

Abstract
Genome‐wide transcriptional expression analysis is a powerful strategy for characterizing the biological activity of anticancer compounds. It is often instructive to identify gene sets involved in the activity of a given drug compound for comparison with different compounds. Currently, however, there is no comprehensive gene expression database and related application system that is; (i) specialized in anticancer agents; (ii) easy to use; and (iii) open to the public. To develop a public gene expression database of antitumor agents, we first examined gene expression profiles in human cancer cells after exposure to 35 compounds including 25 clinically used anticancer agents. Gene signatures were extracted that were classified as upregulated or downregulated after exposure to the drug. Hierarchical clustering showed that drugs with similar mechanisms of action, such as genotoxic drugs, were clustered. Connectivity map analysis further revealed that our gene signature data reflected modes of action of the respective agents. Together with the database, we developed analysis programs that calculate scores for ranking changes in gene expression and for searching statistically significant pathways from the Kyoto Encyclopedia of Genes and Genomes database in order to analyze the datasets more easily. Our database and the analysis programs are available online at our website (http://scads.jfcr.or.jp/db/cs/). Using these systems, we successfully showed that proteasome inhibitors are selectively classified as endoplasmic reticulum stress inducers and induce atypical endoplasmic reticulum stress. Thus, our public access database and related analysis programs constitute a set of efficient tools to evaluate the mode of action of novel compounds and identify promising anticancer lead compounds.
Cancer chemotherapy has gradually improved with the development of new anticancer agents. In particular, recent progress in the development of molecular cancer therapeutics has revealed new types of anticancer agents that directly target abnormal proteins in cancer cells.1, 2 These agents are effective against certain types of cancer where the target protein plays a predominant role in the growth and survival of the cancer cells, but are generally less successful against other types of tumor.
To improve the present status of cancer chemotherapy, it is essential to search for novel compounds that selectively target new classes of molecular targets in cancer or induce cancer‐specific cell death with new modes of action. It has been shown that compounds that selectively interfere with cellular biological targets abrogate specific signaling pathways and modulate the expression of individual subsets of signature genes. Therefore, gene signature‐based analysis is a powerful strategy for characterizing the mechanism of action of drug candidates. Recently, Lamb et al.3, 4 has developed a systematic approach named C‐map to find connections among small molecules sharing a mechanism of action, chemicals and physiological processes, and diseases and drugs. They used a reference collection of gene expression profiles from cultured human cells treated with bioactive small molecules. However, the system was not specifically designed for anticancer drugs and lacks several standard agents.
Here, we obtained comprehensive gene expression datasets of anticancer drugs, consisting of standard anticancer agents, molecularly targeted drugs, and related inhibitors. The datasets include some compounds that are not contained in the C‐map database. To develop a comprehensive database, we used the Affymetrix GeneChip HG‐U133 Plus 2.0 arrays (Affymetrix, Santa Clara, CA, USA), which contained more probes (54 675 probe sets) than the arrays that were mainly used for the C‐map database (HT_HG‐U133A arrays; 22 283 probe sets). We further developed a calculation program that enables us to rapidly compare gene signatures of test compounds with those of antitumor agents to predict likely modes of action. Using our established systems, we successfully show that ER stress is differentially involved in the effect of antitumor agents.
Materials and Methods
Cell line and compounds
We used human colon adenocarcinoma HT‐29 cells for the analysis. The cells were cultured in a humidified atmosphere of 5% CO2 and 95% air at 37°C. The anticancer compounds used in our analysis are listed in Table 1. These compounds were added to culture medium, with the solvent being <0.5% of the medium's volume.
Table 1.
