

Abstract
Chemotherapy for colorectal cancer has become more complicated and diversified with the appearance of molecular‐targeting agents. 5‐Fluorouracil (5‐FU) has been a mainstay of chemotherapy for colorectal cancer, but it is still unknown whether the combining of 5‐FU with novel molecular‐targeting agents is effective. Thymidylate synthase (TS) is a direct target of 5‐FU, and the low TS level has been generally supposed to sensitize 5‐FU's efficacy. We therefore hypothesized that RB‐reactivating agents could enhance the efficacy of 5‐FU, because the RB‐reactivating agents could suppress the function of transcription factor E2F of TS gene promoter. We used three RB‐reactivating agents, trametinib/GSK1120212 (MEK inhibitor), fenofibrate (PPARα agonist), and LY294002 (PI3K inhibitor), with 5‐FU against human colon cancer HT‐29 and HCT15 cells. Trametinib induced p15 and p27 expression and reduced cyclin D1 levels in HT‐29 cells. Fenofibrate also dephosphorlated ERK1/2 and reduced cyclin D1 levels in HT‐29 cells. LY294002 induced p27 expression in HCT15 cells. All three agents caused dephosphorylation of RB protein and G1‐phase arrest with a reduction of TS expression. As a consequence, the combination of 5‐FU with each of the agents resulted in a significant decrease of colony numbers in HT‐29 or HCT15 cells. These results suggest “RB‐reactivation therapy” using molecular‐targeting agents to be a new strategy for 5‐FU‐based chemotherapy. In particular, we strongly expect trametinib, which was discovered in Japan and was recently submitted to FDA for approval, to be used together with established regimens for colorectal cancer.
5‐Fluorouracil (5‐FU) has been widely used for the treatment of advanced colorectal cancers. In recent years, combined treatments based on 5‐FU have improved the outcome of patients with advanced colorectal cancers.1, 2 However, 5‐FU has problems with chemosensitivity or toxicity, including considerable bone marrow suppression in some cases.3
5‐Fluorouracil inhibits thymidylate synthase (TS), which plays a central role in DNA biosynthesis. Thymidylate synthase expression is crucial to the efficacy of 5‐FU.4, 5 Actually, the level of TS expression is inversely correlated with the clinical response to 5‐FU‐based chemotherapy for colorectal and other cancers.6. Therefore, the downregulation of TS expression might be a promising way to enhance the efficacy of 5‐FU.7 On the other hand, TS is a target of E2F1, whose transcriptional ability is repressed by direct binding to hypophosphorylated RB protein.8 We then hypothesized that clinically used molecular‐targeting agents that reactivate RB might be useful to improve the efficacy of 5‐FU.
In the present study, we used three RB‐reactivating agents; the MEK inhibitor trametinib/GSK1120212 (also called JTP‐74057),9 PPARα agonist fenofibrate, and PI3K inhibitor LY294002. Trametinib is a novel allosteric MEK1/2 inhibitor, originally identified as a p15 inductive compound by us,9 shown to have broad antitumor effects in various tumor xenograft models.10, 11 Of note, trametinib improved progression‐free and overall survival of patients with B‐Raf mutated melanoma in phase 3 clinical trial,12, 13 and was submitted to FDA for approval. Fenofibrate, is a PPARα agonist widely used in the treatment of hyperlipidemia.14 Recently, several reports have showed anticancer effects of fenofibrate in various cancer cell lines.15, 16, 17, 18, 19, 20 The third agent, LY294002, is known to reactivate RB function by inhibiting the PI3K‐Akt pathway.21, 22
We found that these three compounds enhanced the efficacy of 5‐FU in human colon cancer cells by downregulating TS expression. This study provides a new treatment strategy for 5‐FU‐based chemotherapy against colon cancer.
Materials and Methods
Cell culture
Human colon adenocarcinoma HT‐29 cells were cultured in DMEM. Human colon adenocarcinoma HCT15 cells were cultured in RPMI medium. Each medium was supplemented with 10% FBS, glutamine (4 mM for DMEM and 2 mM for RPMI), and antibiotics (50 U/mL penicillin and 100 μg/mL streptomycin). Cells were incubated at 37°C in a humidified atmosphere of 5% CO2.
Reagents
5‐FU was obtained from Nacalai Tesque (Kyoto, Japan). Trametinib was kindly provided by GlaxoSmithKline (Brentford, Middlesex, UK). Fenofibrate was obtained from Cayman Chemical (Ann Arbor, MI, USA). LY294002 was obtained from Cell Signaling Technology (Beverly, MA, USA). They were dissolved in DMSO.
Cell viability assay
The number of viable cells was measured by a Cell Counting Kit‐8 assay according to the manufacturer's instructions (Dojindo, Kumamoto, Japan). After the incubation of HT‐29 cells or HCT15 cells for 72 h with the indicated concentrations of various drugs, kit reagent WST‐8 was added to the medium and incubated for 4 h. The absorbance at 450 nm of the samples was measured using a multi‐plate reader (DS Pharma Biomedical, Osaka, Japan).
