

Abstract
Proteasome inhibitors represent a novel class of anticancer agents that are used in the treatment of hematologic malignancies and various solid tumors. However, mechanisms underlying their anticancer actions were not fully understood. It has been reported that strong 14‐3‐3 protein expression is observed and associated with tumor genesis and progression of astrocytoma. In addition, global inhibition of 14‐3‐3 functions with a general 14‐3‐3 antagonist difopein induces apoptosis of human astrocytoma cells, validating 14‐3‐3 as a potential molecular target for anticancer therapeutic management. In the current study, for the first time we demonstrated that proteasome inhibitors downregulated 14‐3‐3ε and 14‐3‐3θ/τ in U87 and SF295 glioma cells. Overexpression of 14‐3‐3ε and 14‐3‐3θ/τ significantly suppressed apoptosis of human glioma cells induced by proteasome inhibitors. We also demonstrated that MG132 activated ASK1 and siASK1 compromised the MG132‐induced apoptosis of glioma cells. Furthermore, overexpression of 14‐3‐3ε and 14‐3‐3θ/τ markedly suppressed activation of ASK1. Collectively, the current study supported that proteasome inhibitors, at least in part, caused cytotoxicity of glioma cells via downregulation of 14‐3‐3ε and 14‐3‐3θ/τ and subsequent activation of ASK1. (Cancer Sci 2013; 104: 55–61)
14‐3‐3 proteins are highly conserved family of 28–33‐kDa acidic proteins and expressed in all eukaryotic cells. There are seven known 14‐3‐3 isoforms (β, σ, ε, ζ, η, θ/τ and γ), which were encoded by separate genes in mammals. 14‐3‐3 proteins, potentially acting both as oncogenes and as tumor suppressors, can associate with a wide variety of proteins involved in signal transduction, cell cycle control, intracellular trafficking/targeting, cytoskeletal structure, transcription, DNA replication and apoptosis.1, 2, 3 To date, over 300 proteins have been identified as 14‐3‐3 binding ligands, and the number is still increasing. Considering the number of binding partners, it is not surprising that 14‐3‐3 proteins play crucial roles in regulating many vital cellular processes such as DNA damage response, metabolism, protein trafficking, signal transduction, transcriptional regulation, apoptosis and cell cycle progression. It has been reported that 14‐3‐3 proteins are overexpressed in some human cancers including brain tumors and are gradually being recognized as important regulators of carcinogenesis and tumor development.4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16
Although 14‐3‐3 proteins have been found in different organisms such as the suprarenal gland, intestines, and liver, the highest concentrations of 14‐3‐3 proteins were found in human brains, comprising about 1% of its total soluble protein.17, 18 All seven 14‐3‐3 proteins have been reported to be distributed mainly in the neurons, only weak expression of ε, ζ and θ/τ was found in some glial cells.15, 19, 20 However, β, ε, ζ, η and θ/τ was found in the majority of astrocytoma samples and its expression was increased markedly with an increase in the pathologic grade of human astrocytoma,15, 16 indicating that the five isoforms may play an important role in the carcinogenesis of human astrocytoma. Furthermore, global inhibition of 14‐3‐3 functions triggers apoptosis, suppresses de novo tumor formation and inhibits growth of established tumors of astrocytoma,21 promising 14‐3‐3 as a potential therapy target for astrocytoma treatment.
Apoptosis signal‐regulating kinase 1 (ASK1) is an ubiquitously expressed serine‐threonine protein kinase that functions as a MAPKKK to activate the JNK and p38 MAPK signaling cascades.22, 23 ASK1 is activated in response to various stresses, including tumor necrosis factor (TNF), serum withdrawal, endoplasmic reticulum stress, Fas ligation, and H2O2, 22, 23, 24, 25, 26 and its activation is a pivotal mechanism in a broad range of cell death paradigms.27 ASK1 activity is regulated at multiple steps, including dimerization, phosphorylation, and protein‐protein interactions.24, 28 Among them, dissociation from 14‐3‐3 proteins is one of the mechanisms underlying activation of ASK1.29, 30, 31
Proteasome inhibitors represent a novel class of anti‐tumor agents with pre‐clinical and clinical evidence of activity against hematologic malignancies and solid tumors, but the mechanisms are not fully elucidated. Both natural and synthetic inhibitors are capable of blocking the proteolytic activity of the proteasome. In the current study, we used four different proteasome inhibitors, including MG132, PSI, lactacystin and epoxomicin to investigate their role and possible mechanism in glioma cells. MG132 and PSI are synthetic peptide aldehydes, which inhibit the proteasome complex's chymotrypsin‐like activity in a potent but reversible manner. Lactacystin and epoxomicin are natural, cell‐permeable, nonpeptide inhibitors that are relatively selective. Lactacystin and epoxomicin inhibit the proteasome activity in an irreversible and reversible manner, respectively. In the current study, we demonstrated that all of these proteasome inhibitors induced apoptosis of glioma SF295 and U87 cells. In addition, we found that these proteasome inhibitors reduced 14‐3‐3ε and 14‐3‐3θ/τ expression in human glioma U87 and SF295 cells. Overexpression of 14‐3‐3ε and 14‐3‐3θ/τ significantly blocked the apoptosis of U87 cells induced by MG132, indicating the involvement of 14‐3‐3 proteins in MG132‐mediated cytotoxicity of human glioma. In addition, we demonstrated that 14‐3‐3 proteins regulated MG132‐mediated ASK1 activation in glioma cells.
