tumors developed in KK‐A y and KK mice were distributed in the middle to distal portion (Fig. 4a–d). Most large tumors developed in KK‐A y mice were red‐colored (Fig. 4a,b), and red blood cells were observed to be abundant in those tumors (Fig. 4f). Histopathological examination revealed most AOM‐induced colorectal tumors to be adenocarcinomas (Fig. 4e–h). Table 2 also summarizes data on multiplicity (number of tumors/mouse). The average number of tumors in KK‐A y mice was more than twice that in KK mice (P < 0.05). The predominant histological types of carcinomas in KK‐A y mice were well‐differentiated.
Table 2.
Incidence and multiplicity of colorectal tumors in five strains of mice treated with azoxymethane
| Strain of mice | No. of mice | Age when killed, weeks | Incidence, n (%) | No. of tumors | |||||
|---|---|---|---|---|---|---|---|---|---|
| Adenoma | Adenocarcinoma | Total | |||||||
| Well‐ differentiated | Moderately differentiated | Poorly differentiated | Mucinous | ||||||
| KK‐A y | 15 | 25 | 13 (87)*** | 0.0 ± 0.0 | 2.3 ± 2.0*,** | 0.4 ± 0.7 | 0.0 ± 0.0 | 0.1 ± 0.3 | 2.7 ± 2.3*,** |
| KK | 15 | 25 | 11 (73)*** | 0.1 ± 0.3 | 0.5 ± 0.5** | 0.4 ± 0.8 | 0.1 ± 0.3 | 0.0 ± 0.0 | 1.2 ± 1.1** |
| ICR | 15 | 25 | 0 (0) | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| A y | 15 | 55 | 0 (0) | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| +/+ | 14 | 55 | 3 (20) | 0.0 ± 0.0 | 0.2 ± 0.4 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.2 ± 0.4 |
Data are expressed as mean ± SD. *P < 0.05 versus KK/TaJcl (KK). **P < 0.01, ***P < 0.001 versus ICR. +/+, C57BL/6J‐Ham‐+/+; A y, C57BL/6J‐Ham‐A y/+; KK‐A y, KK‐A y/TaJcl.
Figure 4.
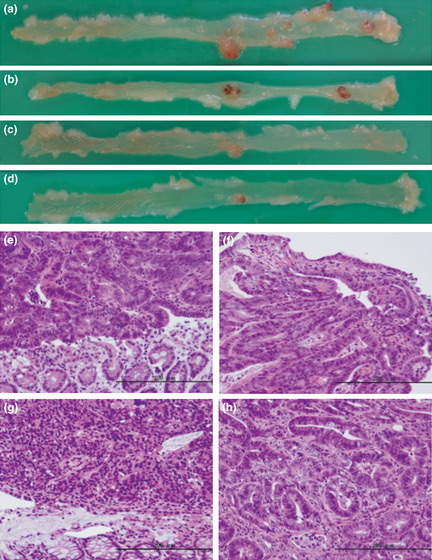
Appearance of colon cancers developed in azoxymethane‐treated mice. Macroscopic views of colon cancers (a–d) and microscopic views with H&E staining (e–h) at the age of 25 weeks in KK‐Ay/TaJcl (KK‐Ay) (a,b,e,f) and KK/TaJcl (KK) (c,d,g,h) mice. The tissue used in (e), (f), (g), and (h) were obtained from the largest tumor in the colon (a), (b), (c) and (d), respectively. Bar = 200 μm.
The number of large tumors (diameter ≥5 mm) was significantly higher in KK‐A y mice than in KK mice, and average tumor volume per mouse in KK‐A y mice was more than threefold that in KK mice (Fig. 5), suggesting tumor promotion due to agouti gene overexpression in KK‐A y mice.
Figure 5.
