

Abstract
The peroxisome proliferator‐activated receptor‐γ (PPARγ) is a ligand‐activated transcription factor belonging to the nuclear receptor superfamily. Peroxisome proliferator‐activated receptor‐γ ligands can inhibit cell growth and increase apoptosis of cancer cell lines, suggesting a potential role for PPARγ as a tumor suppressor. Whereas the related studies between PPARγ and cancer cell invasion are still poor. Our previous study indicates that β‐estradiol (E2) suppresses hepatocellular carcinoma (HCC) cell invasion. We report here that E2 can activate PPARγ of HCC cells, and activated PPARγ suppresses cell invasion by upregulating the expression level of plasminogen activator inhibitor‐1 (PAI‐1). We found that PPARγ plays an important role in the E2‐induced HCC cell invasion process. Using PPARγ agonist GW1929, a reduced invasion effect was found in HCC cell lines, and this inhibition of cell invasion was dosage‐dependent. However, cell invasion was restored by treatment with PPARγ antagonist GW9662. The activated PPARγ upregulated the expression of cell migration‐related protein PAI‐1. Furthermore, knockdown of PPARγ in HCC cells decreased the level of PAI‐1 and advanced cell invasion in response to GW1929. On the contrary, overexpression of PPARγ in HCC cells elevated the level of PAI‐1 and inhibited cell invasion. These findings suggest that PPARγ activation inhibits HCC cell invasion via the upregulation of PAI‐1 and implicate that PPARγ is a target for the treatment and prevention of HCC cell invasion.
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third most common cause of cancer‐related death in the world. An estimated 748 300 new liver cancer cases and 695 900 cancer deaths occurred worldwide in 2008. Half of these cases and deaths were estimated to occur in China.1 Hepatitis B and C virus infection, aflatoxin B1 (AFB) exposure, alcohol‐related cirrhosis and insulin resistance syndrome mainly account for the liver cancer.2 The high mortality rate is due to its diagnosis at a stage when the disease is already incurable. To date, surgical resection is clinically the most effective treatment for HCC. In addition, sorafenib is the only targeted therapy approved by the US Food and Drug Administration to therapy HCC. However, sorafenib is associated with some side‐effects such as liver cirrhosis with impaired metabolic function and dose‐limiting toxicities.3 Thus, there is still a compelling need for a novel strategy that will improve the treatment of HCC and ultimately increase the survival of patients with HCC.
The nuclear receptor peroxisome proliferator‐activated receptor‐γ (PPARγ) is a ligand‐activated transcription factor that functions as an obligate heterodimer with RXRs.4 Peroxisome proliferator‐activated receptor‐γ has been suggested to behave as a tumor suppressor gene.5 Peroxisome proliferator‐activated receptor‐γ−deficient (PPARγ+/−) mice were more susceptible to diethylnitrosamine (DEN) ‐induced HCC than wild‐type (PPARγ+/+) mice.6 Clinical evidence shows that PPARγ protects against colorectal cancer in human. Patients with PPARγ‐positive tumors have significantly lower overall mortality than patients with PPARγ‐negative tumors.7
The 15‐deoxy‐Δ12,14‐prostaglandin J2 is the most potent endogenous ligand for PPARγ. In addition, some unsaturated fatty acids also are natural ligands of the PPARγ. Synthetic PPARγ ligands, known as antidiabetic drugs thiazolidinediones (TZDs), include rosiglitazone, pioglitazone, and troglitazone.8 Peroxisome proliferator‐activated receptor‐γ ligands inhibit the growth of HCC cells and induce apoptosis. Peroxisome proliferator‐activated receptor‐γ could be a regulator of cell survival and growth in HCC.9, 10 Peroxisome proliferator‐activated receptor‐γ activation not only inhibits tumor cells growth, but also represses invasion and metastasis. Studies on breast cancer indicate that PPARγ is expressed at significant levels in metastatic tumor cell, but ligand activation of PPARγ causes less malignant state of cells.11 The metastasis of non‐small‐cell lung cancer cells (NSCLC) that overexpress PPARγ decreases apparently, and survival rate of tumor‐bearing animals also is improved.12
In the process of cancer development, direct invasion is a very important mode. This process can make the mass larger, which causes many severe complications.13 Degradation of the extracellular matrix (ECM) components is important for tumor from invasion to metastasis. Glycosidase, matrix metalloproteinases, cathepsin B and plasminogen activation system14 play a critical role in this process. Thus, research on catabolic enzymes of ECM has been a focal point in tumor field. Plasminogen activator inhibitor‐1 (PAI‐1), a member of the serpin family of serine protease inhibitors, inactivates urokinase‐type plasminogen activator (uPA) and inhibits degradation of extracellular matrix.14 There is increasing evidence for involvement of PAI‐1 in cell migration, tumor invasion, and metastasis.15 Growth of some tumors can be attenuated by PAI‐1.16 In addition, Sawai have found that activation of PPARγ by ligands decreases pancreatic cancer cell invasion through increasing PAI‐1 and decreasing the uPA level. The results reveal that PAI‐1 can be the downstream gene of nuclear receptor PPARγ.17 Therefore, our interests are attracted by the function of PPARγ in HCC cell invasion and regulation of PPARγ activation on PAI‐1 expression.