Materials and Methods
Culture of cancer cell lines
SF295 and U87 glioma cell lines were maintained in DMEM (Sigma‐Aldrich, Saint Louis, MO, USA) supplemented with 10% FBS (ExCell Biology, Shanghai, China).
Tissue samples and culture of primary human glioma cells
This study was approved by the institutional ethics committees of China Medical University and informed consent was obtained from all participants involved in this study. Tumor samples from five human glioma patients were removed by craniotomy, stored in sterilized pre‐cooled PBS and transported to the lab at 4°C. Tumor tissues were washed three times with pre‐cooled PBS, and cut into pieces. Tissue pieces were digested in PBS‐buffered collagenase/dispase cocktail, which contained 1 mg/mL collagenase, 2 mg/mL dispase, 70 U/mL DNase, 0.1% BSA. Digestion was performed for 30 min in a 37°C rotator oven. Samples were gently triturated several times with 5 mL stripette. The digested tissue mixture was transferred into a 40 μM filter to exclude remaining tissue pieces. Pass through was centrifuged and cells were collected and seeded into culture plate.
Chemicals
MG132, epoxomycin, PSI and lactacystin were purchased from Calbiochem (La Jolla, CA, USA). 0.02% DMSO was used as vehicle control.
Cell viability assays
For cell viability assays, cells were plated in 96‐well dishes (1 × 104 cells per well) and the next day were treated with or without apoptosis inducing agents in 10% FBS‐containing media and grown over a 24‐h period. Cell viability was assessed using the MTT assay (Chemicon, Bedford, MA, USA) according to the manufacturer's instruction.
Detection of apoptotic cell death
For apoptotic cell death assays, cells were washed twice in PBS and then stained with Annexin V‐FITC (Biovision, Mountainview, CA, USA) and propidium iodide (PI, Sigma–Aldrich, St Louis, MO, USA) according to the manufacturer's instructions. After staining with Annexin V‐FITC and PI, samples were analyzed by fluorescence‐activated cell scanner (FACScan) flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
RNA isolation and real‐time reverse transcription‐polymerase chain reaction
RNA isolation and real‐time RT‐PCR were performed as previously reported.32 5′‐ATATGATGATATGGCTGCAG‐3′ and 5′‐AGAACATCATTGCAGATGTC‐3′, 5′‐AATTGAGACGGAGCTAAGAG‐3′ and 5′‐ATGCTTGTTGTGACTGATCG‐3′, 5′‐AAGAATGTGATTGGAGCTAG‐3′ and 5′‐ATTGCAATATCACTAGCAGC‐3′, 5′‐AGATTGAGAAGGAGCTGGAG‐3′ and 5′‐TTCAGAAGCTTCGACCACAC‐3′, 5′‐TCATCAGTAGCATTGAGCAG‐3′ and 5′‐TGTGCTCTTTGCTGATCTCG‐3′, 5′‐ATGAGGACATGGCAGCCTTC‐3′ and 5′‐AGTCACCCTTCATCTTCAGG‐3′, 5′‐GAAGTTGCAGCTGATTAAGG‐3′ and 5′‐TCAAATGCCTCTTGGTAAGC‐3′ primer pairs were used to amplify 14‐3‐3β, ε, γ, ζ, η, σ and θ/τ, respectively. For β‐actin, the forward primer was 5′‐GAGACCTTCAACACCCCAGCC‐3′ and the reverse was 5′‐GGATCTTCATGAGGTAGTCAG‐3′. Results were normalized against those of β‐actin and presented as arbitrary unit.
Western blot analysis
Cells were lysed in lysis buffer (20 mM Tris‐HCl, 150 mM NaCl, 2 mM EDTA, 1% Triton‐×100 and protease inhibitor cocktail (Sigma–Aldrich). Cell extract protein amounts were quantified using the BSA protein assay kit. Equivalent amounts of protein (25 μg) were separated using 12% SDS‐PAGE and transferred to PVDF membrane (Millipore Corporation, Billerica, MA, USA). The following antibodies were used: antibodies against 14‐3‐3 proteins (Santa Cruz Biotechnology, Santa Cruz, CA, USA), ASK1, phospho‐JNK1/2 and phosphor‐p38 (ExCell Biology), JNK1/2, p38 and phospho‐ASK1 (Cell Signaling Technology, Danvers, MA, USA).
Construction of 14‐3‐3 plasmids and cell transfection
14‐3‐3 expression vectors were generously provided by Professor Haian Fu (Rollins Research Center). A cDNA encoding human ASK1 was generated by PCR from human brain cDNA library (Invitrogen, Carlsbad, CA, USA) and subcloned into the eukaryotic expression plasmid pcDNA3. Cells were transfected with Lipofectamine 2000 reagent (Invitrogen) as instructed by the supplier.
Small interfering RNA
The siRNA sequences used here were as follows: siRNA against ASK1 (siASK1), AUGUGCUCCACACGCCGGCUGUU. The scramble nonsense siRNA (scramble; CCGUAUCGUAAGCAGUACU) that has no homology to any known genes was used as control. siRNA against 14‐3‐3ε and 14‐3‐3θ/τ was purchased from Santa Cruz. Transfection of siRNA oligonucleotide was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations.
Statistics
The statistical significance of the difference was analyzed by anova and post hoc Dunnett's test. Statistical significance was defined as P < 0.05. All experiments were repeated three times, and data were expressed as the mean ± SD from a representative experiment.
