

Abstract
Head and neck squamous cell carcinoma (HNSCC) is an aggressive cancer with a 5‐year survival rate of ~50%. With the use of a custom cDNA‐capture system coupled with massively parallel sequencing, we have now investigated transforming mechanisms for this malignancy. The cDNAs of cancer‐related genes (n = 906) were purified from a human HNSCC cell line (T3M‐1 Cl‐10) and subjected to high‐throughput resequencing, and the clinical relevance of non‐synonymous mutations thus identified was evaluated with luciferase‐based reporter assays. A CASP8 (procaspase‐8) cDNA with a novel G‐to‐C point mutation that results in the substitution of alanine for glycine at codon 325 was identified, and the mutant protein, CASP8 (G325A), was found to activate nuclear factor‐κB (NF‐κB) signaling to an extent far greater than that achieved with the wild‐type protein. Moreover, forced expression of wild‐type CASP8 suppressed the growth of T3M‐1 Cl‐10 cells without notable effects on apoptosis. We further found that most CASP8 mutations previously detected in various epithelial tumors also increase the ability of the protein to activate NF‐κB signaling. Such NF‐κB activation was shown to be mediated through the COOH‐terminal region of the second death effector domain of CASP8. Although CASP8 mutations associated with cancer have been thought to promote tumorigenesis as a result of attenuation of the proapoptotic function of the protein, our results now show that most such mutations, including the novel G325A identified here, separately confer a gain of function with regard to activation of NF‐κB signaling, indicating another role of CASP8 in the transformation of human malignancies including HNSCC.
Head and neck squamous cell carcinoma (HNSCC) is one of the most common types of human cancer, with an annual incidence of more than 500 000 cases worldwide.1, 2 The major risk factors for HNSCC are tobacco use, alcohol consumption, and infection with human papilloma virus.3 It is an aggressive cancer with a propensity for local invasion and metastasis, which directly leads to disease‐ or treatment‐related morbidity. The goals of HNSCC treatment are therefore not only to improve survival outcome but also to preserve vital physiological functions such as speech, breathing, swallowing, and hearing.
Most patients with HNSCC, however, present with advanced disease at the time of first evaluation and have a 5‐year survival rate of only ~50%. Although advances in surgery and chemoradiation treatment have helped to preserve organ function in such individuals, they have resulted in only a moderate improvement in patient survival during the past 30 years. Characterization of the molecular mechanisms of HNSCC oncogenesis is expected to provide important information for the development of novel anticancer agents and the identification of biomarkers.
The recent advent of massively parallel sequencers, or next‐generation sequencers, has rendered resequencing of the cancer genome manageable in private laboratories.4 We have recently shown that a custom cDNA‐capture system coupled with massively parallel sequencing provides a feasible and relatively simple approach for the simultaneous detection of point mutations, insertions/deletions (indels), and gene fusions among the captured genes.5 Here we show that such high‐throughput resequencing of targeted cDNAs from an oral squamous cell carcinoma cell line led to the identification of a missense mutation in caspase‐8 (CASP8), a member of the cysteine–aspartic acid protease (caspase) family. Unexpectedly, CASP8 with this amino acid substitution (glycine‐325 to alanine, or G325A) was found to activate signaling by the antiapoptotic transcription factor nuclear factor‐κB (NF‐κB) to an extent markedly greater than that observed with the wild‐type protein. Of interest, most CASP8 mutants previously identified in human cancers were also found to activate the NF‐κB pathway. As far as we are aware, a direct antiapoptotic effect of CASP8 in cancer has not previously been demonstrated.
Materials and Methods
Cell lines and plasmids
Human embryonic kidney 293T (HEK293T) cells, human oral squamous cell carcinoma T3M‐1 Cl‐10 cells, and human esophageal squamous cell carcinoma OE21 cells were obtained from RIKEN Cell Bank (Tsukuba, Japan), ATCC (Manassas, VA, USA), and European Collection of Cell Cultures (Salisbury, UK), respectively. All cells were maintained in DMEM–F12 supplemented with 10% FBS and 2 mM l‐glutamine (all of which were from Invitrogen, Carlsbad, CA, USA). A full‐length cDNA for the G325A mutant form of CASP8 was isolated by RT‐PCR from T3M‐1 Cl‐10 cells and inserted into the retroviral plasmid pMXS.6 Expression vectors for wild‐type and previously identified mutant forms of CASP8 were generated by PCR‐based mutagenesis. The nucleotide sequences of all constructs were confirmed by Sanger sequencing.
