

Abstract
Malignant pleural mesothelioma is a poorly responsive tumor known to overexpress the phase II detoxification enzyme glutathione‐S‐transferase, which catalyzes the conjugation between glutathione and platinum(II)‐containing drugs. Therefore, we evaluated the effect of the strong glutathione S‐transferase inhibitor NBDHEX on human mesothelioma cell lines (MSTO‐211H, MPP89, MM‐B1 and Mero 48a) featuring the most common mesothelioma phenotypes: epithelioid and biphasic. Even though a different response to NBDHEX was observed, the molecule was very effective on all cell lines tested, triggering a sustained activation of both JNK and p38, followed by caspase activation and apoptosis. NBDHEX also caused severe oxidative stress in the MPP89 cells and, to a lesser extent, in the MMB1 cells, while it did not cause a significant redox imbalance in the other cell lines. The efficacy of the drug was found to be comparable or even higher than that of cisplatin. Moreover, it showed synergistic or additive effects when used in combination with cisplatin. In conclusion, NBDHEX was effective on mesothelioma cell lines, with IC 50 values in the low micromolar range (IC 50 between 1 and 4 μM). These findings indicate that NBDHEX, alone or in combination with cisplatin, is a promising new strategy for treating this rare and aggressive malignancy.
Mesothelioma is a malignant tumor that arises from pluripotent mesothelial cells of pleura or peritoneum.1, 2 Mesothelioma cells show different phenotypes, including the epithelioid, sarcomatoid and the mixed type. Epithelioid is the most common form (60–70% of cases). The sarcomatoid form represents 10–20% of mesotheliomas, and the mixed (or biphasic) form, with epithelioid and sarcomatoid areas, represents 30–40% of all mesotheliomas.3 There is strong evidence for a causal relation between the development of mesotelioma and asbestos exposure.4 Although its use has been widely abandoned in the developed world, the mortality rate will continue to increase over the forthcoming years because of the long latency period between the exposure to asbestos and the onset of tumor.5 Malignant pleural mesothelioma (MPM) responds poorly to chemotherapy.6, 7 Several clinical studies have been performed using the platinum(II)‐containing compound CDDP in combination with antifolates (pemetrexed and raltitrexed), nucleoside analogues (gemcitabine), topoisomerase inhibitors (irinotecan) or mitotic inhibitors (vinorelbine) to improve the response rate of MPM to treatment.8, 9, 10, 11, 12 The combination of pemetrexed plus CDDP is the approved “standard of care” for patients with unresectable MPM;13, 14, 15, 16 however, only a partial benefit has been shown in terms of survival.15, 17 Among the mechanisms of CDDP resistance, the most studied are those dependent on the elevated expression of glutathione S‐transferases (GST)18, 19 and of the export pumps MRP1 and MRP2 (multidrug resistance‐associated protein). A coordinated action between these pumps and GST enables excretion from the cells of a wide range of glutathione‐conjugated compounds, leading to a reduction in the effectiveness of drug treatment.20 This has led to the idea that the intracellular CDDP can be neutralized by the GST‐catalyzed reaction, and expelled from the cell.21 However, Peklak‐Scott et al.22 report that such catalysis has a minor role in the protection from CDDP toxicity. In contrast, a number of drugs, including CDDP, cause apoptosis through the activation of the MAPK pathway,23, 24 and GST might play an important role in the resistance to these anticancer agents by interfering with the activation of this pro‐apoptotic pathway. Indeed, several published investigations report that the GST isoenzyme GSTP1‐1 binds directly to the MAPK JNK, inhibiting its activation.25, 26, 27 Recent studies have shown that the GSTP1‐1 and GSTM1‐1 isoenzymes are able to inhibit the activity of the apoptosis signal‐regulating kinase (ASK1), which, in turn, is responsible for the activation of JNK and p38 kinases.28 We have designed and synthesized a class of nitro‐benzoxadiazole derivatives, which act as efficient and specific inhibitors of the GST catalytic and anti‐apoptotic activity.29 The most promising derivative is 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX), a non‐glutathione peptidomimetic molecule that is known to induce the dissociation of the GSTP1‐JNK complex, leading to the activation of JNK and to the programmed cell death thereafter.29, 30 An important property of this compound is that it is not recognized as a substrate by the ABC transporters P‐glycoprotein and MRP1 and is highly cytotoxic against tumor cells that overexpress these transporters.31, 32, 33 Given the promising results already observed in several different tumors,31, 32, 33, 34, 35, 36, 37, 38 in the present study we evaluate the effect of NBDHEX in four human malignant mesothelioma cell lines, MSTO‐211H, MPP89, MM‐B1 and Mero 48a, featuring the two most common phenotypes, epithelioid and biphasic. The mechanism of cell death triggered by NBDHEX and the in vitro efficacy of NBDHEX in combination with CDDP are investigated.
Materials and Methods
Drugs
Cisplatin (CDDP) was purchased from Teva Pharma B.V. (Mijdrecht, the Netherlands); glutathione and 1‐chloro‐2,4‐dinitrobenzene were obtained from Sigma‐Aldrich (St. Louis, MO, USA); and NBDHEX was synthesized as reported by Ricci and colleagues and dissolved in DMSO.29 Just before use, stock solutions of NBDHEX were diluted to the appropriate concentration in complete cell medium with final DMSO concentration never exceeding 0.01% (v/v), a dosage at which it has no cytotoxic effect on our cell lines.