In the present study, we demonstrate the plasminogen activator system plays a critical role in the HCC cell invasion. We observe that PPARγ activation decreases liver cancer cell invasion by specific PPARγ‐dependent regulation of PAI‐1. Our data also suggest the potential role of PPARγ agonists therapeutic agents in HCC.
Materials and Methods
Cell culture and drug treatment
Murine Hepa1‐6 and human HepG2 cell lines were obtained from Shanghai Institutes for Biological Sciences, the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in DMEM medium supplemented with 10% FBS (Invitrogen, Carlsbad, CA, USA), 2 mM L‐glutamine in a humidified atmosphere of 5% CO2 at 37°C. E2 (Sigma‐Aldrich St. Louis, MO, USA) was dissolved in ethanol and then diluted in medium containing 10% FBS to a final ethanol concentration of 0.1%. GW1929/GW9662 (Sigma‐Aldrich) were dissolved in DMSO and then diluted in medium containing 10% fetal bovine serum to a final DMSO concentration of 0.1%.
Matrigel invasion assay
The invasion of cells across matrigel was evaluated objectively in invasion chambers (Costar, Cambridge, MA, USA) with polycarbonate membranes (8.0‐µm pore size) coated with 100 μL Matrigel (BD Biosciences, Bedford, MA, USA) on the top side of the membrane. The upper surface of the matrix was plated with HCC cells, and cells were kept in serum‐free medium containing 0.1% BSA. The lower chamber contained medium supplemented with 10% serum. After 16 h incubation, non‐invasive cells and Matrigel on the upper surface of the membrane were removed carefully with a cotton swab. Cells invaded onto the lower surface were fixed with 4% paraformaldehyde and stained with crystal violet solution (Beyotime, Nantong, China). Enumeration of the cells that invaded through the matrix was accomplished by visually counting at five randomly chosen areas. Each experiment was performed in triplicate wells per time and repeated three times.
Western blot
Cell samples were lysed with radio immunoprecipitation assay (RIPA) lysis buffer (Beyotime) containing freshly added protease inhibitor tablets (Roche Applied Science, Mannheim, Germany) and the protein concentrations were determined using a BCA kit (Beyotime). The whole cell lysates were subjected to reducing 10% SDS‐ PAGE, transferred onto PVDF membranes (Millipore, Bedford, MA, USA), blocked with 5% skim milk for 1 h at room temperature, and immunoblotted at 4°C overnight with the primary antibody for PAI‐1 (Santa Cruz, Delaware, CA, USA), PPARγ (Santa Cruz). The blots were visualized using an enhanced chemiluminescent method kit (Cell Signaling Technology, Danvers, MA, USA). As an internal control for equal protein loading, blots were stripped and probed with antibodies against glyceraldehye‐3‐phosphate dehydrogenase (GAPDH) (Kangchen, Shanghai, China)
Electrophoretic mobility shift assay
Nuclear extracts of cells were prepared using nuclear and cytoplasmic protein extraction kit (Beyotime). A consensus double‐stranded PPARγ oligonucleotide 5′‐CAAAACTAGGTCAAAGGTCA‐3′ was 5′ end‐labeled with biotin. Electrophoretic mobility shift assay (EMSA) for PPARγ was performed using the chemiluminescent EMSA kit (Thermo Fisher Scientific, Waltham, MA, USA). Biotin end‐labeled DNA containing the binding site of interest was incubated with nuclear extracts. Protein‐oligonucleotide complexes were separated from the free oligonucleotide by electrophoresis on 6% polyacrylamide gels, and then transferred to a nylon membrane (Roche Applied Science). The biotin end‐labeled DNA was detected using the Streptavidin‐Horseradish Peroxidase Conjugate and Chemiluminescent Substrate. Cold competition was done by adding a 100‐fold excess of specific unlabeled double‐stranded probe to the reaction mixture.
Luciferase assay
We PCR‐amplified murine and human pai‐1 promoter (−1300 to +30) from total DNA isolated from Hepa1‐6 and HepG2 cells with specific primers, respectively. The amplified DNA was cloned into the pGL‐Basic vector (Promega, Madison, WI, USA). As an internal control reporter, the plasmid pRL containing Renilla luciferase cDNA was used. Cells were seeded on 24‐well cell culture plates (Costar) in triplicate and allowed to grow overnight to reach 90–95% confluence. The following day, the cells were transfected with pGL‐P‐mPAI‐1/pGL‐P‐hPAI‐1 promoter construct and pRL‐control using Lipofectamine 2000 transfection reagent (Invitrogen). Twenty‐four hours later, luciferase activities were measured using the Dual‐Luciferase reporter assay system (Promega). Renilla luciferase activity was normalized to firefly luciferase activity.
Overexpression of protein
Human and mouse PPARγ coding sequences were amplified from RNA isolated from HepG2 and Hepa1‐6 cells. The amplified cDNAs were digested with Xho I/Kpn I, and cloned in Xho I/Kpn I sites at the multiple cloning site of pIRES2‐EGFP plasmid (BD Biosciences). The cells were transfected with the PPARγ constructs using Lipofectamine 2000 according to the manufacturer's protocol. After 48 h, the expression levels of PPARγ were evaluated with fluorescence microscope.