Resequencing coupled with a cDNA‐capture system
Resequencing coupled with a custom cDNA‐capture system was carried out as described previously.5 In brief, RNA capture probes (Agilent Technologies, Santa Clara, CA, USA) designed to cover cDNAs of 906 human protein‐coding genes were hybridized with cDNA fragments prepared from T3M‐1 Cl‐10 cells according to the protocols for the SureSelect Target Enrichment system (Agilent Technologies). Purified cDNA fragments were then subjected to deep sequencing for 76 bases from both ends with a Genome Analyzer IIx (GAIIx; Illumina, San Diego, CA, USA). Reads with a Q‐value ≥20 at every base were selected, and mapped to the reference cDNA sequences as well as the human genome sequence (GRCh37) with the Bowtie algorithm.7 After removing single nucleotide polymorphisms (dbSNP build 32; http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi), non‐synonymous mutations (≥30% mutation ratio at ≥30× coverage) for the target cDNAs were isolated by our in‐house pipeline.
Luciferase‐based reporter assays
The HEK293T cells were transfected with a CASP8 expression vector, the pGL‐TK plasmid (Promega, Madison, WI, USA), and a luciferase‐based reporter plasmid for signaling by c‐Fos (pFL700),8 c‐Myc (pHXL),9 β‐catenin (TOP‐flash; Upstate Biotechnology, Lake Placid, NY, USA), JNK (AP1; Panomics, Santa Clara, CA, USA), TP53,10 Notch (pGa981‐6),11 Rho (pSRE.L),12 MAPK (ELK1; Panomics), Gli (Genentech, South San Francisco, CA, USA), or NF‐κB (Agilent Technologies). Luciferase activities were then assayed with a Dual‐Luciferase Reporter Assay System (Promega), and the activity of firefly luciferase was normalized by that of Renilla luciferase.
Apoptosis and cell proliferation assays
The T3M‐1 Cl‐10 cells, which express CASP8(G325A), and OE21 cells, which express wild‐type CASP8, were infected with a retrovirus generated from the pMXS‐CASP8‐ires‐EGFP vector (Clontech, Mountain View, CA, USA), which allows simultaneous expression of CASP8 and enhanced green fluorescent protein (EGFP). The cells were then collected and assayed for apoptosis by staining with annexin V and propidium iodide (eBioscience, San Diego, CA, USA) followed by flow cytometry (FACSCanto II instrument; BD Biosciences, San Jose, CA, USA). Cell apoptosis was quantified by the Click‐iT TUNEL Alexa Fluor Imaging Assay (Invitrogen). Cell proliferation was assayed by flow cytometric determination of the cell fraction positive for EGFP.
Statistical analysis
Quantitative data are presented as means ± SD and were compared with Student's t‐test. A P‐value of <0.05 was considered statistically significant.
Results
Identification of a CASP8 mutation in T3M‐1 Cl‐10 cells
To identify oncogenes for oral squamous cell carcinoma, we selected cDNA fragments for cancer‐related genes (n = 906) from T3M‐1 Cl‐10 oral squamous cell carcinoma cells with the use of our custom cDNA‐capture system.5 Deep sequencing of such fragments with a GAIIx sequencer yielded 91 961 299 independent high‐quality reads that mapped to 850 cDNAs with a mean coverage of 1202 reads/bp. Screening for missense mutations, indels, and gene fusions with our in‐house computational pipeline resulted in the identification of 12 non‐synonymous mutations that were further confirmed by Sanger sequencing (Table 1). We did not detect any indels or gene fusions that were confirmed by the capillary sequencing.
Table 1.
Non‐synonymous mutations detected in T3M‐1 Cl‐10 cells
| Gene | GenBank accession no. | Read coverage | Mismatch reads (%) | Nucleotide change | Amino acid change |
|---|---|---|---|---|---|
| CASP8 | NM_033355 | ×469 | 98.5 | 1183G>C | G325A |
| ELF4 | NM_001421 | ×188 | 39.8 | 1016C>A | L211M |
| GSG2 | NM_031965 | ×119 | 100.0 | 1238T>C | V402A |
| HRAS | NM_005343 | ×613 | 25.1 | 370A>T | Q61L |
| IRAK2 | NM_001570 | ×155 | 49.0 | 591C>T | S172L |
| NUAK2 | NM_030952 | ×162 | 51.2 | 1427G>A | A434T |
| PDPK1 | NM_002613 | ×226 | 37.6 | 1663G>C | E507Q |
| PRKCZ | NM_002744 | ×61 | 49.1 | 306C>T | R49C |
| PXK | NM_017771 | ×82 | 57.3 | 364A>G | I89V |
| RHOA | NM_001664 | ×1127 | 52.9 | 394G>C | E40Q |
| TP53 | NM_000546 | ×234 | 97.8 | 1035A>G | R280G |
| TTBK2 | NM_173500 | ×100 | 51.0 | 1402G>C | L321F |
The 12 missense mutations include a novel G‐to‐C change at position 1183 of CASP8 cDNA (GenBank accession number, NM_033355.3), which results in a glycine‐to‐alanine substitution at codon 325 of the encoded protein (Fig. 1a), as well as known HNSCC‐related mutations such as those in TP53 and HRAS. In our deep sequencing data, this substituted position of CASP8 cDNA was read at a depth of ×469 and showed a mutation ratio of 98.5%, indicative of loss of heterozygosity at this locus.