Results
Proteasome inhibitors‐induced cytotoxicity of U87 and SF295 glioma cells
U87 and SF295 glioma cells were treated with proteasome inhibitor MG132 at concentrations ranging from 0 to 5 μM for 24 h, the cell viability was determined using the MTT assay. MG132 induced growth inhibition in U87 and SF295 glioma cells in a dose‐dependent manner (Fig. 1A). After 24 h of treatment, the inhibitory rate of MG132 (5 μM) on viability of U87 and SF295 cells had reached 43.5 ± 1.87% and 48.6 ± 1.91%, respectively. To confirm an apoptotic mechanism in MG132‐induced death of U87 and SF295 cells, apoptotic cell death was analyzed using Annexin V and PI double staining followed by flow cytometry. Flow cytometry demonstrated that MG132 induced apoptotic cell death of U87 and SF295 glioma cells in a dose dependent manner (Fig. 1B). To confirm the suppression of proteasomal activity by MG132, ubiquitinated proteins were measured using Western blot analysis. MG132 resulted in accumulation of ubiquitinated proteins in a dose‐dependent manner in both U87 and SF295 glioma cells (Fig. 1C). Time course showed that 5 μM of MG132 caused time‐dependent suppression of both U87 and SF295 cell viability (Fig. 1D). Obvious reduction of cell viability was observed after 8 h of MG132 exposure and cell viability was further decreased afterwards. Cell viability was reduced abruptly after 24 h, and less than 10% viable cells were observed after 48 h of MG132 exposure (Fig. 1D). Three other proteasome inhibitors PSI, epoxomicin (epoxo) and lactacystin (lacta) also caused growth inhibition (Fig. 1E) and apoptotic cell death (Fig. 1F) in U87 and SF295 cells, as assessed by MTT assay and flow cytometry, respectively. To investigate whether proteasome inhibitors could induce cytotoxicity in primary glioma cells, we isolated primary human glioma cells from five glioma patients, and found that MG132 significantly decreased proliferation in 3 of 5 primary glioma cells (Fig. 1G).
Figure 1.
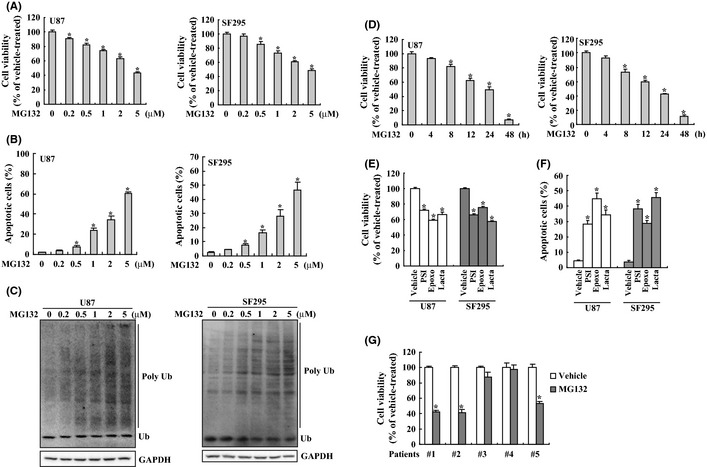
Cytotoxicity of glioma cells induced by proteasome inhibitors. (A) U87 and SF295 cells were treated with the indicated concentration of MG132 for 24 h, and cell viability was measured using 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay. (B) U87 and SF295 cells were treated with the indicated concentration of MG132 for 24 h, and apoptotic cell death was measured using Annexin V/propidium iodide (PI) double staining followed by flow cytometry. (C) U87 and SF295 cells were treated with the indicated concentration of MG132 for 24 h, and Western blot was performed to measure the level of ubiquitinated proteins. (D) U87 and SF295 cells were treated with 5 μM of MG132 for the indicated time, and cell viability was analyzed using MTT assay. (E) U87 and SF295 cells were treated with vehicle, PSI, epoxomycin (epoxo) or lactacystin (lacta) for 24 h, and cell viability was measured using MTT assay. (F) U87 and SF295 cells were treated with the indicated proteasome inhibitor for 24 h, and apoptotic cell death was measured using flow cytometry. (G) Primary human glioma cells were treated with vehicle or MG132 for 24 h, and cell viability was measured using MTT assay. *P < 0.01.
Reduction of 14‐3‐3ε and 14‐3‐3θ/τ expression by proteasome inhibitors in U87 and SF295 glioma cells
We first performed real‐time RT‐PCR to study regulation of 14‐3‐3 mRNA expression by MG132 in U87 and SF295 glioma cells. Incubation with MG132 decreased 14‐3‐3ε and 14‐3‐3θ/τ mRNA levels in U87 and SF295 cells in a dose‐dependent manner (Fig. 2A). MG132 had no obvious effect on the mRNA expression levels of other 14‐3‐3 isoforms (data not shown). Western blot analysis confirmed a marked decrease of 14‐3‐3ε and 14‐3‐3θ/τ proteins in U87 and SF295 cells upon MG132 exposure (Fig. 2B). Real‐time RT‐PCR (Fig. 2C) and Western blot analysis (Fig. 2D) confirmed that PSI, epoxomycin and lactacystin also reduced expression of 14‐3‐3ε and 14‐3‐3θ/τ in U87 cells. Western blot analysis demonstrated that MG132 also reduced 14‐3‐3ε and 14‐3‐3θ/τ expression in some primary glioma cells isolated from glioma tissues of human patients (Fig. 2E). To confirm whether proteasome inhibition downregulated 14‐3‐3ε and 14‐3‐3θ/τ expression via suppression of its transcription, newly synthesized RNA was labeled and isolated from control and MG132‐treated U87 cells using Click‐iT Nascent RNA Capture (Life Technology, Carlsbad, CA, USA) and real‐time PCR was performed to measure 14‐3‐3ε and 14‐3‐3θ/τ mRNA levels (Fig. 2F). Nascent 14‐3‐3ε and 14‐3‐3θ/τ mRNA levels were significantly reduced in MG132‐treated cells (Fig. 2F), indicating that proteasome inhibition decreased 14‐3‐3ε and 14‐3‐3θ/τ expression via suppression of transcription. To further clarify the potential role of 14‐3‐3ε and 14‐3‐3θ/τ downregulation in cytotoxicity of glioma cells mediated by proteasome inhibition, we used specific siRNAs against 14‐3‐3ε or 14‐3‐3θ/τ to knockdown their expression (Fig. 2G). Importantly, knockdown of 14‐3‐3ε or 14‐3‐3θ/τ significantly suppressed cell viability (Fig. 2H) and induced apoptosis (Fig. 2I) of both U87 and SF295 cells.