Analyses of cell cycle and apoptosis
Cells were incubated with various drugs for 24 or 72 h, and then harvested by trypsinization. After centrifugation, the cells were suspended in PBS containing 0.1% Triton X‐100, 150 μg/mL RNase A (Sigma, St Louis, MO, USA), and 25 μg/mL propidium iodide. The stained cells were analyzed using FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA). The data were analyzed using Modifit LT software and Cell Quest software (Becton Dickinson).
Protein isolation and western blot analysis
Cells were lysed with a buffer containing 50 mM Tris‐HCl, 1% SDS, 1 mM DTT, 0.43 mM 4‐(2‐aminoethyl) benzenesulfonyl fluoride hydrocholoride (ABSF), and Phosphatase Inhibitor Cocktail (Nacalai Tesque). The lysate was sonicated and centrifuged at 15 000 g for 20 min at 4°C, and the supernatant was collected. Equal amounts of the protein extract were subjected to SDS‐PAGE, and transferred to a PVDF membrane (Millipore, Bedford, MA, USA). The following were used as the primary antibody: rabbit anti‐human p15 polyclonal antibody (C‐20, Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti‐human p21 polyclonal antibody (C‐19, Santa Cruz Biotechnology), rabbit anti‐human p27 polyclonal antibody (C‐19, Santa Cruz Biotechnology), mouse anti‐human cyclin D1 monoclonal antibody (MBL, Nagoya, Japan), mouse anti‐human thymidylate synthase monoclonal antibody (Abcam, Cambridge, UK), rabbit anti‐human Akt, phospho‐Akt (Ser473), p42/44 MAPK, phospho‐p42/44 MAPK, phospho‐Rb (Ser780), and phospho‐Rb (Ser807/811) (Cell Signaling Technology). The signals were detected with a Chemi‐Lumi One L (Nacalai Tesque) or an Immobilon Western Chemiluminescent HRP Substrate (Millipore).
RNA isolation and real‐time quantitative RT‐PCR
Total RNA was isolated from cells treated with agents for 24 h using Sepasol‐RNAI(Nacalai Tesque) according to the manufacturer's instructions. Total RNA (10 μg) was reverse transcribed to cDNA in a 20 μL reaction volume, with MMTV‐reverse transcriptase (Promega, Madison, WI, USA) and oligo (dT) primers (Toyobo, Osaka, Japan). The reaction mixture was incubated at 37°C for 1 h. An equivalent volume (1 μL) of cDNA solution was used for the quantitative RT‐PCR. cDNA was amplified by PCR using TaqMan Probes (Applied Biosystems, Foster, CA, USA) and an ABI 7300 real‐time PCR system (Applied Biosystems). The expression of TS mRNA was normalized to that of β2‐microgloblin (β2MG) mRNA in the same sample.
Colony formation assay
HT‐29 or HCT15 cells were seeded at a density of 200 cells per well in 6‐well plates. After incubating for 24 h, cells were treated with 5‐FU (0.5 μM) with or without trametinib (10 nM), fenofibrate (50 μM) or LY294002 (50 μM) for 24 h. The medium was then replaced with one containing 5‐FU (0.5 μM) only. After incubating for 24 h, the medium was replaced with fresh medium, and the cells were cultured for 7–10 days. The colonies fixed in 10% formalin were stained in crystal violet. The images of stained colonies were obtained using a Keyence fluorescence stereomicroscope attached to a cooled CCD camera (VB‐G05, VB‐7010/7000, Keyence, Osaka, Japan). The number of colonies in each well was counted with the software BZ‐H1C (Keyence).
Statistical analysis
All data are presented as the mean ± SD. The statistical significance of the differences of means between three or more groups was tested using a one‐way anova. That of comparison between two groups was tested using an unpaired Student's t‐test.
Results
Trametinib induces G1 arrest by reactivating RB protein
B‐Raf mutant cancer cells are known to be sensitive to MEK inhibitors.23, 24, 25 In fact, tramenitib, a selective allosteric inhibitor of MEK1/2,9, 10, 11 showed a subnanomolar IC50 value for 72 h in a Cell Counting Kit‐8 assay of human colon cancer HT‐29 cells, harboring constitutively active B‐Raf mutation (Fig. 1a). We then performed a cell cycle analysis and measured the sub‐G1 population to quantify apoptotic cells, using flow cytometry. The treatment with trametinib for 24 h dose‐dependently increased the G1 phase with a decrease in the S phase (Fig. 1b,c), and 72 h treatment induced apoptosis in a dose‐dependent manner together with G1 arrest (Fig. S1a,b).
Figure 1.
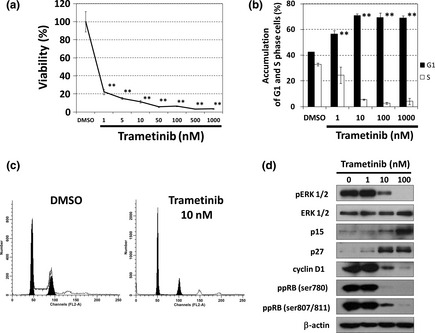
Trametinib induces G1‐phase arrest by reactivating retinoblastoma gene (RB) protein in HT‐29 cells. (a) Growth inhibitory effect of trametinib on HT‐29 cells. Cells were treated with trametinib at the indicated concentrations for 72 h, and cell viability was measured with a Cell Counting Kit‐8 assay. The data obtained with the solvent dimethyl sulfoxide (DMSO) was taken as 100%. Points, means (n = 3); bars, standard deviation (SD). **P < 0.01, compared with the DMSO‐treated control. (b) and (c) Cell cycle analysis of HT‐29 cells treated with trametinib. Cells were treated with trametinib at the indicated concentrations for 24 h. The DNA contents of the cells were analyzed by flow cytometry. The percentages in G1 and S phases of the cell cycle are shown in (b), and representative histograms are shown in (c). Columns, means (n = 3); bars, SD. **P < 0.01, compared with the DMSO‐treated control. (d) The effects of trametinib at the indicated concentrations for 24 h on cell‐cycle regulatory proteins analyzed by western blotting. β‐actin was used as a loading control.