Figure 1.
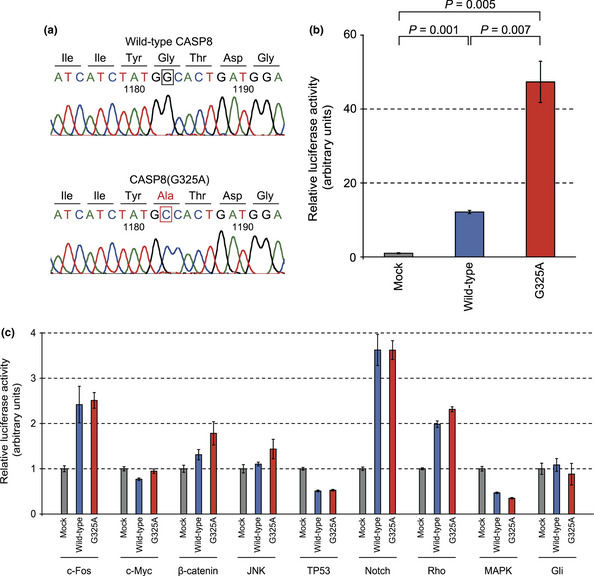
Mutant protein CASP8(G325A) selectively activates the nuclear factor‐κB (NF‐κB) signaling pathway. (a) Electrophoretograms of CASP8 cDNA identified an 1183G > C substitution, which results in a G325A amino acid substitution, in T3M‐1 Cl‐10 cells. (b,c) HEK293T cells were transfected with an expression vector for CASP8 or CASP8(G325A) or with the corresponding empty vector (Mock) together with pGL‐TK and reporter plasmids for NF‐κB (b) or other (c) signaling pathways, after which the cells were lysed and assayed for luciferase activities. The activity of firefly luciferase was normalized by that of Renilla luciferase then expressed relative to the corresponding value for mock‐transfected cells. Data are means ± SD from three independent experiments. P‐values were calculated using Student's t‐test.
The CASP8 gene encodes the inactive (pro) form of CASP8, which plays an essential role in the execution of apoptosis.13 Caspase‐8 is composed of a COOH‐terminal catalytic domain and an NH2‐terminal prodomain region that contains two tandem death effector domains (DEDs) (Fig. S1). Activation of CASP8 requires autoproteolysis that generates a heterodimer consisting of large (p20) and small (p10) protease subunits. The G325A mutation of CASP8 is located near the catalytic site in the p20 subunit.
Mutant CASP8(G325A) activates the NF‐κB signaling pathway
To evaluate the biological relevance of the CASP8(G325A) mutant, we carried out a reporter assay for a wide range of intracellular signaling pathways. Wild‐type CASP8 markedly increased reporter activity for the NF‐κB pathway (Fig. 1b), consistent with previous observations.14, 15 The G325A mutant of CASP8, however, increased such reporter activity to an extent far greater than that observed with the wild‐type protein. In contrast, the effects of the wild‐type and mutant forms of CASP8 on other signaling pathways, including those mediated by c‐Fos, c‐Myc, β‐catenin, JNK, TP53, Notch, Rho, MAPK, and Gli, did not differ significantly (Fig. 1c), indicating that the G325A mutation influences NF‐κB signaling specifically.
Catalytic activity of CASP8 and its mutants was also examined. The wild‐type CASP8, CASP8(G325A), CASP8(C360A) (an amino acid substitution at the catalytic center),16 CASP8(D210A/D216A) (double mutations at the autoprocessing region), or CASP8(D210A/D216A/D223A) (triple mutations at the autoprocessing region), was introduced into HEK293 cells that were then subjected to an enzymatic assay for CASP8. As expected, wild‐type CASP8 is catalytically active in HEK293, but a mutation at its catalytic center almost abolished its processing potency (Fig. 2a). Interestingly, the G325A substitution severely hampered CASP8 activity. In contrast, CASP8 with mutations at the autoprocessing region carry a decreased, but apparent, processing ability.