Figure 2.
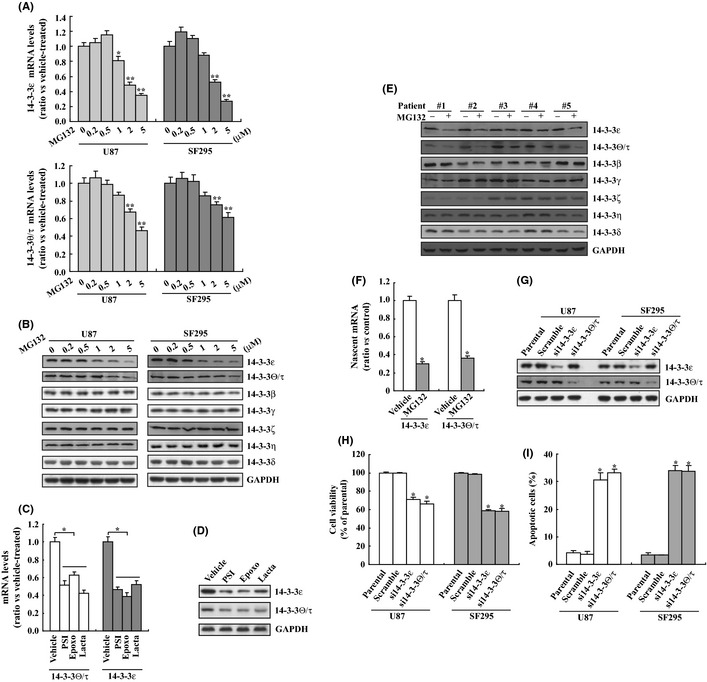
Reduction of 14‐3‐3ε and 14‐3‐3θ/τ by proteasome inhibition in glioma cells. (A) U87 and SF295 cells were treated with the indicated concentration of MG132 for 8 h, and real‐time polymerase chain reaction (PCR) was performed to analyze 14‐3‐3 mRNAs expression. (B) U87 and SF295 cells were treated with the indicated concentration of MG132 for 24 h, and Western blot analysis was performed to investigate 14‐3‐3 proteins expression. (C) U87 cells were treated with the indicated proteasome inhibitor, and 14‐3‐3 mRNA levels were analyzed using real‐time PCR. (D) U87 cells were treated with the indicated proteasome inhibitor, and 14‐3‐3 protein levels were measured using Western blot analysis. (E) Primary human glioma cells were treated with MG132 and Western blot analysis was performed. (F) U87 cells were treated with vehicle or MG132 for 8 h, then nascent RNA was labeled with EU using Click‐iT nascent RNA Capture kit (Life Technology) for an additional 4 h. EU‐nascent RNA was isolated and real‐time PCR was performed. (G) U87 or SF295 cells were transfected with scramble small interfering RNA (siRNA) against 14‐3‐3ε (si14‐3‐3ε) or 14‐3‐3θ/τ (si14‐3‐3θ/τ) for 24 h and Western blot was performed. (H) U87 or SF295 cells were transfected with scramble siRNA, siRNA against 14‐3‐3ε (si14‐3‐3ε) or 14‐3‐3θ/τ (si14‐3‐3θ/τ) for 24 h and cell viability was measured using 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay. (I) U87 or SF295 cells were transfected with scramble siRNA, siRNA against 14‐3‐3ε (si14‐3‐3ε) or 14‐3‐3θ/τ (si14‐3‐3θ/τ) for 24 h and apoptotic cells were analyzed using flow cytometry.
Suppression of MG132‐mediated U87 and SF295 glioma cell death by overexpression of 14‐3‐3 proteins
We next examined functional roles of 14‐3‐3 proteins in MG132‐medaited apoptosis. Overexpression of 14‐3‐3ε and 14‐3‐3θ/τ caused significant increases of their protein expression under basal conditions in U87 cells (Fig. 3A). In addition, transfection with 14‐3‐3ε and 14‐3‐3θ/τ blocked MG132‐mediated reduction of their proteins in U87 cells (Fig. 3A). To determine the effect of 14‐3‐3ε and 14‐3‐3θ/τ overexpression on MG132‐mediated cell death, 14‐3‐3ε and 14‐3‐3θ/τ alone or combination transfected cells were cultured for 24 h in the presence of MG132, labeled with annexin V‐FITC/PI and subjected to flow cytometry. Cells transfected with 14‐3‐3ε or 14‐3‐3θ/τ alone displayed an abrupt reduction in cell death with MG132 treatment (Fig. 3B). When compared with transfection with 14‐3‐3ε or 14‐3‐3θ/τ alone, cotransfection demonstrated a tendency to further reduce apoptotic cell death mediated by MG132, but no statistical significance was observed (Fig. 3B). Similar like in U87 cells, overexpression of 14‐3‐3ε or 14‐3‐3θ/τ also dramatically inhibited MG132‐mediated apoptosis of SF295 cells (Fig. 3C). In addition, overexpression of 14‐3‐3ε or 14‐3‐3θ/τ significantly decreased apoptosis of U87 cells (Fig. 3D) or SF295 cells (Fig. 3E) mediated by other proteasome inhibitors including PSI, epoxo and lacta.