We next analyzed the expression of cell‐cycle regulatory proteins by western blotting. The treatment with trametinib for 24 h induced the expression of cyclin‐dependent kinase (CDK) inhibitors, p15 and p27, and reduced cyclin D1 expression, accompanied by the dephosphorylation of ERK1/2 and RB (Fig. 1d).
Trametinib reduces TS expression
Thymidylate synthase is a transcriptional target of E2F, which is activated by phosphorylation of RB. We assumed that trametinib downregulated TS expression through the RB/E2F pathway. As shown in Figure 2(a), trametinib at 10 nM or more reduced TS protein expression with dephosphorylation of RB protein. Furthermore, we performed quantitative real‐time RT‐PCR, and found that trametinib similarly decreased TS mRNA levels (Fig. 2b). These results suggest that MEK inhibition causes suppression of TS expression through reactivation of RB protein.
Figure 2.
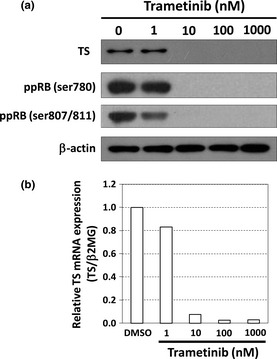
Trametinib reduces thymidylate synthase (TS) expression in HT‐29 cells. (a) The effect of trametinib at the indicated concentrations for 24 h on the expression of TS and the phosphorylation status of retinoblastoma gene (RB) analyzed by western blotting. β‐actin was used as a loading control. (b) The expression of TS mRNA measured by a real‐time reverse transcription‐polymerase chain reaction (RT‐PCR). HT‐29 cells were treated with trametinib at the indicated concentrations for 24 h. TS mRNA was normalized to β2MG mRNA, and the data obtained with dimethyl sulfoxide (DMSO) (control) was taken as 1.0. Columns, means (n = 3).
Trametinib enhances the efficacy of 5‐FU on HT‐29 cells
We treated HT‐29 cells with a combination of 5‐FU (0.5 μM) and/or trametinib (10 nM), according to the time schedule shown in Figure S2(a), and analyzed cell‐cycle and sub‐G1 population by flow cytometry. As shown in Figure S2(b,c), the 5‐FU treatment caused S phase arrest, and the combined treatment with 5‐FU and trametinib synergistically induced apoptosis. We next performed a colony formation assay using HT‐29 cells by the same treatment schedule (Fig. 3a) as that in Figure S2(a). Combined treatment with 5‐FU (0.5 μM) and trametinib (10 nM) markedly decreased the number of colonies compared with each drug alone (Fig. 3b,c). These results indicate that trametinib can sensitize cells to 5‐FU.
Figure 3.
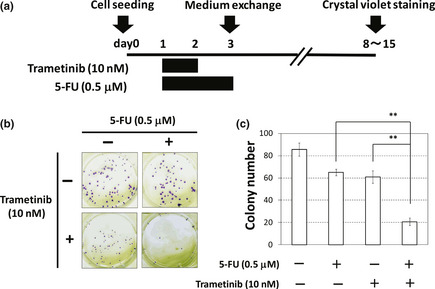
Trametinib enhances the efficacy of 5‐fluorouracil (5‐FU) in HT‐29 cells. (a) A schematic representation of the protocol for the colony formation assay. HT‐29 cells were treated with 5‐FU (0.5 μM) for 48 h with or without trametinib (10 nM) for 24 h as described in Materials and Methods. (b) and (c) The effect of co‐treatment with 5‐FU and trametinib on clonogenic growth. (b) Pictures of the colonies, treated with 5‐FU (0.5 μM), trametinib (10 nM), or a combination of both. (c) The number of each colony. Columns, means (n = 3); bars, standard deviation (SD). **P < 0.01.
Fenofibrate induces G1 arrest by blocking the MAPK cascade
Fenofibrate has been attracting attention as an antitumor drug. Actually, fenofibrate at 50 μM or more markedly inhibited the growth of HT‐29 cells (Fig. 4a). Furthermore, the treatment with fenofibrate for 24 h dose‐dependently increased the population of G1 phase (Fig. 4b), and also increased the populations of G1 and sub‐G1 after 72 h (Fig. S3a,b).
Figure 4.