Figure 2.
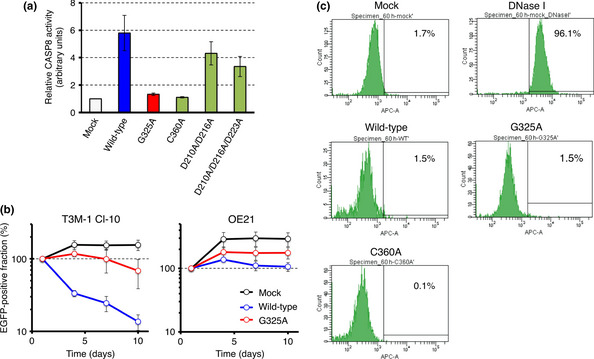
Caspase‐8 (CASP8) suppresses the proliferation of T3M‐1 Cl‐10 cells but not that of OE21 cells. (a) Proteolytic activity of CASP8 was measured with the Caspase‐Glo8 assay for HEK293 cells expressing wild‐type CASP8, CASP8(G325A), CASP8(C360A), CASP8(D210A/D216A), or CASP8(D210A/D216A/D223A), and is shown relative to the value for mock‐transfected cells. Data are means ± SD from three independent experiments. (b) T3M‐1 Cl‐10 cells (left panel) or OE21 cells (right panel) were infected with a retrovirus encoding enhanced green fluorescent protein (EGFP) either alone (Mock) or together with wild‐type or G325A mutant forms of CASP8. The number of EGFP‐positive cells was then measured by flow cytometry at 1, 4, 7, and 10 days after infection and is expressed as a percentage of that at 1 day after infection. Data are means ± SD from three independent experiments. (c) Fragmented DNA in apoptotic cells was quantified by TUNEL assay for T3M‐1 Cl‐10 cells infected with an empty virus (Mock), or virus expressing wild‐type, G325A mutant, or C360A mutant of CASP8. Fractions of cells with fragmented DNA are indicated as percentages. T3M‐1 Cl‐10 cells treated with DNase I were used as a positive control of apoptosis.
To examine whether the G350A mutation contributes directly to malignant transformation, we infected T3M‐1 Cl‐10 cells harboring the mutant CASP8 gene with a retrovirus encoding both EGFP and either wild‐type CASP8 or the G325A mutant, then assayed the proliferation of EGFP‐positive cells. Surprisingly, forced expression of wild‐type CASP8 resulted in a marked reduction in the number of EGFP‐positive cells, whereas the G325A mutant had only a slight effect on cell number (Fig. 2b). This suppression of cell growth by wild‐type CASP8 was not observed in another squamous cell carcinoma cell line, OE21, which harbors the wild‐type CASP8 gene (Fig. 2b).
Interestingly, while annexin V‐positive fraction was marginally increased in T3M‐1 Cl‐10 cells overexpressing wild‐type CASP8 (Fig. S2), neither CASP8 nor CASP8 (G325A) induced notable apoptosis in T3M‐1 Cl‐10, as judged by the TUNEL assay (Fig. 2c). It is, therefore, possible that CASP8 regulation of cell growth in cancer may be independent, in part, of its apoptosis‐inducing function.
We further depleted the CASP8 message in T3M‐1 Cl‐10 by the use of siRNA, and examined its effects on the expression of NF‐κB targets. As shown in Figure S3, decrease in the CASP8 message led to a marked suppression in BCL2 expression, supporting the positive role of CAPS8 in NF‐κB signaling.
CASP8 mutations in human tumors
Non‐synonymous mutations in CASP8 have been previously reported in various epithelial tumor types including gastric cancer (GC), colorectal cancer, hepatocellular carcinoma, and HNSCC.16, 17, 18, 19 These mutations include seven missense, one nonsense, and six frameshift mutations as well as one in‐frame deletion (Fig. S1, Table S1). Whereas such mutations have been thought to contribute to carcinogenesis through a loss of the proapoptotic function of CASP8, we unexpectedly found that most of the mutants markedly activated NF‐κB signaling (Fig. 3), suggestive of a gain of function with regard to such signaling. Of note, all of the three CASP8 mutants (GC1, GC4, and GC7) that failed to activate NF‐κB signaling harbor non‐synonymous mutations within the DEDs, suggesting that these domains may be essential for NF‐κB activation.