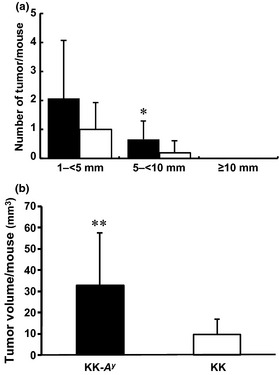
Tumor size difference observed in azoxymethane‐treated KK‐Ay/TaJcl (KK‐Ay) and KK/TaJcl (KK) mice at the age of 25 weeks. (a) Azoxymethane‐developed tumors were divided into three groups by longitudinal diameter, 1–<5 mm, 5–<10 mm, and ≥10 mm. (b) Tumor volume was calculated using the formula, V = length × diameter (short) × diameter (long). ■, KK‐Ay mice; □, KK mice. Data are mean ± SD. *P < 0.05, **P < 0.01 vs tumor volume of KK mice.
Activation of macrophages involved in cancer promotion is suggested to be the difference between KK‐A y and KK mice
To evaluate the activation of β‐catenin signaling in AOM‐induced colorectal cancer in KK‐A y and KK mice, nuclear localization of β‐catenin (active form of β‐catenin) was examined by immunohistochemistry (Fig. 6). Nuclear localization of β‐catenin was not observed in the tumor surrounding normal parts of colorectal mucosa. β‐catenin was clearly activated only in cancerous parts of the colorectum.
Figure 6.
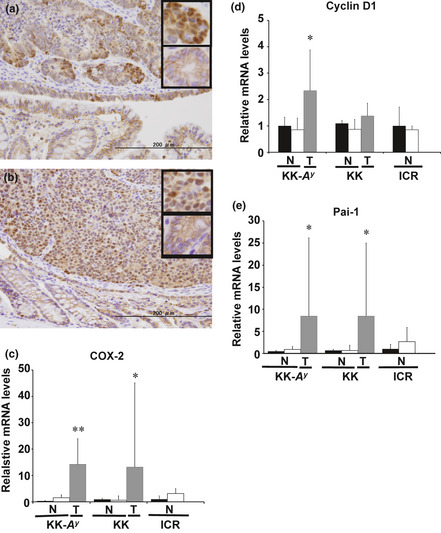
β‐Catenin activation and relative expression levels of mRNA downstream of β‐catenin signaling. Representative examples of colorectal cancer tissue immunostained with anti‐β‐catenin antibodies (n = 3) of KK‐Ay/TaJcl (KK‐Ay) (a) and KK/TaJcl (KK) (b) mice are shown with enlarged images of β‐catenin staining in the small panels (upper panel, cancerous part; lower panel, non‐cancerous part). Bar = 200 μm. Cyclooxygenase‐2 (COX‐2) (c), cyclin D1 (d), and plasminogen activator inhibitor‐1 (Pai‐1) (e) mRNA in non‐cancerous (N) and cancerous (T) parts of colorectum at 25 weeks were examined by RT‐PCR. Data from the mucosa of the saline‐treated group were set as 1. β‐Actin mRNA was used to normalize the data. ■, Mucosa from the saline‐treated group (n = 3); □, mucosa from azoxymethane‐treated group (n = 2–4);  , cancerous part (n = 5–6). Data are mean ± SD. *P < 0.05, **P < 0.01 vs mucosa from saline‐treated group. KK‐Ay, KK‐Ay/TaJcl; KK, KK/TaJcl.
, cancerous part (n = 5–6). Data are mean ± SD. *P < 0.05, **P < 0.01 vs mucosa from saline‐treated group. KK‐Ay, KK‐Ay/TaJcl; KK, KK/TaJcl.
Translocation of β‐catenin to the nucleus, as shown in Figure 6(a,b), suggested transcriptional activation of β‐catenin‐responsive genes. Thus, we carried out semiquantitative RT‐PCR to confirm the activation of β‐catenin signaling pathways. Cyclooxygenase‐2, cyclin D1, and Pai‐1 mRNA, the β‐catenin target genes, in the non‐cancerous part and cancerous part of the colorectum were examined at 25 weeks. There was an increase of all the genes in the cancerous part, except for cyclin D1 in KK mice, in which there was no significant difference in the non‐cancerous part of mucosa with or without AOM treatment.