Cell culture and treatments
In the present study we used a panel of four adherent MPM cell lines featuring different morphology. The MPP89 cell line, with epitheliod phenotype, and the MCF7 (breast carcinoma cell line) were provided by the Cell Factory Facility from the Istituto Nazionale per la Ricerca sul Cancro, IST, Genoa, Italy; the MSTO‐211H, with biphasic phenotype, was obtained from the American Type Culture Collection; the biphasic Mero 48a was a gift from Dr Marjan Versnel, from the Department of Immunology, Erasmus Medical Center, Rotterdam, the Netherlands; and the biphasic MM‐B1 was provided by the Department of Clinical Sciences and Translational Medicine, Tor Vergata University, Rome, Italy.
Cells were maintained at 37°C and 5% CO2 in a humidified atmosphere in RPMI (MSTO 211‐H and MPP89; Lonza) or DMEM (Mero 48a, MM‐B1) (Sigma–Aldrich) medium, supplemented with 10% FBS (v/v; Gibco (Paisley, UK) and Lonza (Basel, Switzerland), respectively), 2 mM l‐glutamine, 100 U/mL of penicillin and 100 mg/mL streptomycin (Sigma–Aldrich). For all the experiments, cells were plated in culture flasks at a density of 25 000 cells/cm2. After 24 h, MSTO‐211H, MM‐B1, Mero 48a and MPP89 cells were exposed to equitoxic concentrations of NBDHEX (10, 8, 8 and 15 μM, respectively). Alternatively, cells were incubated with either the caspase (40 μM Z‐VAD‐fmk; Sigma–Aldrich) or the JNK (10 μM SP600125; Sigma–Aldrich) inhibitor, added 1 h before the incubation with NBDHEX. Cells were collected at different time points and subjected to analysis.
Cell viability and combination studies
An evaluation of cell viability after 48 h of treatment with different drug (NBDHEX or CDDP) concentrations was determined by sulforodamine B (Sigma–Aldrich) assay,39 as previously reported.30 The dose‐response profile fulfills the IC50 value (the concentration used to obtain 50% of cell growth inhibition) for each compound. To study the efficacy of NBDHEX as an adjuvant of CDDP, a combination study was performed to measure the interaction in terms of synergism, additivity or antagonism. Cells were simultaneously incubated with equitoxic concentrations of the two compounds, ranging from 10% to 60% of the values of cell growth inhibition. Alternatively, the MSTO‐211H cell line was subjected to the sequential administration of NBDHEX and CDDP. The combination index was calculated according to the Chou and Talalay median effect using the CalcuSyn software (Biosoft, Cambridge, UK).40, 41
Flow cytometry
The percentage of viable early and late apoptotic and necrotic cells was determined after 15 h of treatment with equitoxic amounts of NBDHEX, by simultaneous staining of cells with propidium iodide (PI) and Annexin V‐FITC (Annexin V‐FITC Apoptosis Detection Kit; Sigma–Aldrich), as previously reported.30 For cell cycle analysis, cells were fixed with 70% ethanol and stained with PI. In both cases, the emission of unstained cells treated with NBDHEX was used as background fluorescence. Stained cells were analyzed using a FACSCalibur instrument (BD Bioscence, San Jose, CA, USA). Flow cytometric data were statistically analyzed using FlowJo 8.8.6 software.
Western blotting
At each time point analyzed, the cell pellet was collected and lysed as previously reported;30 the protein concentration of the sample was determined using the Lowry method.42 Proteins (40 μg) were loaded on 12% SDS‐polyacrylamide gel and transferred onto a PVDF membrane (GE Healthcare, Chalfont St. Giles, UK). Anti‐P‐JNK (Thr183 Tyr185), anti‐JNK, anti‐P‐p38 (Thr180 Tyr182), anti‐p38, anti‐caspase‐8, anti‐caspase‐9, anti‐GSTP1‐1, anti‐Bcl‐2 (Cell Signaling, Beverly, MA, USA) and anti‐β actin (Sigma–Aldrich) were used as primary antibodies. Anti‐rabbit or anti‐mouse secondary antibody (Cell Signaling) was revealed using the ECL LiteAblot Extend (Euroclone, Milan, Italy).
Enzyme activities
Glutathione S‐transferase and caspase activities were measured at 25°C in total cell lysates as previously described.30 GST‐specific activity was expressed as U/mg of total proteins, 1 unit of enzymatic activity being defined as the amount of enzyme that catalyzes the formation of 1 μmol/min of product. The caspase activity is expressed as ΔF/min/cell number.
HPLC determination of various forms of glutathione
For free glutathione determination, cell lysates were diluted with 12% sulfosalicylic acid and the analyte content determined on the acid‐soluble fraction. The protein bound glutathione (GS‐Pro) was assayed in the protein pellet dissolved in 0.1 N NaOH. For oxidized glutathione (GSSG) determination, the cells were sonicated in the presence of 5 mM N‐ethylmaleimide and the analyte content was determined in cell lysates, as described above. Protein concentration was quantified by BCA‐protein assay (BCA‐protein assay, Pierce, Rockford, IL, USA). The derivatization and chromatography procedures were performed, with little modifications, as previously reported.43
Statistical analysis
All the experiments were repeated at least three times; results are presented as means ± SD. Statistical evaluation was done using the Student's t‐test. The criterion for statistical significance used was P < 0.05.
Results
Glutathione S‐transferase content in mesothelioma cells
We found a high expression of GSTP1‐1 in all mesothelioma cell lines, except for MCF7 cells used as negative control (Fig. 1a).44 Moreover, the enzymatic activity paralleled the GSTP1‐1 protein content, suggesting that the major contribution to the activity was given by this GST isoform (Fig. 1b).
Figure 1.