Figure 3.
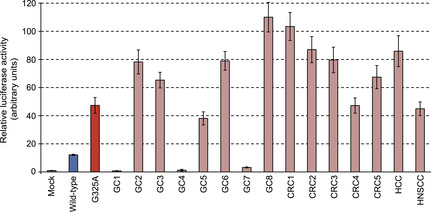
Tumor‐associated caspase‐8 (CASP8) mutants activate the nuclear factor‐κB pathway. HEK293T cells were transfected with a luciferase reporter plasmid for nuclear factor‐κB, with pGL‐TK, and with expression vectors for wild‐type or the indicated mutant forms of CASP8. Normalized firefly luciferase activity was then determined and expressed relative to the value for mock‐transfected cells. Data are means ± SD from three independent experiments. CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; HNSCC, head and neck squamous cell carcinoma.
In addition, screening as of January 2013 for non‐synonymous mutations in CASP8 among public databases for cancer genome mutations (COSMIC version 62, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/; The Cancer Genome Atlas, https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp; and the International Cancer Genome Consortium, http://icgc.org) list 76 independent missense/nonsense mutations, seven frame‐shift indels within CASP8, many of which had been confirmed to be somatic changes. Interestingly, amino acid substitutions at Gly‐325 (including G325A) were identified in multiple cancer specimens (such as those for large intestine carcinoma and cervical squamous cell carcinoma), suggesting that missense mutations at this position are recurrent.
Caspase‐8 domains linked to NF‐κB activation
To investigate further how CASP8 controls the NF‐κB pathway, we generated a series of CASP8 mutants (Fig. 4a). As shown in Figure 4(b), the D210A/D216A mutant is still able to activate NF‐κB signaling by an extent similar to that achieved with the wild‐type protein. Similarly, the addition of both D210A and D216A substitutions to CASP8(G325A) did not substantially affect its ability to activate the NF‐κB pathway.
Figure 4.
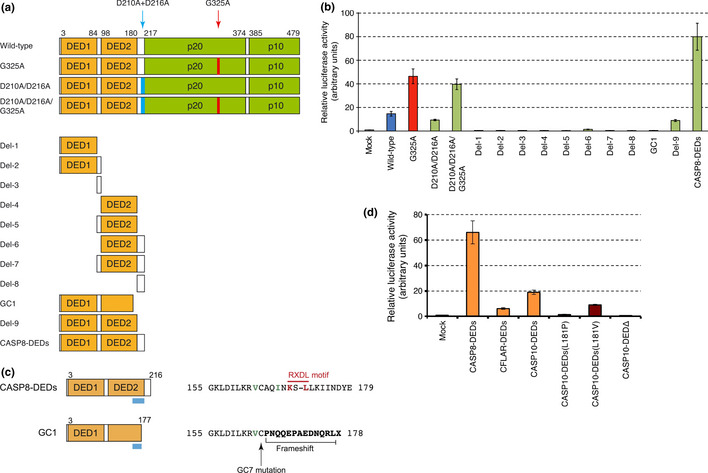
Death effector domains (DEDs) activate nuclear factor‐κB (NF‐κB) signaling. (a) The protein structure of caspase‐8 (CASP8) mutants is shown schematically with amino acid numbers indicated at the top. In addition to the wild‐type and D210A/D216A mutant forms with or without the G325A substitution, various constructs for the DED and Hinge regions (Del‐1 to Del‐9) were generated. The structure of the mutant from cancer specimen GC1 and a mutant encompassing both DEDs and Hinge regions (CASP8‐DEDs) is also shown. (b) Expression plasmids for the CASP8 mutants in (A) were introduced into HEK293T cells for the NF‐κB reporter assay. Normalized firefly luciferase activity is expressed relative to the value for mock‐transfected cells. Data are means ± SD from three independent experiments. (c) Amino acid sequences of the COOH‐terminal regions (depicted by light blue bars in the left panel) of CASP8‐DEDs and the CASP8 mutant from specimen GC1 are shown at the right. The conserved RXDL motif is indicated in red, and Val163 and Ile167 residues that contribute to the conserved hydrophobic patch are indicated in green. In the GC1 mutant, a frameshift deletion changes the amino acid sequence after Cys164 and generates a termination codon. The GC7 mutant has a C164Y substitution that markedly attenuates CASP8‐induced NF‐κB activation. (d) The ability to activate the NF‐κB pathway was examined for CASP8‐DEDs and the corresponding regions of CFLAR (amino acid residues 1–196) and CASP10 (residues 1–219), as in (b). For CASP10, we also examined the DED region with a L181P or L181V substitution, or RXDL‐deleted DEDΔ encompassing only amino acid residues 1–175.