In the next examination, we focused on inflammatory status, especially in macrophage activation, which may explain the tumorigenic difference between KK‐A y and KK mice. In the cancerous tissue, CSF1, F4/80, IL‐1β, MCP1, and OPN mRNA levels were significantly increased in KK‐A y mice compared with mucosa without AOM treatment (Fig. 7). The data of immunohistochemical staining for CSF1 and the macrophage marker F4/80 confirmed the higher amount of macrophages, especially activated macrophages, in the tumor tissue in KK‐A y mice compared to those in KK mice (Fig. 8). An increase in the number of macrophages was represented by the F4/80 level, and their proliferation, differentiation, and infiltration were represented by CSF1 and MCP1 levels. Activation of macrophages is shown by an increase of IL‐1β and OPN mRNA levels. Among these mRNAs, only MCP1 and OPN mRNA levels were significantly increased in KK mice compared with mucosa without AOM treatment. Expression levels of CSF1, F4/80, IL‐1β, and OPN in cancerous tissues were significantly higher in KK‐A y mice than in KK mice, clearly showing characteristics of cancers developed in KK‐A y mice.
Figure 7.
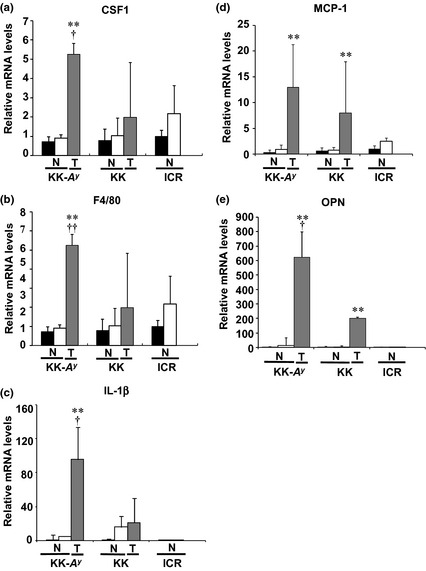
Relative expression levels of mRNA regarding macrophage activation. Messenger RNA levels of colony‐stimulating factor 1 (CSF1) (a), F4/80 (b), interleukin‐1β (IL‐1β) (c), monocyte chemotactic protein 1 (MCP1) (d), and osteopontin (OPN) (e) in non‐cancerous (N) and cancerous (T) parts of colorectum in KK‐Ay/TaJcl (KK‐Ay), KK/TaJcl (KK), and ICR mice aged 25 weeks were examined by RT‐PCR analysis. Data from the mucosa from the saline‐treated group were set as 1. β‐Actin mRNA level was used to normalize the data. ■, Mucosa from saline‐treated group (n = 3); □, mucosa from azoxymethane‐treated group (n = 2–4); , cancerous part (n = 5–6). Data are mean ± SD. **P < 0.01 vs tumor tissue from KK/TaJcl (KK) mice. †P < 0.05, ††P < 0.01 vs mucosa from saline‐treated group.
Figure 8.
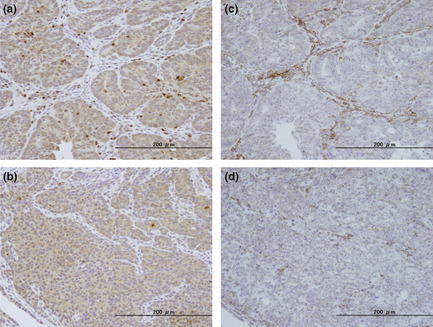
Accumulation of macrophages in the colon tumor tissue of KK‐Ay/TaJcl (KK‐Ay) mice. Represen‐tative examples of colorectal cancer tissue immuno‐stained with anti‐colony‐stimulating factor 1 antibodies (a,b) and anti‐F4/80 antibodies (c,d) of KK‐Ay (a,c) and KK/TaJcl (KK) (b,d) mice are shown. Bar = 200 μm.