Figure 3.

Implication of downregulation of 14‐3‐3 proteins in cytotoxicity of glioma cells mediated by proteasome inhibitors. (A) U87 cells were transfected with 14‐3‐3ε and 14‐3‐3θ/τ alone or in combination for 24 h, and then treated with vehicle or MG132 for additional 24 h. Western blot analysis was performed using the indicated antibodies. (B) U87 cells were transfected with 14‐3‐3ε and 14‐3‐3θ/τ alone or in combination for 24 h, and then treated with vehicle or MG132 for additional 24 h. Apoptotic cells were analyzed using Annexin V and propidium iodide (PI) double staining followed by flow cytometry. (C) SF295 cells were transfected with 14‐3‐3ε and 14‐3‐3θ/τ alone or in combination for 24 h, then treated with vehicle or MG132 for additional 24 h. Apoptotic cells were analyzed using Annexin V and PI double staining followed by flow cytometry. (D) U87 cells were transfected with 14‐3‐3ε or 14‐3‐3θ/τ for 24 h, then treated with the indicated proteasome inhibitor for additional 24 h. Cells were labeled using Annexin V and PI, subsequently analyzed by flow cytometry to investigate apoptotic cells. (E) SF295 cells were transfected with 14‐3‐3ε or 14‐3‐3θ/τ for 24 h, then treated with the indicated proteasome inhibitor for additional 24 h. Cells were labeled using Annexin V and PI, subsequently analyzed by flow cytometry to investigate apoptotic cells. *P < 0.01.
Implication of ASK1 activation in MG132‐mediated U87 and SF295 cell death
We have previously reported that ASK1 was involved in MG132‐mediated thyroid cancer cell death.33 Similar to in thyroid cancer cells, MG132 activated endogenous ASK1, as well as its downstream effectors p38 and JNK in U87 and SF295 glioma cells (Fig. 4A), indicating that MG132 also activated the ASK1‐p38 and the ASK1‐JNK pathways in glioma cells. We next examined whether ASK1 is required for apoptosis of U87 or SF295 glioma cells mediated by MG132 using siRNA against ASK1 (siASK1) to knockdown the endogenous ASK1. siASK1 successfully reduced the expression of total and phosphorylated ASK1 (Fig. 4B). In addition, siASK1 also reduced phosphorylation of p38 and JNK mediated by MG132 (Fig. 4B). Importantly, MG132‐mediated apoptosis of U87 or SF295 glioma cells was markedly inhibited by siASK1 (Fig. 4C), indicating that ASK1 was also implicated in U87 and SF295 glioma cell death induced by proteasome inhibition. To further confirm the involvement of ASK1‐mediated activation of p38 and JNK in glioma cytotoxicity mediated by proteasome inhibition, pharmacological inhibitors SB203580 and SP600125 were used to inhibit activation of p38 and JNK, respectively. Unexpectedly, contrary to siASK1, both SB20358 and SP600125 enhanced responsiveness of U87 and SF295 cells to MG132 treatment (Fig. 4D).
Figure 4.
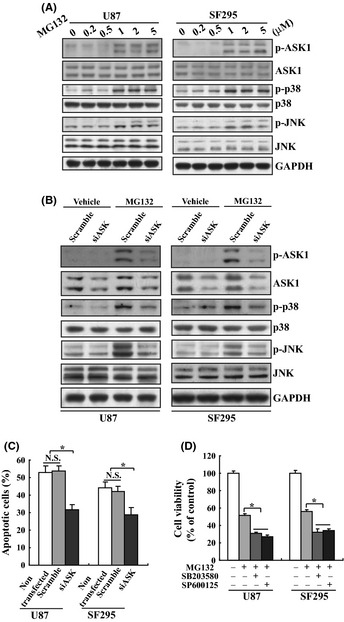
Involvement of ASK1 activation in apoptosis of glioma cell induced by MG132. (A) U87 and SF295 cells were treated with the indicated concentration of MG132 for 24 h, and Western blot analysis was performed using the indicated antibodies. (B) U87 and SF295 cells were transfected with scramble or siRNA against ASK1 (siASK1) for 24 h, then treated with vehicle or MG132 for additional 24 h. Western blot analysis was performed using the indicated antibodies. (C) U87 and SF295 cells were nontransfected, or transfected with scramble or siASK1 for 24 h, then treated with vehicle or MG132 for additional 24 h. Apoptotic cells were measured using Annexin V and PI double staining followed by flow cytometry. (D) U87 and SF295 cells were treated with MG132 in the absence or presence of SB203580 or SP600125 for 24 h, and cell viability was measured using 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay. *P < 0.01.