We also generated expression constructs for the NH2‐terminal DED (DED1) or COOH‐terminal DED (DED2) either alone or together with the Hinge regions between DED1 and DED2 (Hinge‐1) or between DED2 and p20 (Hinge‐2) (Fig. 4a). None of these deletion mutants activated the NF‐κB pathway (Fig. 4b). In contrast, a deletion mutant consisting of the entire prodomain (CASP8‐DEDs) activated NF‐κB signaling to a level even higher than that induced by CASP8(G325A). Deletion of Hinge‐2 from CASP8‐DEDs (the Del‐9 mutant) markedly reduced the stimulatory effect on NF‐κB signaling. A CASP8 cDNA previously identified in the specimen designated GC1 has a 2‐bp deletion (Table S1) that results in premature termination within DED2 (Fig. 4c). This truncation almost completely abrogated the ability of CASP8 to activate NF‐κB signaling (Figs 3,4b). Both Hinge‐2 and the COOH‐terminal end of DED2 thus likely play an essential role in the regulation of NF‐κB signaling by CASP8. This notion was reinforced by the observation that a Cys164‐to‐Tyr substitution at the COOH‐terminal end of DED2 previously identified in the GC7 specimen (Table S1, Fig. 4c) also largely abolished the ability of CASP8 to activate NF‐κB signaling (Fig. 3).
Members of the DED family of proteins possess a key hydrophobic patch (for DED–DED interactions) that is exposed at the surface of each molecule and includes the conserved RXDL motif (corresponding to “KS – L” in DED2 of CASP8) in the COOH‐terminal region of the DED.20, 21, 22 Our findings are thus consistent with the idea that this conserved region contributes to the regulation of NF‐κB.
Prodomains of CASP8‐related proteins are able to activate NF‐κB signaling
Given that the prodomain of CASP8 is sufficient to fully active NF‐κB signaling, we tested whether the prodomains of the CASP8‐related proteins CFLAR (also known as cFLIPL) and caspase‐10 (CASP10) might have similar effects. CFLAR is structurally similar to CASP8 but does not possess functional caspase activity, given that it does not contain the conserved catalytic cysteine residue found in all functional caspases.23 Caspase‐10 is highly homologous to CASP8 and is also recruited to, and becomes activated by, death receptors.24, 25, 26 We found that the entire prodomains of CFLAR and CASP10 each markedly increased the level of NF‐κB signaling (Fig. 4d) as already shown for CASP10 by other groups.27 Although conservation of the RXDL motif is less clear in CASP10 compared to the other members (Fig. S4), substitution of the conserved Leu181 in CASP10 to either Pro or Val residues attenuated its NF‐κB‐activating potential (Fig. 4d). Furthermore, the CASP10‐DEDs protein lacking the putative RXDL region completely lost such ability, confirming the essential role of the DEDs COOH‐terminus in CASP10 activation of the NF‐κB pathway.
Discussion
One of the most proximal caspases in the apoptosis cascade, CASP8 is driven by the death‐inducing signaling complex in response to ligation of death receptors13 such as tumor necrosis factor receptor 1, CD95 (Fas, or Apo1), and tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL, or Apo2L) receptor. Activation of CASP8 requires dimerization and consequent autocleavage of the procaspase‐8 zymogen,28 and it initiates the extrinsic apoptosis cascade through activation of downstream effectors such as CASP3, CASP6, and CASP7.29
In addition, CASP8 has the potential to activate the antiapoptotic transcription factor NF‐κB through its tandem DED region,14, 15, 30 and IκB kinase γ (IKKγ), an essential regulatory subunit of the IKK complex, participates in this CASP8‐mediated activation of NF‐κB.31 The tandem DEDs of CFLAR also directly interact with and recruit IKKγ and thereby activate NF‐κB.32 However, we failed to detect a direct association between the tandem DEDs of CASP8 and IKKγ (data not shown). How the tandem DEDs of CASP8 mediate NF‐κB activation thus remains unclear. Given the essential role of the COOH‐terminal region of DED2 and the Hinge‐2 region of CASP8 in the activation of NF‐κB, it will be of interest to profile the cellular proteins that associate with these regions. Importantly, whereas somatic non‐synonymous mutations in CASP8 are detected relatively frequently in human tumors, the mutant proteins have been assumed to accelerate carcinogenesis as a result of a loss of proapoptotic function.16, 18, 19
In contrast, our data now suggest that many of the somatic mutations within CASP8 in human cancer provide simultaneously inactivation of its proapoptotic function and activation of NF‐κB signaling. Additionally, restoration of CASP8 expression in a CASP8‐mutant cell line (T3M‐1 Cl‐10) clearly suppressed cell proliferation without apparent effects on apoptosis, further confirming the relevance of CASP8 mutation on carcinogenesis. It should be noted, however, that our data does not prove a direct linkage between an enhanced NF‐κB signaling and cell growth. It may be possible that CASP8 mutants exert cancer‐promoting functions other than the activation of NF‐κB pathway.