Discussion
In the present study, obese diabetic KK‐A y and KK mice were found to be highly susceptible to AOM‐induced ACF compared to those of A y, +/+, C57BL/6J, and ICR mice. The KK‐A y and KK mice, but not other mice, showed hyperinsulinemia, hypertriglyceridemia, and hyperglycemia. Other obese and diabetic mice, induced by monosodium glutamate, are also susceptible to AOM‐induced colon ACF and β‐catenin‐accumulated crypts.9 Regarding the development of ACF, it has been shown that hyperinsulinemia plays an important role. Long‐term treatment with long‐acting insulin on C57BL/KsJ‐db/db mice, which lack a leptin receptor, revealed that insulin increases colonic epithelial cell proliferation, and formation of ACF.10, 11, 12 Moreover, the anti‐insulin resistant medicine pioglitazone prevented ACF development in KK‐A y mice through improvement of serum insulin levels, and increase of p27 and p53 mRNA levels in the colorectal mucosa.13 Hence, it seems likely that hyperinsulinemia in the KK‐A y and KK mice enhanced the development of AOM‐induced ACF in the present study. In addition, hyperglycemia and hypertriglyceridemia may also contribute to the development of ACF.14, 15, 16
To clarify the strain difference regarding susceptibility to AOM‐induced colorectal cancer, we treated mice with AOM six times to develop cancerous lesions. Precancerous lesions, both in rodents and humans, have several genetic abnormalities, such as K‐ras and APC gene mutations.17, 18, 19 Accumulation of β‐catenin protein in the nucleus and cytoplasm are regarded as putative precursors to colorectal adenomas.20
In this study, obese diabetic KK‐A y and KK mice developed colorectal carcinomas within very short periods after AOM injection, but not in A y or ICR mice. Although diabetic conditions were similar between KK‐A y and KK mice (Fig. 2), tumor incidence, multiplicity, and sizes were greater in the KK‐A y mice compared to those of KK mice. Thus, in the next experiments, we attempted to identify the phenotypic differences in the induced lesions in the two strains, KK‐A y and KK. We first carried out immunohistostaining for β‐catenin. β‐catenin mutations seem to be involved in generating dysplastic lesions. β‐Catenin mutations stabilize β‐catenin protein in the cytoplasm and activate β‐catenin/Tcf signaling to induce target genes, such as cyclin D1, COX‐2, and Pai‐1.21, 22, 23 Accumulation of β‐catenin protein in the nucleus of cancer cells was observed in both KK‐A y and KK mice, along with similar induction of downstream targets of β‐catenin, cyclin D1, COX‐2, and Pai‐1. K‐ras mutations contribute to hyperplastic changes. Mutated K‐ras activates the MAPK and phosphoinositide‐3 kinase/Akt pathways, and leads to overexpression of cyclin D1 and COX‐2.
As COX‐2 and Pai‐1 are also good parameters for showing inflammatory status, we further evaluated other inflammation‐associated genes. Among them, molecules that are associated with macrophages, CSF1, F4/80, IL‐1β, MCP1, and OPN,24, 25 were shown to be higher in cancerous parts of KK‐A y mice than in those of KK mice. Indeed, the number of macrophages was observed to be abundant in the tumor tissue of KK‐A y mice compared to that in KK mice. These results suggest that the activation of macrophages may cause promotion of tumor formation in KK‐A y mice. Colony‐stimulating factor 1 is a major chemoattractant of macrophages released from tumor cells and recruits macrophages to the tumor periphery where they secrete motility and angiogenic factors that facilitate tumor cell invasion and metastasis.26 As one of the factors, tumor‐associated macrophage‐derived IL‐1β stimulates Wnt signaling and growth of colon cancer cells27 and protects colon cancer cells from apoptosis through stabilization of Snail.28 Indeed, it has been reported that the number of intratumoral tumor‐associated macrophages correlates with tumor progression in colorectal cancer.29 As far as we know, the impact of A y overexpression on molecules associated with macrophage activation has not been elucidated. However, this issue should be clarified in the near future.