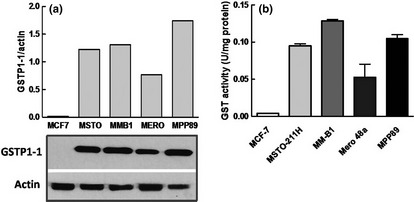
Glutathione S‐transferase (GST) content in mesothelioma cells. (a) Western blot and densitometric analysis of GSTP1‐1 expression in the human mesothelioma cell lines. The MCF‐7 breast cancer cell line has been utilized as a negative control for GSTP1‐1 expression. The experiment is representative of three that gave similar results. (b) GST‐specific activity was expressed as U/mg of total proteins, 1 unit of enzymatic activity being defined as the amount of enzyme that catalyzes the formation of 1 μmol/min of product (mean ± SD).
Cytotoxicity assay
The cytotoxic effects of either NBDHEX or CDDP were measured in the mesothelioma cell lines after 48 h of treatment. The dose‐response profile with NBDHEX gave IC50 values between 1.0 and 4.0 μM, while the IC50 values of CDDP were between 2.0 and 7.0 μM (Table 1). NBDHEX exhibited higher cytotoxic activity than CDDP in the Mero 48a and MM‐B1 cells, while equivalent cytotoxicity of both drugs was observed in MSTO‐211H and in MPP89 cell lines. Moreover, the NBDHEX antitumor efficacy in these mesothelioma cell lines was comparable to that observed in other tumor cells.31, 36
Table 1.
IC50 values (μM) and combination index (CI) of 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX) and CDDP
| Cell lines | IC50 NBDHEX | IC50 CDDP | CI50 | CI75 | CI90 |
|---|---|---|---|---|---|
| MSTO‐211H | 1.5 ± 0.4 | 2.0 ± 0.3 | 1.09¶ , †† | 0.77§ , †† | 0.56‡ , †† |
| MM‐B1 | 1.3 ± 0.4 | 5.7 ± 1.0 | 0.28† | 0.25† | 0.22† |
| Mero 48a | 1.3 ± 0.5 | 6.5 ± 0.7 | 0.84§ | 0.59‡ | 0.45‡ |
| MPP89 | 3.9 ± 0.9 | 3.6 ± 0.5 | 0.48‡ | 0.27† | 0.16† |
†Strong synergism, ‡synergism, §moderate synergism, ¶nearly additive, ††NBDHEX was added first. IC50: the concentration used to obtain 50% of cell growth inhibition. CI50, CI75, CI90: The NBDHEX‐CDDP combination index at 50%, 75% and 90% inhibition of growth.
Analysis of the cell death induced by 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol
The growth curves of each cell line, incubated in the absence and in the presence of equitoxic concentrations of NBDHEX, are shown in Figure 2(a). While the untreated cells underwent active replication, the number of NBDHEX‐treated cells remained essentially constant during the first 8–10 h, to decrease afterward.
Figure 2.
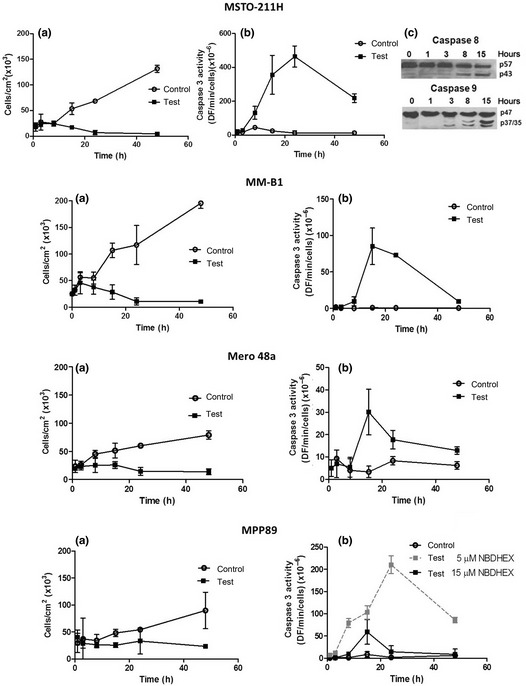
6‐(7‐Nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX) inhibits cell growth and induces caspase activation. (a) Growth curves of MSTO‐211H, MM‐B1, Mero 48a and MPP89 cells incubated in the absence ( ) or in the presence (
) or in the presence ( ) of equitoxic concentrations of NBDHEX (every experimental dot represents the mean of three independent experiments ±SD). (b) The kinetic of caspase activation was measured on MSTO‐211H, MMB1, Mero 48a and MPP89 cells incubated in the absence (
) of equitoxic concentrations of NBDHEX (every experimental dot represents the mean of three independent experiments ±SD). (b) The kinetic of caspase activation was measured on MSTO‐211H, MMB1, Mero 48a and MPP89 cells incubated in the absence ( ) or in the presence (
) or in the presence ( ) of equitoxic concentrations of NBDHEX (mean ± SD). The effect of 5 μM NBDHEX (dashed line) on MPP89 cells is also shown. (c) Immunoblotting analysis of MSTO‐211H at different incubation times with NBDHEX. The panel shows the cleavage of procaspase‐9 (p47) and procaspase‐8 (p57) to the active caspase‐9 (p37/35) and caspase‐8 (p43).
) of equitoxic concentrations of NBDHEX (mean ± SD). The effect of 5 μM NBDHEX (dashed line) on MPP89 cells is also shown. (c) Immunoblotting analysis of MSTO‐211H at different incubation times with NBDHEX. The panel shows the cleavage of procaspase‐9 (p47) and procaspase‐8 (p57) to the active caspase‐9 (p37/35) and caspase‐8 (p43).