Importantly, a recent large‐scale exome sequencing of HNSCC specimens (n = 74) detected somatic mutations of CASP8 in 8% of tumors,33 suggestive of an unexpected and transforming role of CASP8 in HNSCC (and maybe also in other epithelial tumors). Whereas activation of NF‐κB is frequently detected in a wide array of human malignancies, little is known about the exact mechanisms underlying such activation. Our results show that somatic mutation of CASP8 may be one such mechanism, and they suggest the possibility of treating CASP8 mutation‐positive tumors with inhibitors of NF‐κB, or targeting other proteins that contribute to the NF‐κB activation pathway.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Non‐synonymous mutations in caspase‐8. CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; HNSCC, head and neck squamous cell carcinoma.
{kind=link}
Fig. S2. Caspase‐8 (CASP8) affects annexin V‐positive fractions in T3M‐1 Cl‐10 head and neck squamous cell carcinoma cells but not in OE21 esophageal squamous cell carcinoma cells.
{kind=link}
Fig. S3. Depletion of CASP8 message leads to a decrease in BCL2 expression.
{kind=link}
Fig. S4. Structure of human death effector domain (DED) family members.
{kind=link}
Table S1. Caspase‐8 (CASP8) non‐synonymous mutations identified in previously published reports.
Acknowledgments
This study was supported in part by a grant for Research on Human Genome Tailor‐made from the Ministry of Health, Labor, and Welfare of Japan, by Grants‐in‐Aid for Scientific Research (B) from the Japan Society for the Promotion of Science, from The Yasuda Medical Foundation, from The Sagawa Foundation for Promotion of Cancer Research, and from The Mitsubishi Foundation.
(Cancer Sci 2013; 104: 1002–1008)
References
- 1. Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell 2004; 5: 311–6. [DOI] [PubMed] [Google Scholar]
- 2. Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med 2008; 359: 1143–54. [DOI] [PubMed] [Google Scholar]
- 3. Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet 2008; 371: 1695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mardis ER. A decade's perspective on DNA sequencing technology. Nature 2011; 470: 198–203. [DOI] [PubMed] [Google Scholar]
- 5. Ueno T, Yamashita Y, Soda M et al High‐throughput resequencing of target‐captured cDNA in cancer cells. Cancer Sci 2012; 103: 131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Onishi M, Kinoshita S, Morikawa Y et al Applications of retrovirus‐mediated expression cloning. Exp Hematol 1996; 24: 324–9. [PubMed] [Google Scholar]
- 7. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu Q, Milfay D, Williams LT. Binding of NCK to SOS and activation of ras‐dependent gene expression. Mol Cell Biol 1995; 15: 1169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takeshita T, Arita T, Higuchi M et al STAM, signal transducing adaptor molecule, is associated with Janus kinase and involved in signaling for cell growth and c‐myc induction. Immunity 1997; 6: 449–57. [DOI] [PubMed] [Google Scholar]
- 10. El‐Deiry WS, Tokino T, Velculescu VE et al WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–25. [DOI] [PubMed] [Google Scholar]
- 11. Kurooka H, Kuroda K, Honjo T. Roles of the ankyrin repeats and C‐terminal region of the mouse notch1 intracellular region. Nucleic Acids Res 1998; 26: 5448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 1995; 81: 1159–70. [DOI] [PubMed] [Google Scholar]
- 13. Muzio M, Chinnaiyan AM, Kischkel FC et al FLICE, a novel FADD‐homologous ICE/CED‐3‐like protease, is recruited to the CD95 (Fas/APO‐1) death–inducing signaling complex. Cell 1996; 85: 817–27. [DOI] [PubMed] [Google Scholar]
- 14. Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L. Activation of the NF‐kappaB pathway by caspase 8 and its homologs. Oncogene 2000; 19: 4451–60. [DOI] [PubMed] [Google Scholar]
- 15. Hu WH, Johnson H, Shu HB. Activation of NF‐kappaB by FADD, Casper, and caspase‐8. J Biol Chem 2000; 275: 10838–44. [DOI] [PubMed] [Google Scholar]