Suppression of MG132‐mediated ASK1 activation by 14‐3‐3ε and 14‐3‐3θ/τ
It has been reported that 14‐3‐3 proteins negatively regulate ASK1 activity via their interaction, and dissociation from 14‐3‐3 proteins causes activation of ASK1,30, 31 therefore, overexpression of 14‐3‐3 proteins may inhibit apoptosis of glioma cells induced by MG132 via its influence on activation of ASK1. To explore this question, we investigated the activation of ASK1 mediated by MG132 in 14‐3‐3ε and 14‐3‐3θ/τ overexpressed U87 or SF295 cells. Overexpression of 14‐3‐3ε and 14‐3‐3θ/τ significantly suppressed activation of ASK1, as well as activation of p38 and JNK mediated by MG132 (Fig. 5A). Cotransfection of 14‐3‐3ε and 14‐3‐3θ/τ demonstrated no further suppression of ASK1, p38 or JNK activation, when compared with transfection 14‐3‐3ε or 14‐3‐3θ/τ alone (Fig. 5A). To clarify whether other types of 14‐3‐3 could block the activation of ASK1 mediated by proteasome inhibition, U87 cells were transfected with various 14‐3‐3 eukaryotic expression vectors (Fig. 5B). Similar to 14‐3‐3ε and 14‐3‐3θ/τ (Fig. 5A), overexpression of 14‐3‐3ζ and 14‐3‐3η also significantly compromised activation of ASK1 mediated by MG132, while other 14‐3‐3 proteins demonstrated no obvious effects on ASK1 activation (Fig. 5B).
Figure 5.
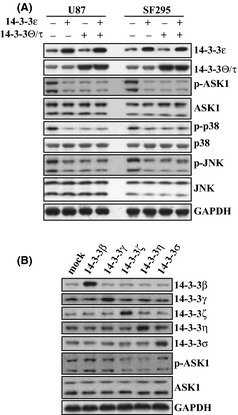
Suppression of MG132‐mediated ASK1 activation by 14‐3‐3 proteins. (A) U87 and SF295 cells were transfected with 14‐3‐3ε or 14‐3‐3θ/τ alone or in combination for 24 h, and then treated with MG132 for an additional 24 h, and Western blot was performed using the indicated antibodies. (B) U87 cells were transfected with the indicated 14‐3‐3 vectors for 24 h, and then treated with MG132 for an additional 24 h, and Western blot was performed using the indicated antibodies.
Discussion
14‐3‐3 plays an important role in a wide range of biological processes through a variety of regulatory mechanisms by its binding to phosphoserine‐containing sequence motifs in diverse partners, such as signal transduction, cell cycle control, vesicular transport, DNA replication, DNA repair and apoptosis.34, 35, 36, 37 Meanwhile, 14‐3‐3 is expressed in most human glioma tissues but not in an overwhelming majority of normal brain tissues.15 Furthermore, global inhibition of 14‐3‐3 functions triggers apoptosis, suppresses de novo tumor formation and inhibits growth of established tumors of glioma,21 validating 14‐3‐3 as a potential therapy target for glioma treatment.
In the current study, we demonstrated that proteasome inhibitors decreased 14‐3‐3ε and 14‐3‐3θ/τ isoforms in U87 and SF295 glioma cells. Real‐time PCR performed on nascent RNA demonstrated that MG132 reduced transcriptional activities of 14‐3‐3ε and 14‐3‐3θ/τ genes, while the exact transcription factors involved in specific downregulation of 14‐3‐3ε and 14‐3‐3θ/τ isoforms remain to be clarified. It has been reported that proteasome inhibitors suppress nuclear factor κB (NFκB) activity, which induces cytotoxicity of various cancer cells. Global screening of promoter regions of 14‐3‐3 isoforms demonstrates that 14‐3‐3ε and 14‐3‐3θ/τ genes contain potential NFκB binding sequences, while other isoforms do not have any such consensus. Therefore, proteasome inhibitors might downregulate 14‐3‐3ε and 14‐3‐3θ/τ expression via suppression of NFκB activation. Further investigation is required to clarify this item in the future. In the current study, we demonstrated that downregulation of 14‐3‐3ε and 14‐3‐3θ/τ isoforms using their specific siRNAs suppressed cell viability and induced apoptosis of U87 and SF295 cells. In addition, overexpression of 14‐3‐3ε and 14‐3‐3θ/τ isoforms dramatically prohibited MG132‐induced glioma cell death. These findings are consistent with the previous report that 14‐3‐3‐targetd strategy using its antagonist, difopein, or siRNA enhances sensitivity of glioma cells to apoptosis,21 offering an attractive opportunity for the development of anticancer agents by inhibiting 14‐3‐3 functions to sensitize glioma to apoptosis. Using in vivo intracranial injection model might further enhance the conclusion in the future.
14‐3‐3 proteins represent an integration point for proliferative, survival, apoptotic and stress signaling pathways. Members of the 14‐3‐3 protein family enhance the activity of many proteins with proliferative and/or survival functions, such as Raf kinases, and antagonize the activity of proteins that promote cell death and senescence, such as Bad, Bim, Bax and ASK1.34 Many proteins involved in cancer cell growth and survival are regulated by proteasomal degradation, and dysregulation of these proteins are supposed to be implicated in cytotoxicity induced by proteaosme inhibitors.38 Therefore, it was reasonable that overexpression of 14‐3‐3 isoforms only partly blocked proteasome inhibitor‐mediated cytotoxicity of glioma cells in the current study. In mammalian cells, ASK1 participates in the JNK and p38 MAP signaling cascades by phosphorylating MKK4/MKK7 and MKK3/MKK6, respectively.22 ASK1 is activated in response to various extracellular and intracellular stimuli, such as lipopolysaccharide, reactive oxygen species (ROS), endoplasmic reticulum (ER) stress and signaling through such as Fas and TNFα.38 ASK1 is well known as a proapoptotic, stress‐activated signaling molecule, and it is under tight regulation at multiple levels, one of which is negatively regulated by association with 14‐3‐3 proteins.39 In the current study, we reported that ASK1 played an essential role in MG132‐mediated glioma cell death, as knockdown ASK1 by siRNA markedly blocked the apoptosis of glioma cells induced by MG132. In addition, we detected that downregulation of 14‐3‐3 proteins played a role in ASK1 activation, as overexpression of 14‐3‐3 proteins reduced MG132‐mediated ASK1 phosphorylation. However, using JNK or p38 inhibitors to block their activation did not suppress, but enhanced the cytotoxicity of glioma cells mediated by MG132, indicating that beside JNK and p38, other downstream effectors might be ascribed to the apoptotic property of ASK1 upon proteasome inhibition.