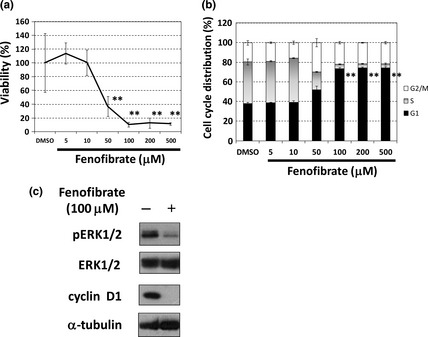
Fenofibrate induces G1‐phase arrest through a blockade of the mitogen‐activated protein kinase (MAPK) cascade in HT‐29 cells. (a) Growth inhibitory effect of fenofibrate on HT‐29 cells. Cells were treated with fenofibrate at the indicated concentrations for 72 h, and viability was measured with a Cell Counting Kit‐8 assay. The data obtained with the solvent dimethyl sulfoxide (DMSO) was taken as 100%. Points, means (n = 3); bars, standard deviation (SD). **P < 0.01, compared with the DMSO‐treated control. (b) Cell cycle analysis of HT‐29 cells treated with fenofibrate. Cells were treated with fenofibrate at the indicated concentrations for 24 h. The DNA contents of the cells were analyzed by flow cytometry. The percentage in each phase of the cell cycle is shown. Columns, means (n = 3); bars, SD. **P < 0.01, compared with the DMSO‐treated control. (c) The effect of fenofibrate at 100 μΜ for 24 h on the phosphorylation status of ERK and the expression of cyclin D1 analyzed by Western blotting. α‐tubulin was used as a loading control.
As fenofibrate interfered with the ERK1/2 and Akt signaling pathways in a mouse melanoma cell line,16 we verified the status of ERK1/2 and Akt. We found that 100 μM fenofibrate after 24 h treatment caused the dephosphorylation of ERK1/2 (Fig. 4c), but not Akt (data not shown). In addition, 100 μM fenofibrate downregulated cyclin D1 expression (Fig. 4c).
Fenofibrate reduces TS expression and enhances the efficacy of 5‐FU in HT‐29 cells
We next examined the phosphorylation status of RB and the expression of TS, and found that fenofibrate decreased phosphorylated RB protein with downregulation of TS protein in a dose‐dependent manner (Fig. 5a). The TS mRNA was also reduced by fenofibrate in a dose‐dependent manner (Fig. 5b).
Figure 5.
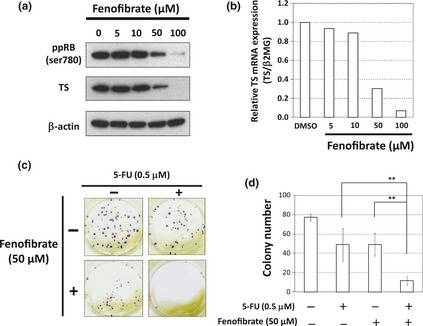
Fenofibrate reduces thymidylate synthase (TS) expression and enhances the efficacy of 5‐fluorouracil (5‐FU) in HT‐29 cells. (a) The effect of fenofibrate at the indicated concentrations for 24 h on the phosphorylation status of retinoblastoma gene (RB), and the expression of TS, analyzed by western blotting. β‐actin was used as a loading control. (b) The expression of TS mRNA measured by a real‐time reverse transcription‐polymerase chain reaction (RT‐PCR). HT‐29 cells were treated with fenofibrate at the indicated concentrations for 24 h. Thymidylate synthase mRNA was normalized to β2MG mRNA, and the data obtained with dimethyl sulfoxide (DMSO) (control) was taken as 1.0. Columns, means (n = 3). (c) and (d) The effect of co‐treatment with 5‐FU and fenofibrate on clonogenic growth. (c) Pictures of the colonies, treated with 5‐FU (0.5 μM), fenofibrate (50 μM), or a combination of both. (d) The number of each colony. Columns, means (n = 3); bars, standard deviation (SD). **P < 0.01.
We then performed flow cytometry and a colony formation assay by the same treatment schedule as that by trametinib (Figs S2a and Fig. 3a). The 5‐FU treatment caused S phase arrest (Fig. S4a), and the combined treatment with 5‐FU and fenofibrate synergistically induced apoptosis (Fig. S4b). Regarding a colony formation assay, the combined treatment with 5‐FU and fenofibrate significantly decreased colony numbers of HT‐29 cells (Fig. 5c,d).
The PI3K inhibitor LY294002 reactivates RB protein and exhibits a combined effect with 5‐FU on HCT15 cells
In human colon cancers, the PI3K‐Akt pathway is frequently activated by a PIK3CA mutation or loss of function of PTEN.26, 27 We then added the PI3K inhibitor LY294002 to human colon cancer HCT15 cells with active‐mutated PIK3CA. As shown in Figure 6(a), LY294002 at 12.5 μM or more increased the G1 phase of the cell cycle. LY294002 upregulated p27 expression with dephosphorylation of Akt, and caused dose‐dependent dephosphorylation of RB protein and a reduction in TS (Fig. 6b). We next performed flow cytometry and a colony formation assay by the same treatment schedule as that by trametinib (Fig. S2a and Fig. 3a). Whereas 5‐FU (0.5 μM) alone had an affect on neither cell cycle nor sub‐G1 population (Fig. S5a,b), the combined treatment with 5‐FU and LY294002 (50 μM) induced modest apoptosis (Fig. S5b) and decreased colony numbers of HCT15 cells (Fig. 6c).