Treatment samples of anticancer compounds, their corresponding control sample, manufacturer, solvent, target/mode of action, and drug concentration for treatment
| Treatment sample ID | Compound | Control sample ID | Manufacturer | Solvent | Target/mode of action | Concentration |
|---|---|---|---|---|---|---|
| GR001 | Cisplatin | CS1 |
Bristol‐Myers Squibb (New York, NY, USA) |
DMSO | DNA cross‐linker | 30 μM |
| GR002 | Trichostatin A | CS1 |
Wako Pure Chemical Industries (Osaka, Japan) |
DMSO | HDAC | 300 nM |
| GR003 | Vorinostat | CS1 |
Cayman Chemical Company (Ann Arbor, MI, USA) |
DMSO | HDAC | 10 μM |
| GR004 | Bortezomib | CS1 |
Millennium Pharmaceuticals (Cambridge, MA, USA) |
DMSO | Proteasome | 100 nM |
| GR005 | MG‐132 | CS1 | Peptide Institute (Osaka, Japan) | DMSO | Proteasome | 1 μM |
| GR006 | Geldanamycin | CS1 | Sigma (St Louis, MO, USA) | DMSO | Hsp90 | 30 nM |
| GR007 | 17‐AAG | CS1 | Wako Pure Chemical Industries | DMSO | Hsp90 | 100 nM |
| GR008 | Vincristine | CS2 | Eli Lilly (Indianapolis, IN, USA) | Distilled water | Tubulin | 30 nM |
| GR009 | Paclitaxel | CS2 | Bristol‐Myers Squibb | DMSO | Tubulin | 30 nM |
| GR010 | Docetaxel | CS2 | Astra Zeneca (London, UK) | DMSO | Tubulin | 30 nM |
| GR011 | 5‐FU | CS2 | Sigma | DMSO | Pyrimidine | 100 μM |
| GR012 | Gemcitabine | CS2 | Eli Lilly | Saline | Pyrimidine | 1 μM |
| GR013 | Melphalan | CS3 | Sigma | 75% DMSO | DNA cross‐linker | 100 μM |
| GR014 | Mitomycin C | CS3 | Sigma | 50% DMSO | DNA alkylator | 10 μM |
| GR015 | Oxaliplatin | CS3 | Yakult (Tokyo, Japan) | DMSO | DNA cross‐linker | 3 μM |
| GR016 | Bleomycin | CS3 | Sigma | Distilled water | DNA cleavage | 30 μg/mL |
| GR017 | Actinomycin D | CS3 | Sigma | DMSO | RNA synthesis | 30 nM |
| GR018 | Neocarzinostatin | CS3 | Sigma | – | DNA cleavage | 3 μg/mL |
| GR019 | Methotrexate | CS3 | Sigma | DMSO | DHFR | 1 μM |
| GR020 | 6‐Mercaptopurine | CS3 | Sigma | DMSO | Purine | 100 μM |
| GR021 | Temsirolimus | CS3 |
Santa Cruz Biotechnology (Santa Cruz, CA, USA) |
DMSO | mTOR | 10 μM |
| GR022 | Everolimus | CS3 | Sigma | DMSO | mTOR | 10 μM |
| GR023 | PP242 | CS3 | Sigma | DMSO | mTOR | 10 μM |
| GR024 | Nimustine | CS4 | Sigma | Distilled water | DNA alkylator | 1 mM |
| GR025 | SN38 (Irinotecan) | CS4 | Yakult | DMSO | Topo I | 3 μM |
| GR026 | Camptothecin | CS4 | Sigma | DMSO | Topo I | 3 μM |
| GR027 | Topotecan | CS4 | Sigma | DMSO | Topo I | 3 μM |
| GR028 | Doxorubicin | CS4 | Sigma | DMSO | DNA intercalator/Topo II | 3 μM |
| GR029 | Etoposide | CS4 | Bristol‐Myers Squibb | DMSO | Topo II | 30 μM |
| GR030 | Mitoxantrone | CS4 | Sigma | DMSO | DNA intercalator/Topo II | 3 μM |
| GR031 | Pemetrexed | CS4 | Santa Cruz Biotechnology | DMSO | DNA/RNA synthesis | 1 μM |
| GR032 | 2‐Deoxyglucose | CS1 | Sigma | Distilled water | ER stress (glycolysis) | 10 mM |
| GR033 | Tunicamycin | CS1 | Nacalai Tesque (Kyoto, Japan) | DMSO | ER stress (N‐glycosylation) | 3 μg/mL |
| GR034 | Thapsigargin | CS1 | Wako Pure Chemical Industries | DMSO | ER stress (SERCA) | 10 nM |
| GR035 | A23187 | CS1 | Wako Pure Chemical Industries | DMSO | ER stress (Ca2+ ionophore) | 3 μM |
| GR036 | Vorinostat (16 h) | CS5 | Cayman Chemical Company | DMSO | HDAC | 10 μM |