- 16. Soung YH, Lee JW, Kim SY et al Caspase‐8 gene is frequently inactivated by the frameshift somatic mutation 1225_1226delTG in hepatocellular carcinomas. Oncogene 2005; 24: 141–7. [DOI] [PubMed] [Google Scholar]
- 17. Mandruzzato S, Brasseur F, Andry G, Boon T, van der Bruggen P. A CASP‐8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J Exp Med 1997; 186: 785–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim HS, Lee JW, Soung YH et al Inactivating mutations of caspase‐8 gene in colorectal carcinomas. Gastroenterology 2003; 125: 708–15. [DOI] [PubMed] [Google Scholar]
- 19. Soung YH, Lee JW, Kim SY et al CASPASE‐8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res 2005; 65: 815–21. [PubMed] [Google Scholar]
- 20. Eberstadt M, Huang B, Chen Z et al NMR structure and mutagenesis of the FADD (Mort1) death‐effector domain. Nature 1998; 392: 941–5. [DOI] [PubMed] [Google Scholar]
- 21. Tibbetts MD, Zheng L, Lenardo MJ. The death effector domain protein family: regulators of cellular homeostasis. Nat Immunol 2003; 4: 404–9. [DOI] [PubMed] [Google Scholar]
- 22. Yu JW, Shi Y. FLIP and the death effector domain family. Oncogene 2008; 27: 6216–27. [DOI] [PubMed] [Google Scholar]
- 23. Budd RC, Yeh WC, Tschopp J. cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol 2006; 6: 196–204. [DOI] [PubMed] [Google Scholar]
- 24. Kischkel FC, Lawrence DA, Tinel A et al Death receptor recruitment of endogenous caspase‐10 and apoptosis initiation in the absence of caspase‐8. J Biol Chem 2001; 276: 46639–46. [DOI] [PubMed] [Google Scholar]
- 25. Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ. Caspase‐10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA 2001; 98: 13884–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sprick MR, Rieser E, Stahl H, Grosse‐Wilde A, Weigand MA, Walczak H. Caspase‐10 is recruited to and activated at the native TRAIL and CD95 death‐inducing signalling complexes in a FADD‐dependent manner but can not functionally substitute caspase‐8. EMBO J 2002; 21: 4520–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shikama Y, Yamada M, Miyashita T. Caspase‐8 and caspase‐10 activate NF‐kappaB through RIP, NIK and IKKalpha kinases. Eur J Immunol 2003; 33: 1998–2006. [DOI] [PubMed] [Google Scholar]
- 28. Pop C, Fitzgerald P, Green DR, Salvesen GS. Role of proteolysis in caspase‐8 activation and stabilization. Biochemistry 2007; 46: 4398–407. [DOI] [PubMed] [Google Scholar]
- 29. Krammer PH. CD95's deadly mission in the immune system. Nature 2000; 407: 789–95. [DOI] [PubMed] [Google Scholar]
- 30. Heyninck K, Beyaert R. Crosstalk between NF‐kappaB‐activating and apoptosis‐inducing proteins of the TNF‐receptor complex. Mol Cell Biol Res Commun 2001; 4: 259–65. [DOI] [PubMed] [Google Scholar]
- 31. Varfolomeev E, Maecker H, Sharp D et al Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor‐related apoptosis‐inducing ligand. J Biol Chem 2005; 280: 40599–608. [DOI] [PubMed] [Google Scholar]
- 32. Golks A, Brenner D, Krammer PH, Lavrik IN. The c‐FLIP‐NH2 terminus (p22‐FLIP) induces NF‐kappaB activation. J Exp Med 2006; 203: 1295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stransky N, Egloff AM, Tward AD et al The mutational landscape of head and neck squamous cell carcinoma. Science 2011; 333: 1157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Non‐synonymous mutations in caspase‐8. CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; HNSCC, head and neck squamous cell carcinoma.
Fig. S2. Caspase‐8 (CASP8) affects annexin V‐positive fractions in T3M‐1 Cl‐10 head and neck squamous cell carcinoma cells but not in OE21 esophageal squamous cell carcinoma cells.
Fig. S3. Depletion of CASP8 message leads to a decrease in BCL2 expression.
Fig. S4. Structure of human death effector domain (DED) family members.
Table S1. Caspase‐8 (CASP8) non‐synonymous mutations identified in previously published reports.