Figure 6.
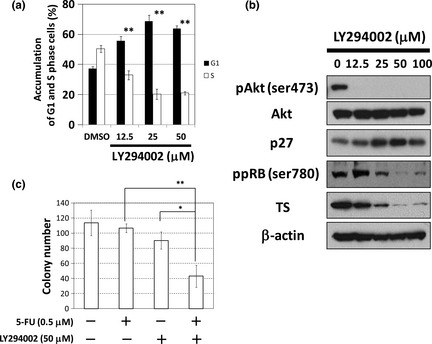
LY294002 reactivates retinoblastoma gene (RB) protein and exhibits a combined effect with 5‐fluorouracil (5‐FU) in HCT15 cells. (a) Cell cycle analysis of HCT15 cells treated with LY294002. Cells were treated with LY294002 at the indicated concentrations for 24 h. The DNA contents of the cells were analysed by flow cytometry. The percentages in G1 and S phases of the cell cycle are shown. Columns, means (n = 3); bars, standard deviation (SD). **P < 0.01, compared with the dimethyl sulfoxide (DMSO)‐treated control. (b) The effect of LY294002 at the indicated concentrations for 24 h on cell‐cycle regulatory proteins analysed by Western blotting. β‐actin was used as a loading control. (c) The effect of co‐treatment with 5‐FU and LY294002 on clonogenic growth. The number of each colony, treated with 5‐FU (0.5 μM), LY294002 (50 μM), or a combination of both, is shown. Columns, means (n = 3); bars, standard deviation (SD). *P < 0.05; **P < 0.01.
Discussion
Cell cycle‐dependent chemotherapy, with vincristine, etoposide, and so on, has been reported to show antagonism with G1‐arrest inducers.28, 29, 30, 31 However, we previously found that CDK inhibitor SU9516 reactivated RB protein and enhanced susceptibility to 5‐FU with a reduction in TS expression.32 Therefore, we raised the possibility that TS reduction following RB reactivation might increase the efficacy of 5‐FU, known to induce DNA damage and cause S arrest and apoptosis. Actually, in the present study, we proved that each of three RB‐reactivating agents, trametinib, fenofibrate, and LY294002, reduced TS expression, and the combined treatment with 5‐FU resulted in induction of apoptosis and decrease in colony formation of human colon cancer cells.
A novel oral MEK inhibitor, trametinib, was originally discovered by high‐throughput screening for a p15 inducer9 and is now being tested in multiple clinical trials without serious adverse events: i.e. phase 3; melanoma with B‐Raf mutation, phase 2; pancreatic cancer, leukemia, non‐small cell lung cancer (http://clinicaltrials.gov/ct2/results?term=GSK1120212).12, 33, 34 We expect this compound to also be effective against colorectal cancers, combined with 5‐FU‐based chemotherapy. Furthermore, in our previous paper,10 we also suggested a combined effect of trametinib with oxaliplatin or SN‐38 (the active metabolite of irinotecan) in HT‐29 cells. Taken together, trametinib might be a good candidate for use with established regimens, FOLFOX and FOLFILI, especially for B‐Raf mutated colorectal cancer.
The second compound fenofibrate, a clinically used anti‐hyperlipidemia agent, also dephospholyrated ERK1/2 and enhanced sensitivity to 5‐FU with a reduction of TS expression. More importantly, the effects were observed at a concentration near that in practical oral administration.16 Therefore, our data show the utility of fenofibrate as a safe and inexpensive 5‐FU sensitizer.
Third, we combined a PI3K inhibitor LY294002 with 5‐FU. About 30% of colon cancers have an active mutated PIK3CA,27, 35 which leads to resistance to MEK inhibitors.36, 37 Even in such cases, LY294002 decreased phosphorlylated RB protein through the induction of p27, and enhanced the efficacy of 5‐FU in HCT‐15 cells. Considering that anti‐apoptotic proteins, Bcl‐xL and MCL1, downstream of Akt, are also related with 5‐FU resistance,38, 39 the use of a PI3K inhibitor with 5‐FU may be favorable, when the PI3K‐Akt pathway is activated.
As mentioned, “RB reactivation therapy” could be a rational chemotherapeutic strategy to sensitize cells to 5‐FU. We speculate that previously reported data on the enhancement of 5‐FU sensitivity by HDAC inhibitors,40, 41, 42, 43 mTOR inhibitors,44, 45, 46 and dietary factors, apigenin,47 quercetin,48 and epigallocatechin‐3‐gallate,49 might be explained by their G1‐arresting ability with downregulation of TS. Interestingly, TS was clarified to be an oncogene.50 Especially in human colon cancer cells, a number of clinical studies have shown a correlation between high TS level and poor prognosis51, 52, 53 or low susceptibility to 5‐FU,54, 55, 56, 57, 58 and we consider that colon cancer cells could be highly addictive to oncogene TS. Therefore, reduction of TS by RB‐reactivating agents could suppress the oncogenic function of TS as well as enhance 5‐FU's efficacy against colon cancer cells.