In conclusion, we have described downregulation of 14‐3‐3 proteins, subsequent activation of ASK1 is another mechanism underlying cytotoxical responses of glioma cells to MG132, validating 14‐3‐3 as a potential therapy target for the development of anticancer agents combat with glioma.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgements
This work was partly supported by National Natural Science Foundation of China (31070697 and 31170727), Program for LNET (LJQ2011083) and Foundation of Liaoning Educational Committee (L2010561) to H‐Q Wang.
(Cancer Sci, doi: 10.1111/cas.12004, 2012)
References
- 1. Berg D, Holzmann C, Riess O. 14–3‐3 proteins in the nervous system. Nat Rev Neurosci 2003; 4: 752–62. [DOI] [PubMed] [Google Scholar]
- 2. Tzivion G, Shen YH, Zhu J. 14–3‐3 proteins; bringing new definitions to scaffolding. Oncogene 2001; 20: 6331–8. [DOI] [PubMed] [Google Scholar]
- 3. Wilker E, Yaffe MB. 14–3‐3 Proteins – a focus on cancer and human disease. J Mol Cell Cardiol 2004; 37: 633–42. [DOI] [PubMed] [Google Scholar]
- 4. Ralhan R, Masui O, Desouza LV, Matta A, Macha M, Siu KW. Identification of proteins secreted by head and neck cancer cell lines using LC‐MS/MS: strategy for discovery of candidate serological biomarkers. Proteomics 2010; 11: 2363–76. [DOI] [PubMed] [Google Scholar]
- 5. Ko BS, Lai IR, Chang TC et al Involvement of 14–3‐3gamma overexpression in extrahepatic metastasis of hepatocellular carcinoma. Hum Pathol 2011; 42: 129–35. [DOI] [PubMed] [Google Scholar]
- 6. Choi JE, Hur W, Jung CK et al Silencing of 14–3‐3zeta over‐expression in hepatocellular carcinoma inhibits tumor growth and enhances chemosensitivity to cis‐diammined dichloridoplatium. Cancer Lett 2011; 303: 99–107. [DOI] [PubMed] [Google Scholar]
- 7. Alaiya AA, Al‐Mohanna M, Aslam M et al Proteomics‐based signature for human benign prostate hyperplasia and prostate adenocarcinoma. Int J Oncol 2011; 38: 1047–57. [DOI] [PubMed] [Google Scholar]
- 8. Arora V, Cheung HH, Plenchette S, Micali OC, Liston P, Korneluk RG. Degradation of survivin by the X‐linked inhibitor of apoptosis (XIAP)‐XAF1 complex. J Biol Chem 2007; 282: 26202–9. [DOI] [PubMed] [Google Scholar]
- 9. Huber E, Vlasny D, Jeckel S, Stubenrauch F, Iftner T. Gene profiling of cottontail rabbit papillomavirus‐induced carcinomas identifies upregulated genes directly Involved in stroma invasion as shown by small interfering RNA‐mediated gene silencing. J Virol 2004; 78: 7478–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tak H, Jang E, Kim SB et al 14–3‐3epsilon inhibits MK5‐mediated cell migration by disrupting F‐actin polymerization. Cell Signal 2007; 19: 2379–87. [DOI] [PubMed] [Google Scholar]
- 11. Qi W, Liu X, Qiao D, Martinez JD. Isoform‐specific expression of 14–3‐3 proteins in human lung cancer tissues. Int J Cancer 2005; 113: 359–63. [DOI] [PubMed] [Google Scholar]
- 12. Sinha P, Kohl S, Fischer J et al Identification of novel proteins associated with the development of chemoresistance in malignant melanoma using two‐dimensional electrophoresis. Electrophoresis 2000; 21: 3048–57. [DOI] [PubMed] [Google Scholar]
- 13. Zang D, Li X, Zhang L. 14–3‐3zeta Overexpression and abnormal beta‐catenin expression are associated with poor differentiation and progression in stage I non‐small cell lung cancer. Clin Exp Med 2010; 10: 221–8. [DOI] [PubMed] [Google Scholar]
- 14. Kuramitsu Y, Baron B, Yoshino S et al Proteomic differential display analysis shows up‐regulation of 14–3‐3 sigma protein in human scirrhous‐type gastric carcinoma cells. Anticancer Res 2010; 30: 4459–65. [PubMed] [Google Scholar]
- 15. Cao L, Cao W, Zhang W et al Identification of 14–3‐3 protein isoforms in human astrocytoma by immunohistochemistry. Neurosci Lett 2008; 432: 94–9. [DOI] [PubMed] [Google Scholar]
- 16. Yang X, Cao W, Lin H et al Isoform‐specific expression of 14–3‐3 proteins in human astrocytoma. J Neurol Sci 2009; 276: 54–9. [DOI] [PubMed] [Google Scholar]
- 17. Baxter HC, Fraser JR, Liu WG et al Specific 14–3‐3 isoform detection and immunolocalization in prion diseases. Biochem Soc Trans 2002; 30: 387–91. [DOI] [PubMed] [Google Scholar]
- 18. Boston PF, Jackson P, Kynoch PA, Thompson RJ. Purification, properties, and immunohistochemical localisation of human brain 14–3‐3 protein. J Neurochem 1982; 38: 1466–74. [DOI] [PubMed] [Google Scholar]