The next step was to analyze the cell death mechanism of NBDHEX. Evident caspase‐3 activation in MSTO‐211H and in MM‐B1 cell lines was induced by the molecule, while lower and negligible effects were observed in the MPP89 and Mero 48a cell lines, respectively (Fig. 2b). The proteolitic activity was clearly detectable after 4–8 h and reached the maximum value at 15–24 h of treatment. The degree of activation of caspase‐8 and caspase‐9, which initiates the cascade leading to the caspase‐3 activation, was also evaluated (Fig. 2c). The caspase‐9 cleavage was visible as early as 3 h after treatment, while the caspase‐8 activation was only detectable after 8 h. These data suggest that NBDHEX activates an intrinsic apoptotic pathway, as reported in other tumor cell lines.33
The MPP89 cells showed low caspase activation when exposed to an equitoxic dose of NBDHEX (15 μM). However, higher caspase activity was detected when the amount of NBDHEX decreased to a dose close to its IC50 value (5 μM). Because high levels of GSSG might cause inhibition of caspase‐3 activity,45 we hypothesize that the data obtained with 15 μM NBDHEX could be the consequence of the strong increase in GSSG levels observed in this cell line after treatment with NBDHEX (see below).
Cytofluorimetric analysis
The ability of NBDHEX to induce an apoptotic process was confirmed by cytofluorimetric analysis after 15 h of treatment with equitoxic concentrations of NBDHEX. The mesothelioma cell lines displayed different distribution of early apoptotic (Annexin V+/PI−) or late apoptotic (Annexin V+/PI+) cell sub‐fractions (Table 2). The MPP89 cell line essentially showed late apoptosis, while both early and late apoptosis were detected in MSTO‐211H and MM‐B1 cell lines. The overall percentage of apoptotic cells was 53%, 59% and 80% in the MPP89, MSTO‐211H and MM‐B1 cell lines, respectively. In the Mero 48a cells only a negligible amount of apoptosis (6%), which well correlated with the low caspase activation, was detected. The compound caused a modest increase (10%) in the fraction of Mero 48a cells in G2/M at 15 h of treatment, while the percentage of cells in G2/M increased to 40% only when NBDHEX treatment was extended up to 48 h (data not shown). These findings are in agreement with the NBDHEX dose‐response profile obtained after 48 h and might explain the discrepancy between the IC50 value and the level of apoptosis observed.
Table 2.
Flow cytometric analysis of cell death
| Cell lines | Annexin V+/PI− | Annexin V‐/PI+ | Annexin V+/PI+ | Early and late apoptosis |
|---|---|---|---|---|
| MSTO‐211H | 25.4 | 1.6 | 33.5 | 59 |
| MM‐B1 | 51.5 | 0.50 | 28.3 | 80 |
| Mero 48a | 2.2 | 0.0 | 3.4 | 6 |
| MPP89 | 0.0 | 5.3 | 53.2 | 53 |
Percentages were obtained after subtraction of background cell death (untreated control cells). A representative experiment of three independent experiments with similar results is presented. Annexin V+/PI−, early apoptotic cells; Annexin V−/PI+, necrotic cells; Annexin V+/PI+, late apoptotic cells.
To prove the involvement of the caspases in the apoptotic cell death induced by NBDHEX in mesothelioma cells, cytofluorimetric analysis was also performed in the presence or absence of the general caspase inhibitor Z‐VAD‐fmk. The effect of the inhibitor was investigated on MSTO‐211H, MM‐B1 and MPP89 sparing the Mero 48a, given the negligible amount of apoptosis observed in this cell line. Z‐VAD‐fmk significantly decreased the amount of subG1 MSTO‐211H and MM‐B1 apoptotic cells treated with NBDHEX (Fig. 3a,c). A protective effect was also observed in the MPP89 cell line (Fig. 3b). These results confirmed that the NBDHEX‐induced cell death was a caspase‐dependent event.
Figure 3.
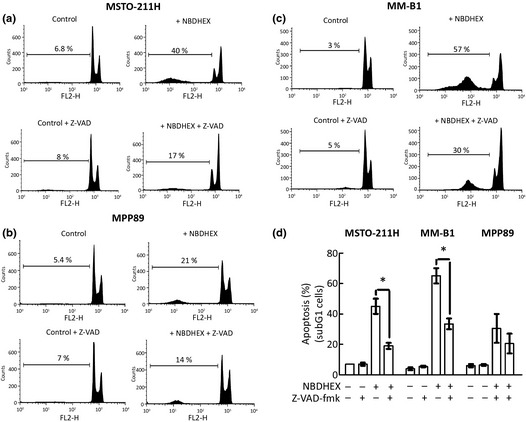
6‐(7‐Nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX) triggers a caspase‐dependent apoptosis. (a) MSTO‐211H, (b) MM‐B1 and (c) MPP89 cells were incubated with the caspase inhibitor Z‐VADfmk (40 μM) 1 h before the addition of equitoxic amounts of NBDHEX. Cytofluorimetric analysis was performed after 15 h of NBDHEX treatment. The amount of subG1 apoptotic cells is shown in (d). The results are the mean of three independent experiments that gave similar results (mean ± SD; *P < 0.05).