Another possibility that RB‐reactivating agents enhanced the 5‐FUs efficacy might be explained as follows. As our treatment schedule indicates in Figures 3(a) or S2(a), when the cells were treated by the combined treatment of 5‐FU and RB‐reactivating agents, RB‐reactivating agents were supposed to cause at least partial G1‐phase arrest, together with S‐phase arrest by 5‐FU on day 2. However, after removing the RB‐reactivating agents, the G1‐arrested cells were expected to progress into S‐phase and were attacked by 5‐FU. We therefore postulate the RB‐reactivating agents could stimulate the efficacy of 5‐FU by the possible mechanisms above. From this standpoint, we have to carefully evaluate a treatment schedule, if we perform a clinical trial using RB‐reactivating agents with 5‐FU.
In conclusion, we found that three RB‐reactivating agents, trametinib, fenofibrate, and LY294002, reduced TS expression and increased sensitivity to 5‐FU in human colon cancer cells. These findings suggest that “RB reactivation therapy” using molecular‐targeting agents is efficacious in the 5‐FU‐based treatment of colorectal cancers. In other words, the addition of RB reactivators can abate the side‐effects of 5‐FU. Other RB reactivators might also be useful as 5‐FU sensitizers. Trametinib, currently in clinical trials, is a promising adjuvant of 5‐FU for patients with colorectal cancers.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Trametinib induces G1‐phase arrest and apoptosis after 72 h treatment in HT‐29 cells.
Fig. S2. 5‐FU treatment caused S phase arrest, while combined treatment with 5‐FU and trametinib synergistically induced apoptosis in HT‐29 cells.
Fig. S3. Fenofirate induces G1‐phase arrest and apoptosis after 72 h treatment in HT‐29 cells.
Fig. S4. Combined treatment with 5‐FU and fenofibrate synergistically induced apoptosis in HT‐29 cells.
Fig. S5. Combined treatment with 5‐FU and LY294002 induced apoptosis in HCT15 cells.
Acknowledgments
This work was partly supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank M. Tomosugi, H. Uchiyama, and R. Nakanishi for technical advice.
(Cancer Sci, doi: 10.1111/cas.12139, 2013)
References
- 1. Douillard JY, Cunningham D, Roth AD et al Irinotecan combined with fluorouracil compared with fluorouracil alone as first‐line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 2000; 355: 1041–7. [DOI] [PubMed] [Google Scholar]
- 2. Goldberg RM, Sargent DJ, Morton RF et al A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2004; 22: 23–30. [DOI] [PubMed] [Google Scholar]
- 3. Hotta T, Takifuji K, Arii K et al Toxicity during l‐LV/5FU adjuvant chemotherapy as a modified RPMI regimen for patients with colorectal cancer. Oncol Rep 2005; 14: 433–9. [PubMed] [Google Scholar]
- 4. Peters GJ, van Groeningen CJ. Clinical relevance of biochemical modulation of 5‐fluorouracil. Ann Oncol 1991; 2: 469–80. [DOI] [PubMed] [Google Scholar]
- 5. Peters GJ, Backus HH, Freemantle S et al Induction of thymidylate synthase as a 5‐fluorouracil resistance mechanism. Biochim Biophys Acta 2002; 1587: 194–205. [DOI] [PubMed] [Google Scholar]
- 6. Leichman CG. Thymidylate synthase as a predictor of response. Oncology (Williston Park). 1998; 12: 43–7. [PubMed] [Google Scholar]
- 7. Di Cresce C, Figueredo R, Ferguson PJ, Vincent MD, Koropatnick J. Combining small interfering RNAs targeting thymidylate synthase and thymidine kinase 1 or 2 sensitizes human tumor cells to 5‐fluorodeoxyuridine and pemetrexed. J Pharmacol Exp Ther 2011; 338: 952–63. [DOI] [PubMed] [Google Scholar]
- 8. DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis‐ and G1/S‐regulatory genes. Mol Cell Biol 1995; 15: 4215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamaguchi T, Yoshida T, Kurachi R et al Identification of JTP‐70902, a p15INK4b‐inductive compound, as a novel MEK1/2 inhibitor. Cancer Sci 2007; 98: 1809–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP‐74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo . Int J Oncol 2011; 39: 23–31. [DOI] [PubMed] [Google Scholar]
- 11. Gilmartin AG, Bleam MR, Groy A et al GSK1120212 (JTP‐74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res 2011; 17: 989–1000. [DOI] [PubMed] [Google Scholar]
- 12. Flaherty KT, Robert C, Hersey P et al Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med 2012; 367: 107–14. [DOI] [PubMed] [Google Scholar]
- 13. Sausville EA. Promises from trametinib in RAF active tumors. N Engl J Med 2012; 367: 171–2. [DOI] [PubMed] [Google Scholar]
- 14. Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta 1996; 1302: 93–109. [DOI] [PubMed] [Google Scholar]