| GR037 | Bortezomib (16 h) | CS5 | Millennium Pharmaceuticals | DMSO | Proteasome | 100 nM |
| GR038 | Vincristine (16 h) | CS5 | Eli Lilly | Distilled water | Tubulin | 30 nM |
| GR039 | Paclitaxel (16 h) | CS5 | Bristol‐Myers Squibb | DMSO | Tubulin | 30 nM |
| GR040 | Docetaxel (16 h) | CS5 | Astra Zeneca | DMSO | Tubulin | 30 nM |
| GR041 | 5‐FU (16 h) | CS5 | Sigma | DMSO | Pyrimidine | 100 μM |
| GR042 | Mitomycin C (16 h) | CS5 | Sigma | 50% DMSO | DNA alkylator | 10 μM |
| GR043 | Vorinostat | CS6 | Cayman Chemical Company | DMSO | HDAC | 10 μM |
| GR044 | Bortezomib | CS6 | Millennium Pharmaceuticals | DMSO | Proteasome | 100 nM |
| GR045 | Vorinostat (16 h) | CS7 | Cayman Chemical Company | DMSO | HDAC | 10 μM |
| GR046 | Bortezomib (16 h) | CS7 | Millennium Pharmaceuticals | DMSO | Proteasome | 100 nM |
| GR047 | Gemcitabine (16 h) | CS8 | Eli Lilly | Saline | Pyrimidine | 1 μM |
| GR048 | Oxaliplatin (16 h) | CS8 | Yakult | DMSO | DNA cross‐linker | 3 μM |
| GR049 | Bleomycin (16 h) | CS8 | Sigma | Distilled water | DNA cleavage | 30 μg/mL |
| GR050 | Neocarzinostatin (16 h) | CS8 | Sigma | – | DNA cleavage | 3 μg/mL |
| GR051 | Methotrexate (16 h) | CS8 | Sigma | DMSO | DHFR | 1 μM |
| GR052 | 6‐Mercaptopurine (16 h) | CS8 | Sigma | DMSO | Purine | 100 μM |
| GR053 | PP242 (16 h) | CS8 | Sigma | DMSO | mTOR | 10 μM |
| GR054 | Etoposide (16 h) | CS8 | Bristol‐Myers Squibb | DMSO | Topo II | 30 μM |
| GR055 | Pemetrexed (16 h) | CS8 | Santa Cruz Biotechnology | DMSO | DNA/RNA synthesis | 1 μM |
17‐AAG, 17‐N‐allylamino‐17‐demethoxygeldanamycin; DHFR, dihydrofolate reductase; ER, endoplasmic reticulum; 5‐FU, 5‐fluorouracil; HDAC, histone deacetylase; Hsp90, heat shock protein 90; mTOR, mammalian target of rapamycin; SERCA, sarco/endoplasmic reticulum Ca2+‐ATPase; Topo, topoisomerase. –, This product was provided as a solution (20 mM MES [2‐Morpholinoethanesulfonic acid, monohydrate] buffer, pH 5.5)
Drug treatment and GeneChip analysis
The HT‐29 cells were seeded in 6‐well plates in RPMI‐1640 medium supplemented with 10% heat‐inactivated FBS and 100 μg/mL kanamycin. After 20 h of culture, we added the anticancer compounds to the cells at various concentrations. Incubation was then continued for a further 6 or 16 h. In each batch, we also prepared untreated cells as a negative control (Table 1, CS1–CS8). Total RNAs were extracted from the drug‐treated cells using an RNeasy Mini kit (Qiagen, Hilden, Germany). The quality of the RNAs was assessed using an Agilent 2100 Bioanalyzer and RNA 6000 Series II Pico Kit (Agilent Technologies, Palo Alto, CA, USA). We then carried out gene expression analyses using the GeneChip 3' IVT Expression Kit and GeneChip Human Genome U133 Plus 2.0 Array (both Affymetrix) according to the protocols provided by the manufacturer. Hybridization was carried out at 45°C for 16 h in a hybridization oven (Affymetrix). The GeneChips were then automatically washed and stained with streptavidin–phycoerythrin conjugate in an Affymetrix GeneChip Fluidics Station. Fluorescence intensities were scanned with a Gene‐Array Scanner (Affymetrix). Affymetrix GeneChip Command Console (AGCC) version 3.1 was used for the data output.