- 19. Kawamoto Y, Akiguchi I, Nakamura S, Budka H. Accumulation of 14–3‐3 proteins in glial cytoplasmic inclusions in multiple system atrophy. Ann Neurol 2002; 52: 722–31. [DOI] [PubMed] [Google Scholar]
- 20. Kawamoto Y, Akiguchi I, Nakamura S, Honjyo Y, Shibasaki H, Budka H. 14–3‐3 proteins in Lewy bodies in Parkinson disease and diffuse Lewy body disease brains. J Neuropathol Exp Neurol 2002; 61: 245–53. [DOI] [PubMed] [Google Scholar]
- 21. Cao W, Yang X, Zhou J et al Targeting 14–3‐3 protein, difopein induces apoptosis of human glioma cells and suppresses tumor growth in mice. Apoptosis 2010; 15: 230–41. [DOI] [PubMed] [Google Scholar]
- 22. Saitoh M, Nishitoh H, Fujii M et al Mammalian thioredoxin is a direct inhibitor of apoptosis signal‐regulating kinase (ASK) 1. EMBO J 1998; 17: 2596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tobiume K, Matsuzawa A, Takahashi T et al ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001; 2: 222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hwang JR, Zhang C, Patterson C. C‐terminus of heat shock protein 70‐interacting protein facilitates degradation of apoptosis signal‐regulating kinase 1 and inhibits apoptosis signal‐regulating kinase 1‐dependent apoptosis. Cell Stress Chaperones 2005; 10: 147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldman EH, Chen L, Fu H. Activation of apoptosis signal‐regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14‐3‐3 dissociation. J Biol Chem 2004; 279: 10442–9. [DOI] [PubMed] [Google Scholar]
- 26. Ouyang M, Shen X. Critical role of ASK1 in the 6‐hydroxydopamine‐induced apoptosis in human neuroblastoma SH‐SY5Y cells. J Neurochem 2006; 97: 234–44. [DOI] [PubMed] [Google Scholar]
- 27. Hsieh CC, Papaconstantinou J. Thioredoxin‐ASK1 complex levels regulate ROS‐mediated p38 MAPK pathway activity in livers of aged and long‐lived Snell dwarf mice. Faseb J 2006; 20: 259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsuzawa A, Nishitoh H, Tobiume K, Takeda K, Ichijo H. Physiological roles of ASK1‐mediated signal transduction in oxidative stress‐ and endoplasmic reticulum stress‐induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid Redox Signal 2002; 4: 415–25. [DOI] [PubMed] [Google Scholar]
- 29. Chen JT, Fong YC, Li TM et al DDTD, an isoflavone derivative, induces cell apoptosis through the reactive oxygen species/apoptosis signal‐regulating kinase 1 pathway in human osteosarcoma cells. Eur J Pharmacol 2008; 597: 19–26. [DOI] [PubMed] [Google Scholar]
- 30. Zhang L, Chen J, Fu H. Suppression of apoptosis signal‐regulating kinase 1‐induced cell death by 14–3‐3 proteins. Proc Natl Acad Sci USA 1999; 96: 8511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lau JM, Jin X, Ren J et al The 14–3‐3tau phosphoserine‐binding protein is required for cardiomyocyte survival. Mol Cell Biol 2007; 27: 1455–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang HQ, Du ZX, Zhang HY, Gao DX. Different induction of GRP78 and CHOP as a predictor of sensitivity to proteasome inhibitors in thyroid cancer cells. Endocrinology 2007; 148: 3258–70. [DOI] [PubMed] [Google Scholar]
- 33. Du ZX, Yan Y, Zhang HY et al Suppression of MG132‐mediated cell death by peroxiredoxin 1 through influence on ASK1 activation in human thyroid cancer cells. Endocr Relat Cancer 2010; 17: 553–60. [DOI] [PubMed] [Google Scholar]
- 34. Morrison DK. The 14–3‐3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol 2009; 19: 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee JA, Park JE, Lee DH et al G1 to S phase transition protein 1 induces apoptosis signal‐regulating kinase 1 activation by dissociating 14–3‐3 from ASK1. Oncogene 2008; 27: 1297–305. [DOI] [PubMed] [Google Scholar]
- 36. Obsilova V, Silhan J, Boura E, Teisinger J, Obsil T. 14–3‐3 proteins: a family of versatile molecular regulators. Physiol Res 2008; 57(Suppl 3): S11–21. [DOI] [PubMed] [Google Scholar]
- 37. Klionsky DJ, Abeliovich H, Agostinis P et al Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4: 151–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1‐MAP kinase cascades in mammalian stress response. J Biochem 2004; 136: 261–5. [DOI] [PubMed] [Google Scholar]
- 39. Thandavarayan RA, Watanabe K, Ma M et al 14–3‐3 protein regulates Ask1 signaling and protects against diabetic cardiomyopathy. Biochem Pharmacol 2008; 75: 1797–806. [DOI] [PubMed] [Google Scholar]