RNA interference
The 21‐nucleotide small interfering RNAs (siRNA) for PPARγ and corresponding control siRNAs were purchased from Invitrogen. The sequences of sense and antisense strands for PPARγ siRNA were 5′‐UGGAAGACCACUCCCACUCTT‐3′ and 5′‐GAGUGGGAGUGGUCUUCCATT‐3′. Transfection of siRNA into the HCC cells was performed using Lipofectamine 2000 (Invitrogen). Cells were transfected at 30–50% confluence using 100 pmol of siRNA in 6‐well plates, and whole‐cell lysates were prepared 48 h after transfection. Knockdown of the expressions of the target mRNAs by the experimental siRNA and the corresponding protein were verified by RT‐PCR and western blot analysis, respectively.
RT‐PCR assay
Total RNA was extracted from the cultured cells using Trizol reagent (Invitrogen) according to the manufacturer's instructions. RNA concentration was assessed spectrophotometrically by absorbance at 260 nm. RNA was reverse‐transcribed into cDNA and 1 μL reaction mixture was used for PCR. Glyceraldehyde 3‐phosphate dehydrogenase RNA was used as an endogenous control. The primers were synthesized by Invitrogen. The primers sequences were: murine PAI‐1 forward, 5′‐AACCCGGCGGCAGATC‐3′, reverse, 5′‐CTTGAGATAGGACAGTGCTT‐3′; human PAI‐1 forward, 5′‐GCTGAATTCCTGGAGCTCAG‐3′, reverse, 5′‐CTGCGCCACCTGCTGAAACA‐3′. The PCR products were separated on 1% agarose gel containing ethidium bromide (Sunshine, Nanjing, China).
uPA activity assay
Serum‐free medium supernatant was collected from cells after 24 h incubation and analyzed for uPA activity by using the uPA Activity Assay kit (Chemicon, Temecula, CA, USA) according to the manufacturer's instructions. Briefly, supernatant aliquots were combined with assay buffer in 96‐well plates. After a 2‐h incubation of the mixture with a chromogenic substrate at 37°C, absorbance was read on a standard microplate reader at 405 nm.
Statistical analysis
Each experiment was repeated at least three times. All data were expressed as mean ± SD. The statistical significance between experimental groups was determined by Student's t‐test using GraphPad Prism software. Differences were considered significant when P < 0.05.
Results
E2‐induced HCC cell invasion
Previous reports have suggested that E2 inhibits liver cancer invasion in vivo and in vitro.18 In this study we tested for the effect of E2 on human HepG2 and murine Hepa1‐6 cell invasion. The HCC cells were added to the upper chamber. The number of cells migrating to the lower surface of the chamber was counted after treatment with 100 nM E2 for 24 h. The invasiveness of HCC cells was markedly suppressed by E2 (Fig. 1).
Figure 1.
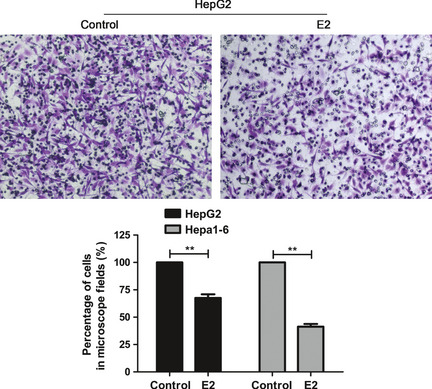
β‐estradiol (E2) inhibited hepatocellular carcinoma (HCC) cell invasion. Matrigel invasion assay examined the effect of E2 on HepG2 cell invasion. Values are expressed as mean (n = 3) ± standard deviation (SD). (**P < 0.01, compared with control; n = 3).
E2 suppresses HCC cell invasion through upregulating PPARγ activity
In animal models we have found that female mice contain higher PPARγ levels than male mice in liver tumors (data not shown). We speculated that E2 could suppress HCC cell invasion by activating PPARγ. To verify the effects of E2 on PPARγ activity, HCC cells were treated with 100 nM E2 for 24 h and the nuclear proteins of cells were prepared for EMSA. Coincident with our supposition, it is evident that E2 activated PPARγ in HepG2 (Fig. 2a) and Hepa1‐6 (Fig. 2b) cells. Moreover, the pretreatment of PPARγ antagonist GW9662 attenuated the above effect. To better examine whether PPARγ plays a critical role in E2‐induced HCC cell invasion, we altered the expression levels of PPARγ by RNA interference and overexpression. After PPARγ levels of HCC cells were downregulated by siRNA (Fig. 3b), E2‐induced descent of cell invasion was recovered compared with negative control (Fig. 3a). In response to PPARγ overexpression (Fig. 3b), E2‐caused inhibition of invasion was enhanced (Fig. 3a). However, this inhibitory effect was not significant. The dose of E2 was not sufficient to activate the overexpressed PPARγ, which might be the main reason.
Figure 2.
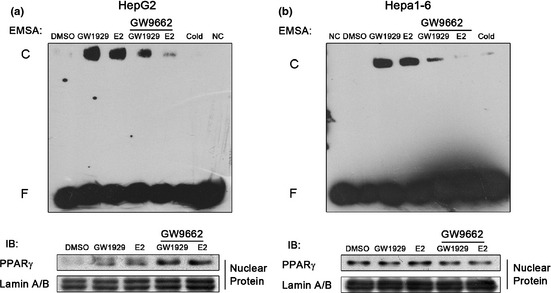
Effect of β‐estradiol (E2) on peroxisome proliferator‐activated receptor‐γ (PPARγ) activity in hepatocellular carcinoma (HCC) cells. (a) In human HepG2 and (b) murine Hepa1‐6 cells, PPARγ activity was determined using electrophoretic mobility shift assay (EMSA). Western blot analyzed nuclear protein PPARγ levels of each group. Lamin A/B served as the loading control. Cold, cold competition reaction; C, protein‐probe complex; F, free probe; NC, no nuclear protein.