Analysis of reduced/oxidized glutathione content
The level of reduced glutathione, which protects cells from oxidative stress and is a substrate for GST, was similar in all cell lines studied (Fig. 4a). We also evaluated the impact of NBDHEX treatment on the intracellular content of both the reduced and the disulfide forms of glutathione. All cell lines were treated with equitoxic amounts of NBDHEX and then analyzed after a short (3 h) and a long incubation time (15 h). NBDHEX induced an intracellular increase of the oxidized species of glutathione in all mesothelioma cell lines and, in particular, in MM‐B1 and MPP89 cell lines (Fig. 4b,c). Extrusion of glutathione in the culture medium was observed only after 15 h of treatment and was likely a consequence of disulfide accumulation (Fig. 4d). However, the depletion of glutathione was balanced by an increase in the basal level of the intracellular glutathione (Fig. 4a). As a result, a mild oxidative imbalance occurred in the mesothelioma cells, with the exception of the MM‐B1 and MPP89 cell lines, where the increase in the oxidized species of glutathione was particularly relevant (Fig. 4e).
Figure 4.
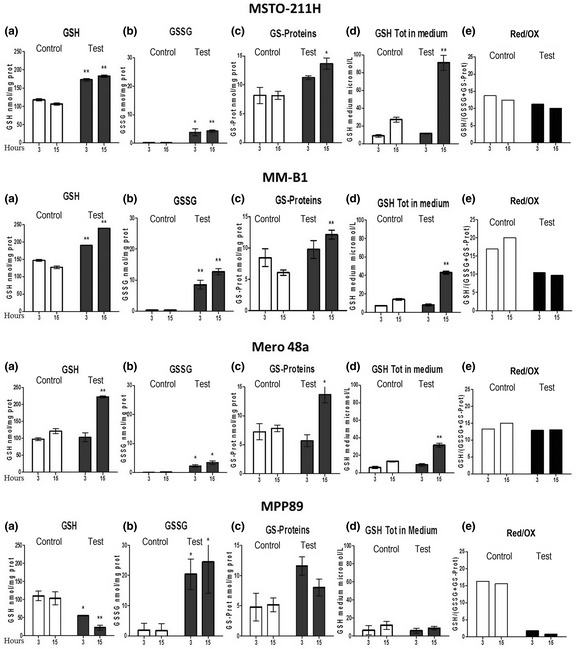
Effect of 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX) on the reduced and oxidized glutathione contents. HPLC analysis of the intracellular levels of: (a) reduced glutathione, (b) oxidized glutathione, (c) glutathyonylated proteins and of (d) glutathione extruded in the medium of MSTO‐211H, MM‐B1, Mero 48a and MPP89 cells after 3 and 15 h of treatment with equitoxic concentrations of NBDHEX (mean ± SD; *P < 0.05, **P < 0.01 versus control). (e) The ratio of reduced/oxidized glutathione is shown for each cell line as indicator of oxidative stress. GSH, glutathione; GSSG, glutathione disulfide.
Analysis of the MAPK involvement
We have previously demonstrated that NBDHEX activates the MAPK signaling pathway in several tumor cell lines.30, 31, 32, 33, 36, 38 In accordance with previous studies, we observed a rapid and sustained increase in the phospho‐active form of JNK and p38 in all mesothelioma cell lines treated with NBDHEX (Fig. 5a). We found elevated basal levels of Bcl‐2 in all mesothelioma cell lines (Fig. 5b), also in accordance with previous findings;46 no significant changes in Bcl‐2 protein levels were observed in the NBDHEX‐treated cells.
Figure 5.
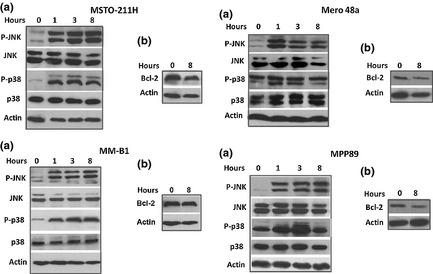
6‐(7‐Nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX) causes MAPK activation. Western blot analysis shows (a) the kinetic of P‐JNK and P‐p38 activation and (b) the levels of Bcl‐2 in MSTO‐211H, MM‐B1, Mero 48a and MPP89 cells treated with equitoxic concentrations of NBDHEX. β actin was used as a loading control.
To prove the involvement of the JNK pathway in the apoptotic cell death induced by NBDHEX in MSTO‐211H, MM‐B1 and MPP89 cells, cytofluorimetric analysis was performed in the presence or absence of the JNK inhibitor SP600125. Pre‐incubation with the JNK inhibitor induced a decrease in the subG1 fraction and an increased G2/M arrest in NBDHEX‐treated MSTO‐211H cells (Fig. 6), supporting the involvement of the JNK pathway in the apoptosis triggered by NBDHEX in this cell line. In contrast, SP600125 did not increase the viability of MM‐B1 and MPP89 cells treated with NBDHEX (data not shown).
Figure 6.
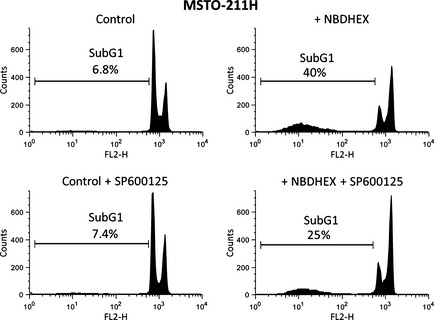
Involvement of the JNK pathway in the MSTO‐211H apoptotic cell death. MSTO‐211H cells were incubated with the JNK inhibitor SP600125 (10 μM) 1 h before the addition of equitoxic amounts of 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX). Cytofluorimetric analysis was performed after 15 h of NBDHEX treatment.