Statistical analysis
All analyses except for original C‐map analysis were carried out using R version 2.15.0 (http://www.r-project.org/) and Bioconductor version 2.10 (http://bioconductor.org/).
Data preprocessing
The R package software of Affymetrix Micro Array Suite 5.0, MAS5, was used to generate signal intensities for each of the HG‐U133 Plus 2.0 arrays in the study. Expression values were normalized to a mean target level of 100.
Identifying gene signatures
We examined gene expression changes in HT‐29 cells after exposure to 35 anticancer compounds (55 treatment samples). Gene sets were extracted and classified as upregulated or downregulated after exposure to the drug. For each treatment sample, we calculated treatment‐to‐control ratio statistics and selected upregulated and downregulated probe sets as gene signatures (see Doc. S1 for details).
Hierarchical clustering
Probe sets for hierarchical clustering were composed of the collection of all gene signatures. We carried out hierarchical clustering using the logarithm of the ratio statistics of 55 treatment samples and the probe set. We used Ward's method for linkage and Pearson's correlation for distance metric.
Connectivity map analysis
For the C‐map analysis, we prepared the up‐ and down‐signature as input query, which consists of the HG‐U133A probe sets. For each treatment sample, up‐signature (ratio >3) and down‐signature (ratio <1/3) were selected from the HG‐U133 Plus 2.0 array data. The probe sets not included in the HG‐U133A array were deleted. We used the online application in the C‐map website developed by Lamb et al.3 (http://www.broadinstitute.org/cmap/).
Ranking gene expression changes
We adopted the connectivity score based on the Kolmogorov–Smirnov statistic as developed by Lamb et al.3 to investigate the relationship between gene signature and compound. In order to calculate the Kolmogorov–Smirnov statistic faster, it was effective to rank the probe sets in descending order of the treatment‐to‐control ratio and save as a database. For each treatment sample, ranking was calculated as described in the supporting online material for Lamb et al.1 We developed a program for calculating connectivity scores. The source code of this program is available on our website (http://scads.jfcr.or.jp/db/cs/).
Finding significant pathways from the KEGG PATHWAY database
The KEGG PATHWAY is a service of KEGG,5 which is a collection of manually drawn pathway maps that represent knowledge on biological networks. We carried out an analysis to identify significant pathways from the KEGG database. For each pathway, probe sets included in the pathway were extracted using the packages hgu133plus2.db and KEGG.db in Bioconductor. Next, gene signatures and probe sets in the pathway were converted to gene symbols. For each pathway, a 2 × 2 contingency table was created for statistical testing (see Doc. S1). We carried out Fisher's exact test for all pathways and extracted significant pathways. According to the multiple testing theory, significance was defined using the criterion that the false discovery rate6 equals 0.15. The pathways were called significant when the values were <0.15. We developed an R program for the analysis in which the package qvalue in Bioconductor was used for the calculation of q‐values.7, 8 The source code of the program is available on our website.