Figure 3.
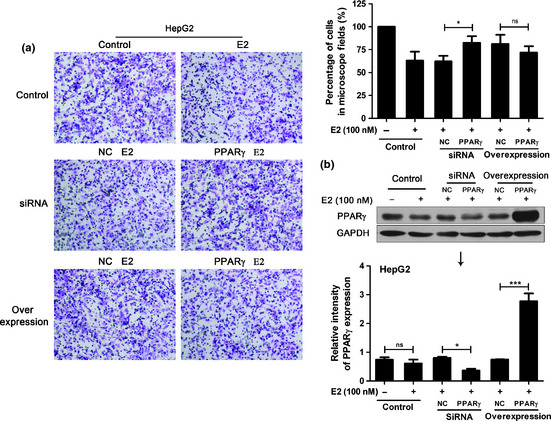
β‐estradiol (E2) inhibited hepatocellular carcinoma (HCC) cell invasion by a peroxisome proliferator‐activated receptor‐γ (PPARγ)‐dependent manner. (a) After PPARγ knockdown or overexpression, Matrigel invasion assay examined HepG2 cell invasion in response to E2. (b) Western blot analyzed the expression levels of PPARγ. Figures showing quantitative analysis include data from at least three independent experiments. Values are expressed as mean ± standard deviation (SD). NC of siRNA, scramble siRNA; NC of overexpression, empty pIRES2‐EGFP plasmid; ns, not significant; *P < 0.05.
PPARγ activation inhibits HCC cell invasion
Previous studies have suggested that PPARγ activation or overexpression inhibits the invasion of diverse tumor cells, even inhibits the tumor metastasis in animal model.11, 12, 17 However, the related research is still deficient in HCC. To address the effects of PPARγ on HCC cell invasion, we introduced the PPARγ agonist GW1929. Hepa1‐6 and HepG2 cells were treated with various concentrations of GW1929 for 24 h. The result shows that PPARγ agonist GW1929 inhibits HCC cell invasion in a dose‐dependent manner. Figure 4(a) shows that PPARγ activity increased gradually over increasing GW1929 concentration, and transcriptional activity was the highest between 1 and 10 μM GW1929. Hepa1‐6 (Fig. 4b) and HepG2 cell (Fig. 4c) invasiveness decreased 16 h after activating PPARγ. Furthermore, the above effect was reversed after 2 h pretreatment with 10 μM antagonist GW9662 (Fig. 4d). These results indicate that PPARγ activation inhibits HCC cell invasion.
Figure 4.
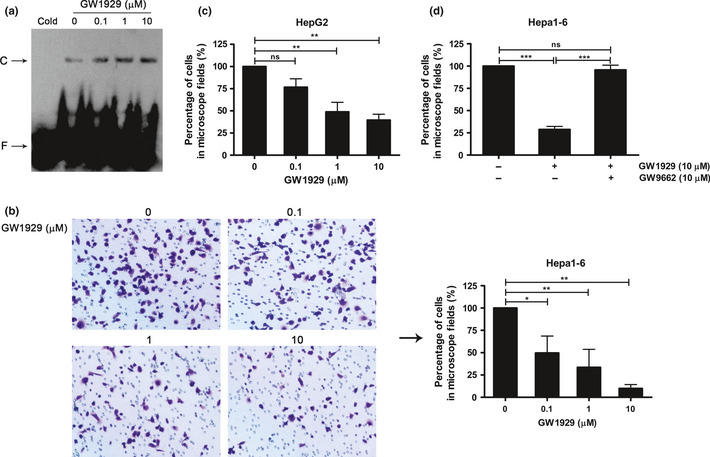
Peroxisome proliferator‐activated receptor‐γ (PPARγ) activation inhibited hepatocellular carcinoma (HCC) cell invasion. (a) After treatment with various concentrations of GW1929, PPARγ activity was analyzed by electrophoretic mobility shift assay (EMSA). (b) Matrigel invasion assay of Hepa1‐6 and (c) HepG2 cell invasion was carried out after treatment with GW1929. (d) Hepatocellular carcinoma cell invasion was examined after pretreatment with PPARγ antagonism GW9662. Cold, cold competition reaction; C, protein‐probe complex; F, free probe. Figures showing quantitative analysis include data from at least three independent experiments. Values are expressed as mean ± standard deviation (SD). ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
PPARγ activation regulates the expression of PAI‐1
PAI‐1 inhibits the degradation of extracellular matrix.14 Researchers have found activation of nuclear receptor PPARγ by ligands increases the PAI‐1 of pancreatic cancer cells and revealed that PAI‐1 can be a PPARγ downstream gene.17 We were interested in the correlation between PPARγ activity and regulation of PAI‐1 expression in HCC cells.
Various concentration of GW1929 treatment resulted in the rising mRNA (Fig. 5a) and protein (Fig. 5b) levels of intracellular PAI‐1. The results suggest that PPARγ activation promotes PAI‐1 expression. Similarly, these effects were also reversed 2 h pretreatment after PPARγ antagonist GW9662 (Fig. 5c,d).
Figure 5.