Combination study between 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol and CDDP
To evaluate the in vitro interactions between NBDHEX and CDDP, human mesothelioma cell lines were incubated with either simultaneous or sequential (in MSTO‐211H cells) administration of the two drugs. As shown in Table 1, the simultaneous administration of equitoxic concentrations of NBDHEX and CDDP resulted in a strong synergistic effect in MM‐B1 and MPP89 cell lines and in a synergistic effect in Mero 48a. An additive‐synergistic effect was observed in MSTO‐211H cells, when NBDHEX was administered prior to CDDP. We have previously shown that the simultaneous exposure to NBDHEX and CDDP produces mostly additive interactions in CDDP‐resistant osteosarcoma cell lines. However, synergistic effects were observed when these cells were treated with NBDHEX immediately followed by CDDP.35 Although the explanation for such behavior still needs to be elucidated, these data should be taken into account in planning treatment schedules.
Discussion
To identify an alternative and specific strategy for the treatment of mesothelioma, we evaluated the effectiveness of the potent GST inhibitor NBDHEX. The four mesothelioma cell lines used in this study are not only characterized by high levels of GSTP1‐1, but also by an elevated expression of the antiapoptotic protein Bcl‐2. Nonetheless, treatment with NBDHEX was effective in these cell lines, with IC50 values in the low micromolar range (IC50 between 1 and 4 μM). Equitoxic concentrations of NBDHEX, ranging between 8 and 15 μM, caused an early and sustained activation of the MAPK p38 and JNK, as well as a time‐dependent increase in caspase activity and Annexin/PI positive cells. The effect of the caspase inhibitor proved that the NBDHEX‐induced cell death was a caspase‐dependent event in all mesothelioma cell lines. The results obtained with the JNK inhibitor suggest that the activation of the JNK pathway is involved in the NBDHEX‐induced apoptosis in MSTO‐211H cells. Of note, the activation of MAPK, and, in particular, of JNK, might suppress the cell survival function of Bcl‐2,47, 48, 49 and might explain why mesothelioma cells undergo NBDHEX‐triggered apoptosis despite expressing high levels of Bcl‐2. However, the response of MPP89 and MMB1 cells to NBDHEX appears to be more complex, possibly involving additional pathways. Interestingly, NBDHEX caused an evident redox imbalance only in these two cell lines. Indeed, although the amount of reduced glutathione was similar in all mesothelioma cell lines studied, the impact of NBDHEX on the redox state was quite different. The NBDHEX caused in all cell lines an increase in the glutathionilated proteins, and in MMB1 and MPP89 a marked accumulation of GSSG. However, in MSTO 211H and in Mero 48a the increase of oxidized species of glutathione was balanced by an increase of reduced glutathione, thus avoiding strong oxidative stress. This response is likely the consequence of an increased synthesis of glutathione mediated by p38. In fact, this kinase is known to promote the activation of the transcription factor Nrf250, which, in turn, controls the expression of γ‐glutamylcysteine synthetase, the rate‐limiting enzyme in the synthesis of glutathione.51
Besides the direct cytotoxic effects of NBDHEX, we also demonstrated a synergistic interaction between NBDHEX and CDDP. The increase of sensitivity towards CDDP is likely a consequence of the NBDHEX‐mediated activation of MAPK; in fact, some data suggest that MAPK pathway plays a major role in the cell response to CDDP, while the resistance to this drug is associated with reduced activation of MAPK.23 The NBDHEX‐mediated disruption of intracellular redox homeostasis may also contribute to the CDDP toxicity; recent data suggest that CDDP induces the formation of reactive oxygen species52 and the synergistic interaction between NBDHEX and CDDP was particularly evident in MM‐B1 and MPP89 cells, which were the more sensitive to oxidative stress.
The present findings indicate that NBDHEX is effective as a single agent, or in combination with CDDP to increase the drug cytotoxicity. So far, much information on the interaction of this compound with conventional drugs had been obtained. We have previously shown that NBDHEX exerts synergistic effects when in combination with temozolomide,53 doxorubicin and vincristine.34, 35 In vivo studies have also shown the effectiveness of NBDHEX in reducing both the tumor growth, using melanoma and Ewing's sarcoma models, and the metastatic ability of U‐2OS osteosarcoma cells. In all these experiments, NBDHEX did not show collateral toxicity and proved to be very well tolerated.30, 34, 36, 53 A clinical implication of these findings is the potential use of NBDHEX to increase the therapeutic index of CDDP in mesothelioma patients. Accordingly, the phospho‐active form of JNK may be considered as a feasible prognostic factor in the response of mesothelioma to therapeutics.
Disclosure Statement
The authors have no conflict of interest to declare.
Acknowledgments
This study was partially supported by grants from the Department of Occupational Medicine of Istituto Nazionale per l'Assicurazione contro gli Infortuni sul Lavoro (formerly ISPESL) and the Associazione Italiana per la Ricerca sul Cancro (Project 10598).