In conclusion, the present studies indicated that KK‐A y mice are the most susceptible to AOM‐induced carcinogenesis compared to strains KK, A y, +/+, C57BL/6J, and ICR. It has become clear that factors such as dyslipidemia and insulin resistance observed in type 2 diabetes could be involved in the susceptibility to colorectal carcinogenesis. Our data indicated that inflammation evoked by macrophages may play important roles in the stages of promotion of colorectal cancer development.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- +/+
C57BL/6J‐Ham‐+/+ mouse strain
- ACF
aberrant crypt foci
- AOM
azoxymethane
- Ay
C57BL/6J‐Ham‐A y/+ mouse strain
- C57BL
C57BL/6J mouse strain
- CSF1
colony‐stimulating factor 1
- IL
interleukin
- KK
KK/TaJcl mouse strain
- KK‐Ay
KK‐A y/TaJcl mouse strain
- MCP1
monocyte chemotactic protein 1
- OPN
osteopontin
- Pai‐1
plasminogen activator inhibitor‐1
Acknowledgments
The authors would like to thank Mr Uchiya and Ms Nakanishi for their expert technical assistance. This work was supported by: a Grant‐in‐Aid for the Third‐Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health, Labor, and Welfare of Japan; a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science; the National Cancer Center Research and Development Fund; a Research Grant of the Princess Takamatsu Cancer Research Fund; and also supported by the National Cancer Center Research Core Facility.
(Cancer Sci 2013; 104: 835–843)
References
- 1. AICR . Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective. Washington, DC: World Cancer Research Fund/American Institute for Cancer Research, 2007. [Google Scholar]
- 2. Otani T, Iwasaki M, Inoue M, Tsugane S. Body mass index, body height, and subsequent risk of colorectal cancer in middle‐aged and elderly Japanese men and women: Japan public health center‐based prospective study. Cancer Causes Control 2005; 16: 839–50. [DOI] [PubMed] [Google Scholar]
- 3. Inoue M, Noda M, Kurahashi N et al Impact of metabolic factors on subsequent cancer risk: results from a large‐scale population‐based cohort study in Japan (JPHC Study). Eur J Cancer Prev 2009; 18: 240–7. [DOI] [PubMed] [Google Scholar]
- 4. Teraoka N, Mutoh M, Takasu S et al High susceptibility to azoxymethane‐induced colorectal carcinogenesis in obese KK‐A y mice. Int J Cancer 2011; 129: 528–35. [DOI] [PubMed] [Google Scholar]
- 5. Kondo K, Nozawa K, Romida T, Ezaki K. Inbred strains resulting from Japanese mice. Bull Exp Anim 1957; 6: 107–12. [Google Scholar]
- 6. Nakamura M, Yamada K. Studies on a diabetic (KK) strain of the mouse. Diabetologia 1967; 3: 212–21. [DOI] [PubMed] [Google Scholar]
- 7. Mutoh M, Watanabe K, Kitamura T et al Involvement of prostaglandin E receptor Subtype EP4 in colon carcinogenesis. Cancer Res 2002; 62: 28–32. [PubMed] [Google Scholar]
- 8. Pozharisski KM. Tumours of the intestines In: Turusov VS, ed. Pathology of Tumours in Laboratory Animals, IARC Scientific Publications no. 1. Lyon: IARC, 1973; 119–40 [PubMed] [Google Scholar]
- 9. Hata K, Kubota M, Shimizu M et al Monosodium glutamate‐induced diabetic mice are susceptible to azoxymethane‐induced colon tumorigenesis. Carcinogenesis 2012; 33: 702–7. [DOI] [PubMed] [Google Scholar]
- 10. Nagel JM, Staffa J, Renner‐Müller I et al Insulin glargine and NPH insulin increase to a similar degree epithelial cell proliferation and aberrant crypt foci formation in colons of diabetic mice. Horm Cancer 2010; 1: 320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tran TT, Naigamwalla D, Oprescu AI et al Hyperinsulinemia, but not other factors associated with insulin resistance, acutelyenhances colorectal epithelial proliferation in vivo . Endocrinology 2006; 147: 1830–7. [DOI] [PubMed] [Google Scholar]