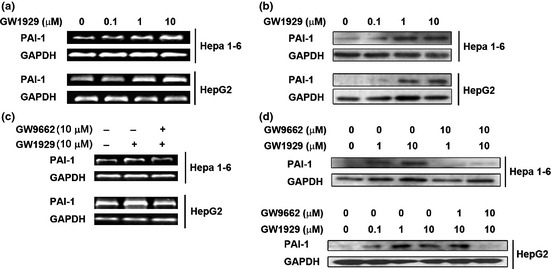
Effect of peroxisome proliferator‐activated receptor‐γ (PPARγ) activation on the expression level of plasminogen activator inhibitor‐1 (PAI‐1). (a) Levels of PAI‐1 mRNA and (b) protein in Hepa1‐6 and HepG2 cells treated with various concentrations of GW1929. (c) Pretreatment of cells with PPARγ antagonism GW9662 restored the level of PAI‐1 mRNA and (d) protein.
Subsequently, we extracted genome DNA of human and murine cells and cloned a fragment of –1300 to +30 of pai‐1 gene into vector pGL3‐Basic. Promoter‐reporter plasmids were successfully constructed, namely, murine plasmid pGL3‐P‐mPAI‐1 and human plasmid pGL3‐P‐hPAI‐1. Cells were cotransfected with promoter‐reporter plasmid and control plasmid pRL. Following the treatment of various concentrations of GW1929, the transfected cells showed the stronger luciferase activity with increasing dose of GW1929 (Fig. 6a). These results indicate that pai‐1 gene expression is positively regulated by activating PPARγ.
Figure 6.
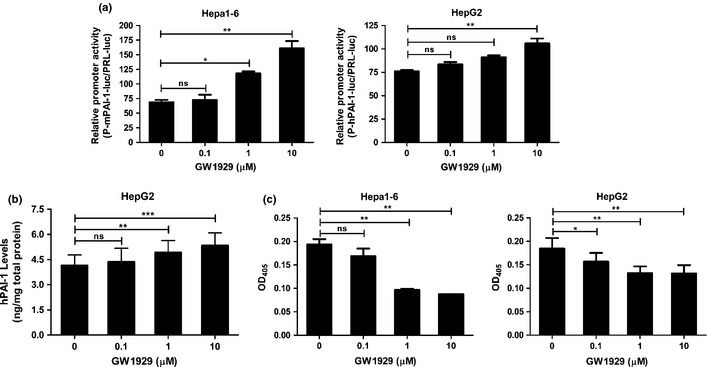
Detection of plasminogen activator inhibitor‐1 (PAI‐1) and urokinase‐type plasminogen activator (uPA). (a) Hepatocellular carcinoma (HCC) cells were transfected with luciferase receptor contrust pGL‐P‐mPAI‐1/pGL‐P‐hPAI‐1 and control plasmid pRL. After treatment of GW1929 for 24 h, cell lysates were collected for luciferase assay. (b) Enzyme linked immunosorbent assay (ELISA) assay showed PAI‐1 levels in HepG2 cell culture supernatant rose. (c) uPA activity in Hepa1‐6 and HepG2 cell culture supernatant was measured. Figures showing quantitative analysis include data from at least three independent experiments. Values are expressed as mean ± standard deviation (SD). ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
uPA is an effector molecule of PAI‐1, and therefore we detected uPA enzyme activity in cell culture supernatant. After 24 h treatment with GW1929, ELISA assay showed PAI‐1 levels in HepG2 cell culture supernatant rose with increasing GW1929 concentration (Fig. 6b). Meanwhile, uPA activity in Hepa1‐6 and HepG2 cell culture supernatant descended (Fig. 6c).
PPARγ activation inhibits HCC cell invasion by regulating PAI‐1 expression
To further explore the relationship between PPARγ and HCC cell invasion, we designed siRNA to PPARγ (Fig. 7a) and constructed PPARγ overexpression plasmid pIRES2‐PPARγ (Fig. 7b). A matrigel‐based transwell assay was carried out in three groups including control, siRNA and overexpression groups. After knockdown of PPARγ agonist GW1929 cannot inhibit HCC cell invasion. Compared to the PPARγ siRNA, in the group of PPARγ overexpression GW1929 inhibited invasiveness (Fig. 7c). After exposure to the transfectin reagents and siRNA or plasmids for 4 h, the cells were incubated with GW1929 for 24 h and cell lysates were obtained. Western blot analysis (Fig. 7d) suggested that cells with reduced PPARγ expression showed decreased levels of PAI‐1. On the contrary, in the PPARγ‐overexpressed cells, the level of PAI‐1 was increased. However, the effect of elevation was not significant. Meanwhile, uPA activity was also different in HCC cell culture supernatant with knockdown or overexpression of PPARγ. As shown in Figure 7(e), transfection with PPARγ siRNA increased the uPA activity in response to GW1929. In the PPARγ‐overexpressed cell supernatant, the uPA activity was lowered compared with control vector‐transfected cells. Although in the group of PPARγ overexpression GW1929 inhibited invasiveness and lowered uPA activity, these differences were not significant. The dosage of GW1929 was not sufficient to activate the overexpressed PPARγ, which is the main reason.
Figure 7.