(Cancer Sci, doi: 10.1111/cas.12061, 2012)
References
- 1. Robinson BW, Lake RA. Advances in malignant mesothelioma. N Engl J Med 2005; 353: 1591–603. [DOI] [PubMed] [Google Scholar]
- 2. Sekido Y. Genomic abnormalities and signal transduction dysregulation in malignant mesothelioma cells. Cancer Sci 2010; 101: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zervos MD, Bizekis C, Pass HI. Malignant mesothelioma 2008. Curr Opin Pulm Med 2008; 14: 303–9. [DOI] [PubMed] [Google Scholar]
- 4. van Meerbeeck JP, Scherpereel A, Surmont VF, Baas P. Malignant pleural mesothelioma: The standard of care and challenges for future management. Crit Rev Oncol Hematol 2011; 78: 92–111. [DOI] [PubMed] [Google Scholar]
- 5. Peto J, Hodgson JT, Matthews FE, Jones JR. Continuing increase in mesothelioma mortality in Britain. Lancet 1995; 345: 535–9. [DOI] [PubMed] [Google Scholar]
- 6. Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet 2005; 366: 397–408. [DOI] [PubMed] [Google Scholar]
- 7. Sugarbaker DJ, Norberto JJ. Multimodality management of malignant pleural mesothelioma. Chest 1998; 113: 61S–5S. [DOI] [PubMed] [Google Scholar]
- 8. Aelony Y. Raltitrexed and pemetrexed studies in mesothelioma have not shown improved quality of life nor prolonged survival compared with effective pleurodesis with thoracoscopic talc poudrage. J Clin Oncol 2006; 24: 4667–8. [DOI] [PubMed] [Google Scholar]
- 9. Fennell DA, Steele JP, Shamash J et al Efficacy and safety of first‐ or second‐line irinotecan, cisplatin, and mitomycin in mesothelioma. Cancer 2007; 109: 93–9. [DOI] [PubMed] [Google Scholar]
- 10. Lazzarini R, Moretti S, Orecchia S, Betta PG, Procopio A, Catalano A. Enhanced antitumor therapy by inhibition of p21waf1 in human malignant mesothelioma. Clin Cancer Res 2008; 14: 5099–107. [DOI] [PubMed] [Google Scholar]
- 11. Maruyama R, Shoji F, Okamoto T et al Triplet chemotherapy with cisplatin, gemcitabine and vinorelbine for malignant pleural mesothelioma. Jpn J Clin Oncol 2005; 35: 433–8. [DOI] [PubMed] [Google Scholar]
- 12. Sorensen JB, Frank H, Palshof T. Cisplatin and vinorelbine first‐line chemotherapy in non‐resectable malignant pleural mesothelioma. Br J Cancer 2008; 99: 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen SE, Pace MB. Malignant pleural mesothelioma. Am J Health Syst Pharm 2012; 69: 377–85. [DOI] [PubMed] [Google Scholar]
- 14. Grosso F, Scagliotti GV. Systemic treatment of malignant pleural mesothelioma. Future Oncol 2012; 8: 293–305. [DOI] [PubMed] [Google Scholar]
- 15. Kindler HL. Systemic treatments for mesothelioma: standard and novel. Curr Treat Options Oncol 2008; 9: 171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mott FE. Mesothelioma: a review. Ochsner J 2012; 12: 70–9. [PMC free article] [PubMed] [Google Scholar]
- 17. Berghmans T, Paesmans M, Lalami Y et al Activity of chemotherapy and immunotherapy on malignant mesothelioma: a systematic review of the literature with meta‐analysis. Lung Cancer 2002; 38: 111–21. [DOI] [PubMed] [Google Scholar]
- 18. Shea TC, Kelley SL, Henner WD. Identification of an anionic form of glutathione transferase present in many human tumors and human tumor cell lines. Cancer Res 1988; 48: 527–33. [PubMed] [Google Scholar]
- 19. Tew KD, Monks A, Barone L et al Glutathione‐associated enzymes in the human cell lines of the National Cancer Institute Drug Screening Program. Mol Pharmacol 1996; 50: 149–59. [PubMed] [Google Scholar]
- 20. Licht T, Fiebig HH, Bross KJ et al Induction of multiple‐drug resistance during anti‐neoplastic chemotherapy in vitro. Int J Cancer 1991; 49: 630–7. [DOI] [PubMed] [Google Scholar]
- 21. Kelland LR. Preclinical perspectives on platinum resistance. Drugs 2000; 59(Suppl 4): 1–8; Discussion 37–8. [DOI] [PubMed] [Google Scholar]
- 22. Peklak‐Scott C, Smitherman PK, Townsend AJ, Morrow CS. Role of glutathione S‐transferase P1–1 in the cellular detoxification of cisplatin. Mol Cancer Ther 2008; 7: 3247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brozovic A, Osmak M. Activation of mitogen‐activated protein kinases by cisplatin and their role in cisplatin‐resistance. Cancer Lett 2007; 251: 1–16. [DOI] [PubMed] [Google Scholar]
- 24. Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal 2000; 12: 1–13. [DOI] [PubMed] [Google Scholar]
- 25. Adler V, Yin Z, Fuchs SY et al Regulation of JNK signaling by GSTp. EMBO J 1999; 18: 1321–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Luca A, Federici L, De Canio M, Stella L, Caccuri AM. New Insights into the Mechanism of JNK1 Inhibition by Glutathione Transferase P1–1. Biochemistry 2012; 51: 7304–12. [DOI] [PubMed] [Google Scholar]
- 27. Yin Z, Ivanov VN, Habelhah H, Tew K, Ronai Z. Glutathione S‐transferase p elicits protection against H2O2‐induced cell death via coordinated regulation of stress kinases. Cancer Res 2000; 60: 4053–7. [PubMed] [Google Scholar]
- 28. Wu Y, Fan Y, Xue B et al Human glutathione S‐transferase P1–1 interacts with TRAF2 and regulates TRAF2‐ASK1 signals. Oncogene 2006; 25: 5787–800. [DOI] [PubMed] [Google Scholar]
- 29. Ricci G, De Maria F, Antonini G et al 7‐Nitro‐2,1,3‐benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S‐transferases. Mechanism of action of potential anticancer drugs. J Biol Chem 2005; 280: 26397–405. [DOI] [PubMed] [Google Scholar]