- 12. Koohestani N, Tran TT, Lee W et al Insulin resistance and promotion of aberrant crypt foci in the colons of rats on a high‐fat diet. Nutr Cancer 1997; 29: 69–76. [DOI] [PubMed] [Google Scholar]
- 13. Ueno T, Teraoka N, Takasu S et al Suppressive effect of pioglitazone, a PPAR gamma ligand, on azoxymethane‐induced colon aberrant crypt foci in KK‐A y mice. Asian Pac J Cancer Prev 2012; 13: 4067–73. [DOI] [PubMed] [Google Scholar]
- 14. Siddiqui AA. Metabolic syndrome and its association with colorectal cancer: a review. Am J Med Sci 2011; 341: 227–31. [DOI] [PubMed] [Google Scholar]
- 15. Herbey II, Ivankova NV, Katkoori VR et al Colorectal cancer and hypercholesterolemia: review of current research. Exp Oncol 2005; 27: 166–78. [PubMed] [Google Scholar]
- 16. Chang CK, Ulrich CM. Hyperinsulinaemia and hyperglycaemia: possible risk factors of colorectal cancer among diabetic patients. Diabetologia 2003; 46: 595–607. [DOI] [PubMed] [Google Scholar]
- 17. Takahashi M, Wakabayashi K. Gene mutations and altered gene expression in azoxymethane‐induced colon carcinogenesis in rodents. Cancer Sci 2004; 95: 475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takayama T, Ohi M, Hayashi T et al Analysis of K‐ras, APC, and beta‐catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology 2001; 121: 599–611. [DOI] [PubMed] [Google Scholar]
- 19. Kukitsu T, Takayama T, Miyanishi K et al Aberrant crypt foci as precursors of the dysplasia‐carcinoma sequence in patients with ulcerative colitis. Clin Cancer Res 2008; 14: 48–54. [DOI] [PubMed] [Google Scholar]
- 20. Mori H, Hata K, Yamada Y et al Significance and role of early‐lesions in experimental colorectal carcinogenesis. Chem Biol Interact 2005; 155: 1–9. [DOI] [PubMed] [Google Scholar]
- 21. Tetsu O, McCormick F. Beta‐catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398: 422–6. [DOI] [PubMed] [Google Scholar]
- 22. Araki Y, Okamura S, Hussain SP et al Regulation of cyclooxygenase‐2 expression by the Wnt and ras pathways. Cancer Res 2003; 63: 728–34. [PubMed] [Google Scholar]
- 23. He W, Tan R, Dai C et al Plasminogen activator inhibitor‐1 is a transcriptional target of the canonical pathway of Wnt/beta‐catenin signaling. J Biol Chem 2010; 285: 24665–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zins K, Abraham D, Sioud M, Aharinejad S. Colon cancer cell‐derived tumor necrosis factor‐alpha mediates the tumor growth‐promoting response in macrophages by up‐regulating the colony‐stimulating factor‐1 pathway. Cancer Res 2007; 67: 1038–45. [DOI] [PubMed] [Google Scholar]
- 25. Rao G, Wang H, Li B et al Reciprocal interactions between tumor‐associated macrophages and CD44 positive cancer cells via osteopontin/CD44 promote tumorigenicity in colorectal cancer. Clin Cancer Res 2013; 19: 785–97. [DOI] [PubMed] [Google Scholar]
- 26. Green CE, Liu T, Montel V et al Chemoattractant signaling between tumor cells and macrophages regulates cancer cell migration, metastasis and neovascularization. PLoS ONE 2009; 8: e6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaler P, Augenlicht L, Klampfer L. Macrophage‐derived IL‐1β stimulates Wnt signaling and growth of colon cancer cells; a crosstalk interrupted by vitamin D3 . Oncogene, 2009; 28: 3892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaler P, Galea V, Augenlichi L et al Tumor associated macrophages protect colon cancer cells from TRAIL‐induced apoptosis through IL‐1β‐dependent stabilization of Snail in tumor cells. PLoS ONE, 2010; 5: e11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang J‐C, Chen J‐S, Lee C‐H et al Intratumoral macrophage counts correlate with tumor progression in colorectal cancer. J Surg Oncol, 2010; 102: 242–8. [DOI] [PubMed] [Google Scholar]