- 15. Saidi SA, Holland CM, Charnock‐Jones DS, Smith SK. In vitro and in vivo effects of the PPAR‐alpha agonists fenofibrate and retinoic acid in endometrial cancer. Mol Cancer 2006; 5: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator‐activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down‐regulation of Akt. Clin Cancer Res 2006; 12: 3028–36. [DOI] [PubMed] [Google Scholar]
- 17. Drukala J, Urbanska K, Wilk A et al ROS accumulation and IGF‐IR inhibition contribute to fenofibrate/PPARalpha‐mediated inhibition of glioma cell motility in vitro. Mol Cancer 2010; 9: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee JJ, Yu JY, Zhang WY et al Inhibitory effect of fenofibrate on neointima hyperplasia via G(0)/G(1) arrest of cell proliferation. Eur J Pharmacol 2011; 650: 342–9. [DOI] [PubMed] [Google Scholar]
- 19. Yamasaki D, Kawabe N, Nakamura H et al Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARalpha‐independent mechanisms. Eur J Cell Biol 2011; 90: 657–64. [DOI] [PubMed] [Google Scholar]
- 20. Urbanska K, Pannizzo P, Grabacka M et al Activation of PPARalpha inhibits IGF‐I‐mediated growth and survival responses in medulloblastoma cell lines. Int J Cancer 2008; 123: 1015–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Katayama K, Nakamura A, Sugimoto Y, Tsuruo T, Fujita N. FOXO transcription factor‐dependent p15(INK4b) and p19(INK4d) expression. Oncogene 2008; 27: 1677–86. [DOI] [PubMed] [Google Scholar]
- 22. Yuan J, Mehta PP, Yin MJ et al PF‐04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther 2011; 10: 2189–99. [DOI] [PubMed] [Google Scholar]
- 23. Solit DB, Garraway LA, Pratilas CA et al BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006; 439: 358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yeh JJ, Routh ED, Rubinas T et al KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther 2009; 8: 834–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jing J, Greshock J, Holbrook JD et al Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther 2012; 11: 720–9. [DOI] [PubMed] [Google Scholar]
- 26. Samuels Y, Wang Z, Bardelli A et al High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554. [DOI] [PubMed] [Google Scholar]
- 27. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006; 441: 424–30. [DOI] [PubMed] [Google Scholar]
- 28. Sakai T, Aoike A, Marui N, Kawai K, Nishino H, Fukushima M. Protection by cycloheximide against cytotoxicity induced by vincristine, colchicine, or delta 12‐prostaglandin J2 on human osteosarcoma cells. Cancer Res 1989; 49: 1193–6. [PubMed] [Google Scholar]
- 29. Blagosklonny MV, Pardee AB. Exploiting cancer cell cycling for selective protection of normal cells. Cancer Res 2001; 61: 4301–5. [PubMed] [Google Scholar]
- 30. Takahara Y, Yogosawa S, Maruyama S et al Lysocellin, a metabolite of the novel drug ‘alopestatin’, induces G1 arrest and prevents cytotoxicity induced by etoposide. Int J Oncol 2006; 28: 823–9. [PubMed] [Google Scholar]
- 31. McDonald GT, Sullivan R, Pare GC, Graham CH. Inhibition of phosphatidylinositol 3‐kinase promotes tumor cell resistance to chemotherapeutic agents via a mechanism involving delay in cell cycle progression. Exp Cell Res 2010; 316: 3197–206. [DOI] [PubMed] [Google Scholar]
- 32. Takagi K, Sowa Y, Cevik OM, Nakanishi R, Sakai T. CDK inhibitor enhances the sensitivity to 5‐fluorouracil in colorectal cancer cells. Int J Oncol 2008; 32: 1105–10. [PubMed] [Google Scholar]
- 33. Infante JR, Fecher LA, Falchook GS et al Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose‐escalation trial. Lancet Oncol 2012; 13: 773–81. [DOI] [PubMed] [Google Scholar]
- 34. Falchook GS, Lewis KD, Infante JR et al Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose‐escalation trial. Lancet Oncol 2012; 13: 782–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parsels LA, Parsels JD, Tai DC, Coughlin DJ, Maybaum J. 5‐fluoro‐2′‐deoxyuridine‐induced cdc25A accumulation correlates with premature mitotic entry and clonogenic death in human colon cancer cells. Cancer Res 2004; 64: 6588–94. [DOI] [PubMed] [Google Scholar]
- 36. Wee S, Jagani Z, Xiang KX et al PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res 2009; 69: 4286–93. [DOI] [PubMed] [Google Scholar]
- 37. Halilovic E, She QB, Ye Q et al PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res 2010; 70: 6804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhu H, Guo W, Zhang L et al Bcl‐XL small interfering RNA suppresses the proliferation of 5‐fluorouracil‐resistant human colon cancer cells. Mol Cancer Ther 2005; 4: 451–6. [DOI] [PubMed] [Google Scholar]
- 39. Graidist P, Phongdara A, Fujise K. Antiapoptotic protein partners fortilin and MCL1 independently protect cells from 5‐fluorouracil‐induced cytotoxicity. J Biol Chem 2004; 279: 40868–75. [DOI] [PubMed] [Google Scholar]
- 40. Tumber A, Collins LS, Petersen KD et al The histone deacetylase inhibitor PXD101 synergises with 5‐fluorouracil to inhibit colon cancer cell growth in vitro and in vivo . Cancer Chemother Pharmacol 2007; 60: 275–83. [DOI] [PubMed] [Google Scholar]