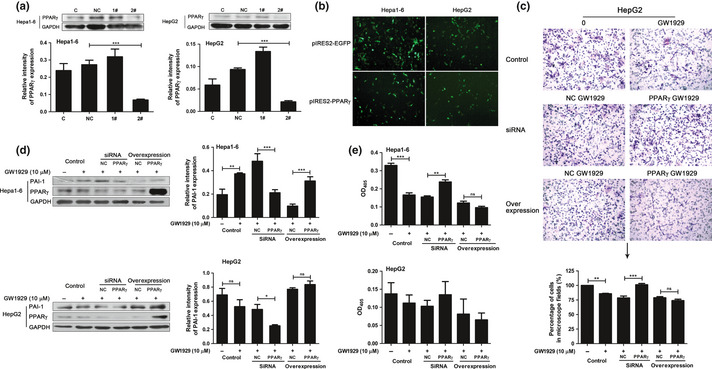
Peroxisome proliferator‐activated receptor‐γ (PPARγ) activation inhibited hepatocellular carcinoma (HCC) cell invasion by regulating plasminogen activator inhibitor‐1 (PAI‐1) expression. (a) Western blot evaluated the effect of PPARγ siRNA. (b) Detection of PPARγ overexpression, about 70% of the total cells expressed the fluorescence. (c) Effect of PPARγ knockdown or overexpression on HepG2 cell invasion, PAI‐1 levels (d), and urokinase‐type plasminogen activator (uPA) activity (e) in response to GW1929. Figures showing quantitative analysis include data from at least three independent experiments. Values are expressed as mean ± standard deviation (SD). 1#, PPARγ siRNA1; 2#, PPARγ siRNA2; NC of siRNA group, scramble siRNA; NC of overexpression group, empty pIRES2‐EGFP plasmid; ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Expression levels of PAI‐1 affect the HCC cell invasion
To investigate whether changes in PAI‐1 levels were associated with HCC cell invasive properties, we examined the effect of knockdown (Fig. 8a) or overexpression (Fig. 8c) of PAI‐1 on HepG2 cell invasion. Our results showed that cell invasion was significantly increased after transfection of PAI‐1 siRNA compared with transfection of scramble siRNA in HepG2 cells (Fig. 8b). Whereas there was a significant decrease for invasiveness in pIRES2‐PAI‐1‐transfected HepG2 cells compared with the empty pIRES2‐EGFP vector (Fig. 8d). These results indicate that PAI‐1 plays an inhibitory role in HCC invasion.
Figure 8.
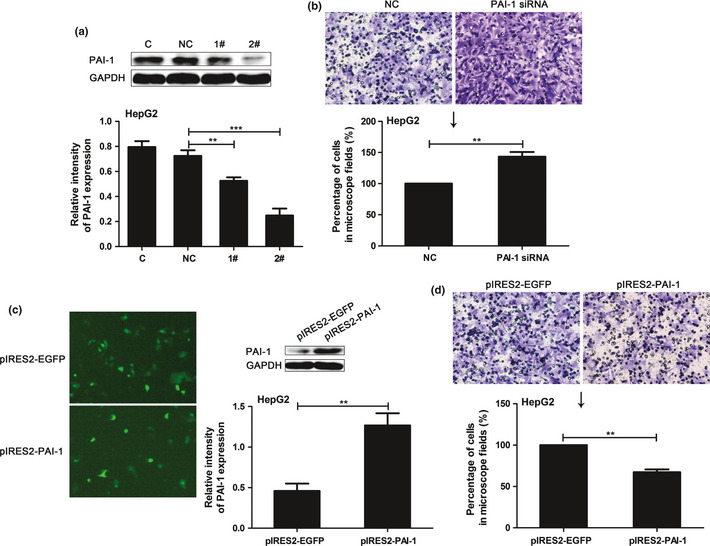
Effect of plasminogen activator inhibitor‐1 (PAI‐1) knockdown or overexpression on hepatocellular carcinoma (HCC) cell invasion. (a) Western blot evaluated the effect of PAI‐1 siRNA. Effect of PAI‐1 knockdown (b) or overexpression (d) on HepG2 cell invasion. (c) Detection of PAI‐1 overexpression, about 65% of the total cells expressed the fluorescence. Figures showing quantitative analysis include data from at least three independent experiments. Values are expressed as mean ± standard deviation (SD). 1#, PAI‐1 siRNA1; 2#, PAI‐1 siRNA2; NC of siRNA group, scramble siRNA; NC of overexpression group, empty pIRES2‐EGFP plasmid. **P < 0.01; ***P < 0.001.
Discussion
Apart from the established metabolic actions, PPARγ also plays a critical role in the treatment of cancer.19 Emerging evidence suggests that activation of PPARγ suppresses tumorigenesis. In this study, we show that PPARγ activation can inhibit HCC cell invasion by upregulating PAI‐1. E2 suppresses HCC cell invasion, and this inhibition is associated with PPARγ activity. GW1929 is selected as PPARγ agonist. Peroxisome proliferator‐activated receptor‐γ activation significantly attenuates HCC cell invasion, and PPARγ overexpression or siRNA also further demonstrates the relationship between PPARγ and HCC cell invasion. In addition, PPARγ activation modulates the expression of PAI‐1, which is an inhibitor of uPA. Finally, we show that PAI‐1 level rises and uPA activity lowers during the inhibition of HCC cell invasion.