- 30. Turella P, Cerella C, Filomeni G et al Proapoptotic activity of new glutathione S‐transferase inhibitors. Cancer Res 2005; 65: 3751–61. [DOI] [PubMed] [Google Scholar]
- 31. Ascione A, Cianfriglia M, Dupuis ML et al The glutathione S‐transferase inhibitor 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol overcomes the MDR1‐P‐glycoprotein and MRP1‐mediated multidrug resistance in acute myeloid leukemia cells. Cancer Chemother Pharmacol 2009; 64: 419–24. [DOI] [PubMed] [Google Scholar]
- 32. Filomeni G, Turella P, Dupuis ML et al 6‐(7‐Nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol, a specific glutathione S‐transferase inhibitor, overcomes the multidrug resistance (MDR)‐associated protein 1‐mediated MDR in small cell lung cancer. Mol Cancer Ther 2008; 7: 371–9. [DOI] [PubMed] [Google Scholar]
- 33. Turella P, Filomeni G, Dupuis ML et al A strong glutathione S‐transferase inhibitor overcomes the P‐glycoprotein‐mediated resistance in tumor cells. 6‐(7‐Nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX) triggers a caspase‐dependent apoptosis in MDR1‐expressing leukemia cells. J Biol Chem 2006; 281: 23725–32. [DOI] [PubMed] [Google Scholar]
- 34. Pasello M, Manara MC, Michelacci F et al Targeting glutathione‐S transferase enzymes in musculoskeletal sarcomas: a promising therapeutic strategy. Anal Cell Pathol (Amst) 2011; 34: 131–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pasello M, Michelacci F, Scionti I et al Overcoming glutathione S‐transferase P1‐related cisplatin resistance in osteosarcoma. Cancer Res 2008; 68: 6661–8. [DOI] [PubMed] [Google Scholar]
- 36. Pellizzari Tregno F, Sau A, Pezzola S et al In vitro and in vivo efficacy of 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX) on human melanoma. Eur J Cancer 2009; 45: 2606–17. [DOI] [PubMed] [Google Scholar]
- 37. Scotlandi K, Remondini D, Castellani G et al Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol 2009; 27: 2209–16. [DOI] [PubMed] [Google Scholar]
- 38. Sau A, Filomeni G, Pezzola S et al Targeting GSTP1‐1 induces JNK activation and leads to apoptosis in cisplatin‐sensitive and ‐resistant human osteosarcoma cell lines. Mol BioSyst 2012; 8: 994–1006. [DOI] [PubMed] [Google Scholar]
- 39. Skehan P, Storeng R, Scudiero D et al New colorimetric cytotoxicity assay for anticancer‐drug screening. J Natl Cancer Inst 1990; 82: 1107–12. [DOI] [PubMed] [Google Scholar]
- 40. Chou TC, Motzer RJ, Tong Y, Bosl GJ. Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: a rational approach to clinical protocol design. J Natl Cancer Inst 1994; 86: 1517–24. [DOI] [PubMed] [Google Scholar]
- 41. Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res 2004; 10: 7994–8004. [DOI] [PubMed] [Google Scholar]
- 42. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951; 193: 265–75. [PubMed] [Google Scholar]
- 43. Pastore A, Massoud R, Motti C et al Fully automated assay for total homocysteine, cysteine, cysteinylglycine, glutathione, cysteamine, and 2‐mercaptopropionylglycine in plasma and urine. Clin Chem 1998; 44: 825–32. [PubMed] [Google Scholar]
- 44. Jhaveri MS, Morrow CS. Methylation‐mediated regulation of the glutathione S‐transferase P1 gene in human breast cancer cells. Gene 1998; 210: 1–7. [DOI] [PubMed] [Google Scholar]
- 45. Huang Z, Pinto JT, Deng H, Richie JP Jr. Inhibition of caspase‐3 activity and activation by protein glutathionylation. Biochem Pharmacol 2008; 75: 2234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shukla A, Bosenberg MW, MacPherson MB et al Activated cAMP response element binding protein is overexpressed in human mesotheliomas and inhibits apoptosis. Am J Pathol 2009; 175: 2197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muscarella DE, Bloom SE. The contribution of c‐Jun N‐terminal kinase activation and subsequent Bcl‐2 phosphorylation to apoptosis induction in human B‐cells is dependent on the mode of action of specific stresses. Toxicol Appl Pharmacol 2008; 228: 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamamoto K, Ichijo H, Korsmeyer SJ. BCL‐2 is phosphorylated and inactivated by an ASK1/Jun N‐terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 1999; 19: 8469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Poommipanit PB, Chen B, Oltvai ZN. Interleukin‐3 induces the phosphorylation of a distinct fraction of bcl‐2. J Biol Chem 1999; 274: 1033–9. [DOI] [PubMed] [Google Scholar]
- 50. Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun 2000; 278: 484–92. [DOI] [PubMed] [Google Scholar]
- 51. Lee JM, Johnson JA. An important role of Nrf2‐ARE pathway in the cellular defense mechanism. J Biochem Mol Biol 2004; 37: 139–43. [DOI] [PubMed] [Google Scholar]
- 52. Brozovic A, Ambriovic‐Ristov A, Osmak M. The relationship between cisplatin‐induced reactive oxygen species, glutathione, and BCL‐2 and resistance to cisplatin. Crit Rev Toxicol 2010; 40: 347–59. [DOI] [PubMed] [Google Scholar]
- 53. Tentori L, Dorio AS, Mazzon E et al The glutathione transferase inhibitor 6‐(7‐nitro‐2,1,3‐benzoxadiazol‐4‐ylthio)hexanol (NBDHEX) increases temozolomide efficacy against malignant melanoma. Eur J Cancer 2011; 47: 1219–30. [DOI] [PubMed] [Google Scholar]