- 41. Lee JH, Park JH, Jung Y et al Histone deacetylase inhibitor enhances 5‐fluorouracil cytotoxicity by down‐regulating thymidylate synthase in human cancer cells. Mol Cancer Ther 2006; 5: 3085–95. [DOI] [PubMed] [Google Scholar]
- 42. Di Gennaro E, Bruzzese F, Pepe S et al Modulation of thymidilate synthase and p53 expression by HDAC inhibitor vorinostat resulted in synergistic antitumor effect in combination with 5FU or raltitrexed. Cancer Biol Ther 2009; 8: 782–91. [DOI] [PubMed] [Google Scholar]
- 43. Noro R, Miyanaga A, Minegishi Y et al Histone deacetylase inhibitor enhances sensitivity of non‐small‐cell lung cancer cells to 5‐FU/S‐1 via down‐regulation of thymidylate synthase expression and upregulation of p21(waf1/cip1) expression. Cancer Sci 2010; 101: 1424–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matsuzaki T, Yashiro M, Kaizaki R et al Synergistic antiproliferative effect of mTOR inhibitors in combination with 5‐fluorouracil in scirrhous gastric cancer. Cancer Sci 2009; 100: 2402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee KH, Hur HS, Im SA et al RAD001 shows activity against gastric cancer cells and overcomes 5‐FU resistance by downregulating thymidylate synthase. Cancer Lett 2010; 299: 22–8. [DOI] [PubMed] [Google Scholar]
- 46. Perotti A, Locatelli A, Sessa C et al Phase IB study of the mTOR inhibitor ridaforolimus with capecitabine. J Clin Oncol 2010; 28: 4554–61. [DOI] [PubMed] [Google Scholar]
- 47. Choi EJ, Kim GH. 5‐Fluorouracil combined with apigenin enhances anticancer activity through induction of apoptosis in human breast cancer MDA‐MB‐453 cells. Oncol Rep 2009; 22: 1533–7. [DOI] [PubMed] [Google Scholar]
- 48. Xavier CP, Lima CF, Rohde M, Pereira‐Wilson C. Quercetin enhances 5‐fluorouracil‐induced apoptosis in MSI colorectal cancer cells through p53 modulation. Cancer Chemother Pharmacol 2011; 68: 1449–57. [DOI] [PubMed] [Google Scholar]
- 49. Qiao J, Gu C, Shang W et al Effect of green tea on pharmacokinetics of 5‐fluorouracil in rats and pharmacodynamics in human cell lines in vitro . Food Chem Toxicol 2011; 49: 1410–5. [DOI] [PubMed] [Google Scholar]
- 50. Bertino JR, Banerjee D. Thymidylate synthase as an oncogene? Cancer Cell 2004; 5: 301–2. [DOI] [PubMed] [Google Scholar]
- 51. Edler D, Kressner U, Ragnhammar P et al Immunohistochemically detected thymidylate synthase in colorectal cancer: an independent prognostic factor of survival. Clin Cancer Res 2000; 6: 488–92. [PubMed] [Google Scholar]
- 52. Popat S, Matakidou A, Houlston RS. Thymidylate synthase expression and prognosis in colorectal cancer: a systematic review and meta‐analysis. J Clin Oncol 2004; 22: 529–36. [DOI] [PubMed] [Google Scholar]
- 53. Ohrling K, Edler D, Hallstrom M, Ragnhammar P, Blomgren H. Detection of thymidylate synthase expression in lymph node metastases of colorectal cancer can improve the prognostic information. J Clin Oncol 2005; 23: 5628–34. [DOI] [PubMed] [Google Scholar]
- 54. Johnston PG, Lenz HJ, Leichman CG et al Thymidylate synthase gene and protein expression correlate and are associated with response to 5‐fluorouracil in human colorectal and gastric tumors. Cancer Res 1995; 55: 1407–12. [PubMed] [Google Scholar]
- 55. Lenz HJ, Hayashi K, Salonga D et al p53 point mutations and thymidylate synthase messenger RNA levels in disseminated colorectal cancer: an analysis of response and survival. Clin Cancer Res 1998; 4: 1243–50. [PubMed] [Google Scholar]
- 56. Aschele C, Debernardis D, Casazza S et al Immunohistochemical quantitation of thymidylate synthase expression in colorectal cancer metastases predicts for clinical outcome to fluorouracil‐based chemotherapy. J Clin Oncol 1999; 17: 1760–70. [DOI] [PubMed] [Google Scholar]
- 57. Wong NA, Brett L, Stewart M et al Nuclear thymidylate synthase expression, p53 expression and 5FU response in colorectal carcinoma. Br J Cancer 2001; 85: 1937–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bertino JR, Banerjee D. Is the measurement of thymidylate synthase to determine suitability for treatment with 5‐fluoropyrimidines ready for prime time? Clin Cancer Res 2003; 9: 1235–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Trametinib induces G1‐phase arrest and apoptosis after 72 h treatment in HT‐29 cells.
Fig. S2. 5‐FU treatment caused S phase arrest, while combined treatment with 5‐FU and trametinib synergistically induced apoptosis in HT‐29 cells.
Fig. S3. Fenofirate induces G1‐phase arrest and apoptosis after 72 h treatment in HT‐29 cells.
Fig. S4. Combined treatment with 5‐FU and fenofibrate synergistically induced apoptosis in HT‐29 cells.
Fig. S5. Combined treatment with 5‐FU and LY294002 induced apoptosis in HCT15 cells.