Nevertheless, the function of PPARγ in tumor development is still controversial. Some evidence suggests that activating PPARγ suppresses tumorigenesis, but some studies indicate that activating PPARγ promotes tumorigenesis.20 The role of PPARγ in the onset and treatment of cancer has been the focus of recent attention. Most research to date indicates that PPARγ agonists can promote terminal differentiation, inhibit cell growth and increase apoptosis of human cancer cell lines, as well as inhibit tumorigenesis in animal models of cancer.21 Genetic studies in mice demonstrate that loss of one allele of PPARγ gene predisposes mice to cancer.6 Our results have showed that PPARγ agonist GW1929 can not inhibit HCC cell proliferation (data not shown), but can suppress HCC cell invasion in a dose‐dependent manner.
By now, the studies on relationship between PPARγ activation and HCC mainly focus on growth and apoptosis of HCC cells.9, 10 Koga et al.22 showed that human hepatoma cell lines exist a concentration‐dependent inhibition of cellular growth and arrest of the cell cycle in G0/1 cell after treatment with troglitazone. Peroxisome proliferator‐activated receptor‐γ agonist inhibits the growth of HCC by inducing apoptosis through caspase 3 activation.9 However, few studies pay attention to HCC invasion or metastasis. Annicotte et al.23 reported that PPARγ activation increases the expression of E‐cadherin and inhibits invasion of prostate cancer. E‐cadherin is one of the major factors that inhibit metastasis and invasion of prostate cancer cells. The studies ultimately showed that E‐cadherin is a bona fide PPARγ target gene. Thus, one possibility to explain the inhibitory of PPARγ agonist on HCC cell invasion is by regulating invasion and metastasis‐related proteins.
Our studies have showed that when PPARγ is activated by specific agonist GW1929, invasiveness of different HCC cells is significantly inhibited. Some evidence suggests that anticancer activity of PPARγ ligands could occur independently of PPARγ. Thiazolidinediones, which are widely used as insulin‐sensitizing agents for the treatment of type 2 diabetes, are synthetic PPARγ ligands. Studies have indicated that TZD inhibits invasiveness of pancreatic cancer cells via PPARγ‐independent (“off‐target”) mechanisms.24 How PPARγ ligands can act independent of PPARγ is under study. To address the issue of PPARγ‐dependent versus PPARγ‐independent mechanisms of action, we used PPARγ overexpression and RNA interference experiments. The results demonstrated that HCC cell invasion is mediated by PPARγ‐ dependent pathway in our studies. On the basis of the above results we continued to explore the associated effector molecules of PPARγ downstream in the invasion.
As is known, invasion and metastasis are critical determinants of cancer mortality. Breaking through the barrier of ECM is the first step of tumor cell invasion. Matrix metalloproteinase and plasminogen activation system play an extensive role in this process. Therefore, we detect the intracellular content of related molecules in two enzyme systems. The results showed that the effect of PPARγ activation on MMP family is not significant, but the effect on inhibitor PAI‐1 of plasminogen activation system is significant. Because PAI‐1 is the main inhibitory factor of uPA in vivo, it can suppress ECM degradation. Peroxisome proliferator‐activated receptor‐γ activation upregulates mRNA and protein levels of PAI‐1, and introduction of PPARγ antagonist reverse this effect. Similarly, PPARγ overexpression and RNA interference tests also have indicated PPARγ activation increases PAI‐1 expression. Our results are consistent with associated research results of human pancreatic cancer,17 which were performed by Sawai et al., who inferred PAI‐1 may be downstream target gene of PPARγ regulation.
In view of the above results and related articles,25, 26 we construct a promoter‐receptor vector of PAI‐1 to further analyze the relationship between PPARγ and PAI‐1. The results confirm our conjecture that activation degree of PAI‐1 promoter‐receptor gene rose along with the increasing concentration of PPARγ agonist. This indicated that PAI‐1 can be the downstream gene of PPARγ. It is well known that extracellular PAI‐1 reacts rapidly with uPA forming a stable complex with a 1:1 stoichiometry, thus inhibits activity of uPA.13 We further validated the effect of increasing PAI‐1 on inhibition of uPA activity by using enzymatic activity analysis assay. Taken together, we draw some conclusions: PPARγ activation upregulates the expression levels of PAI‐1. Increasing secretion of PAI‐1 inhibits uPA activity, and thus suppresses the whole plasminogen activation system, ultimately causing the descent of HCC cell invasion.
In conclusion, the present study demonstrated PPARγ agonist upregulates expression levels of PAI‐1 by activating PPARγ, increased PAI‐1 inhibits extracellular uPA activity, and ultimately HCC cell invasion is suppressed. In addition to classical PPARγ target genes, our findings suggest that PAI‐1 can be another target gene of PPARγ. Although some clinical studies show that administration of PPARγ agonists is accompanied by some side‐effects, whether these side‐effects represent agonist‐specific or off‐target effects, remains uncertain. This study may provide a molecular mechanism for the development of ‘non‐agonist’ modulators of PPARγ, which can target PPARγ without causing the side‐effects.27
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Project Nos 31070764, 91013015 and 81273527) and Natural Science Foundation of Jiangsu Province (BK2010049).
(Cancer Sci, doi: 10.1111/cas.12143, 2013)
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clinic 2011; 61: 69–90. [DOI] [PubMed] [Google Scholar]
- 2. El‐Serag HB. Epidemiology of hepatocellular carcinoma in USA. Hepatol Res 2007; 37: S88–94. [DOI] [PubMed] [Google Scholar]
- 3. Worns MA, Weinmann A, Pfingst K et al Safety and efficacy of sorafenib in patients with advanced hepatocellular carcinoma in consideration of concomitant stage of liver cirrhosis. J Clin Gastroenterol 2009; 43: 489–95. [DOI] [PubMed] [Google Scholar]
- 4. Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X‐receptor interacts with nuclear receptors in retinoic acid, thyroid‐hormone and vitamin‐d3 signaling. Nature 1992; 355: 446–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPAR gamma. Ann Rev Biochem 2008; 77: 289–312. [DOI] [PubMed] [Google Scholar]
- 6. Yu J, Shen B, Chu ES et al Inhibitory role of peroxisome proliferator‐activated receptor gamma in hepatocarcinogenesis in mice and in vitro . Hepatology 2010; 51: 2008–19. [DOI] [PubMed] [Google Scholar]
- 7. Ogino S, Shima K, Baba Y et al Colorectal cancer expression of peroxisome proliferator‐activated receptor γ (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology 2009; 136: 1242–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koeffler HP. Peroxisome proliferator‐activated receptor γ and cancers. Clin Cancer Res 2003; 9: 1–9. [PubMed] [Google Scholar]
- 9. Toyoda M, Takagi H, Horiguchi N et al A ligand for peroxisome proliferator activated receptor gamma inhibits cell growth and induces apoptosis in human liver cancer cells. Gut 2002; 50: 563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu J, Qiao L, Zimmermann L et al Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo . Hepatology 2006; 43: 134–43. [DOI] [PubMed] [Google Scholar]
- 11. Mueller E, Sarraf P, Tontonoz P et al Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell 1998; 1: 465–70. [DOI] [PubMed] [Google Scholar]
- 12. Bren‐Mattison Y, Van Putten V, Chan D, Winn R, Geraci MW, Nemenoff RA. Peroxisome proliferator‐activated receptor‐gamma (PPAR gamma) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non‐small‐cell lung cancer cells (NSCLC). Oncogene 2005; 24: 1412–22. [DOI] [PubMed] [Google Scholar]
- 13. Duffy M, Duggan C. The urokinase plasminogen activator system: a rich source of tumour markers for the individualised management of patients with cancer. Clin Biochem 2004; 37: 541–8. [DOI] [PubMed] [Google Scholar]
- 14. Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer 2003; 3: 489–501. [DOI] [PubMed] [Google Scholar]
- 15. Mazar AP, Henkin J, Goldfarb RH. The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogenesis 1999; 3: 15–32. [DOI] [PubMed] [Google Scholar]
- 16. Bajou K, Masson V, Gerard RD et al The plasminogen activator inhibitor PAI‐1 controls in vivo tumor vascularization by interaction with proteases, not vitronectin. J Cell Biol 2001; 152: 777–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sawai H, Liu J, Reber HA, Hines OJ, Eibl G. Activation of peroxisome proliferator‐activated receptor‐gamma decreases pancreatic cancer cell invasion through modulation of the plasminogen activator system. Mol Cancer Res 2006; 4: 159–67. [DOI] [PubMed] [Google Scholar]
- 18. Xu H, Wei Y, Zhang Y et al Oestrogen attenuates tumour progression in hepatocellular carcinoma. J Pathol 2012; 228: 216–29. [DOI] [PubMed] [Google Scholar]
- 19. Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator‐activated receptor gamma agonists. Lancet Oncol 2004; 5: 419–29. [DOI] [PubMed] [Google Scholar]
- 20. Yang K, Fan KH, Lamprecht SA et al Peroxisome proliferator‐activated receptor gamma agonist troglitazone induces colon tumors in normal C57BL/6J mice and enhances colonic carcinogenesis in Apc1638 N/+ Mlh1 + /‐ double mutant mice. Int J Cancer 2005; 116: 495–9. [DOI] [PubMed] [Google Scholar]
- 21. Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator‐activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer 2012; 12: 181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koga H, Sakisaka S, Harada M et al Involvement of p21WAF1/Cip1, p27Kip1, and p18INK4c in troglitazone‐induced cell‐cycle arrest in human hepatoma cell lines. Hepatology 2001; 33: 1087–97. [DOI] [PubMed] [Google Scholar]
- 23. Annicotte JS, Iankova I, Miard S et al Peroxisome proliferator‐activated receptor gamma regulates E‐cadherin expression and inhibits growth and invasion of prostate cancer. Mol Cell Biol 2006; 26: 7561–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galli A, Ceni E, Crabb DW et al Antidiabetic thiazolidinediones inhibit invasiveness of pancreatic cancer cells via PPARgamma independent mechanisms. Gut 2004; 53: 1688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fink T, Kazlauskas A, Poellinger L, Ebbesen P, Zachar V. Identification of a tightly regulated hypoxia‐response element in the promoter of human plasminogen activator inhibitor‐1. Blood 2002; 99: 2077–83. [DOI] [PubMed] [Google Scholar]
- 26. Buchwalter G, Gross C, Wasylyk B. The ternary complex factor net regulates cell migration through inhibition of PAI‐1 expression. Mol Cell Biol 2005; 25: 10853–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Choi JH, Banks AS, Kamenecka TM et al Antidiabetic actions of a non‐agonist PPARγ ligand blocking Cdk5‐mediated phosphorylation. Nature 2011; 477: 477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]