Results
Development of a gene expression database of antitumor agents
For our analyses, we chose 35 compounds consisting of clinically‐used standard anticancer agents and related drugs (Table 1). The p53‐mutant human colon cancer HT‐29 cell line was used in this study because it represents a typical type of solid tumor and is relatively resistant to cell cycle arrest and apoptosis. We examined the growth inhibitory effect of these agents on the cells and determined effective dosages of the drugs (Fig. S1). We used a concentration of drug that was 3–10‐fold greater than the GI50 value and caused >80% growth inhibition after 48 h of treatment (Table 1). Drug treatment conditions were carefully chosen to enable primary changes in gene expression to be monitored before a secondary cellular response had emerged. Cells were treated for a relatively short time (6 h) for acquisition of gene expression data. As shown in Table 2, the majority of agents caused significant gene expression changes after treatment. However, for the agents that did not show a dramatic effect on gene expression after 6 h of treatment, we also analyzed the gene expression data after a longer exposure time (16 h) (Table 2).
Table 2.
Number of upregulated and downregulated gene signatures for 35 anticancer compounds (55 treatment samples)
| Treatment sample ID | Compound | Upregulated × 3† (×2‡) | Downregulated × 3† (×2‡) |
|---|---|---|---|
| GR001 | Cisplatin | 19 (256) | 52 (697) |
| GR002 | Trichostatin A | 232 (1427) | 181 (1238) |
| GR003 | Vorinostat | 233 (1389) | 173 (1245) |
| GR004 | Bortezomib | 94 (494) | 68 (659) |
| GR005 | MG‐132 | 63 (428) | 51 (583) |
| GR006 | Geldanamycin | 0 (29) | 0 (32) |
| GR007 | 17‐AAG | 7 (68) | 0 (41) |
| GR008 | Vincristine | 0 (45) | 0 (6) |
| GR009 | Paclitaxel | 0 (41) | 0 (9) |
| GR010 | Docetaxel | 1 (37) | 0 (8) |
| GR011 | 5‐FU | 3 (131) | 14 (154) |
| GR012 | Gemcitabine | 2 (26) | 5 (49) |
| GR013 | Melphalan | 85 (716) | 242 (1553) |
| GR014 | Mitomycin C | 3 (69) | 9 (252) |
| GR015 | Oxaliplatin | 0 (11) | 9 (81) |
| GR016 | Bleomycin | 0 (6) | 1 (5) |
| GR017 | Actinomycin D | 26 (294) | 188 (1384) |
| GR018 | Neocarzinostatin | 0 (57) | 2 (138) |
| GR019 | Methotrexate | 1 (28) | 8 (62) |
| GR020 | 6‐Mercaptopurine | 2 (38) | 2 (52) |
| GR021 | Temsirolimus | 10 (132) | 0 (24) |
| GR022 | Everolimus | 16 (162) | 0 (13) |
| GR023 | PP242 | 98 (788) | 37 (530) |
| GR024 | Nimustine | 7 (131) | 13 (318) |
| GR025 | SN38 (Irinotecan) | 75 (602) | 512 (2445) |
| GR026 | Camptothecin | 102 (735) | 809 (3151) |
| GR027 | Topotecan | 28 (576) | 190 (1268) |
| GR028 | Doxorubicin | 49 (459) | 184 (1323) |
| GR029 | Etoposide | 2 (41) | 7 (133) |
| GR030 | Mitoxantrone | 47 (312) | 179 (1408) |
| GR031 | Pemetrexed | 2 (16) | 5 (34) |
| GR032 | 2‐Deoxyglucose | 130 (586) | 16 (439) |
| GR033 | Tunicamycin | 209 (768) | 63 (673) |
| GR034 | Thapsigargin | 69 (323) | 3 (119) |
| GR035 | A23187 | 266 (986) | 86 (931) |
| GR036 | Vorinostat (16 h) | 434 (2142) | 478 (2057) |
| GR037 | Bortezomib (16 h) | 268 (1379) | 299 (1882) |
| GR038 | Vincristine (16 h) | 28 (293) | 77 (335) |
| GR039 | Paclitaxel (16 h) | 21 (263) | 60 (281) |
| GR040 | Docetaxel (16 h) | 18 (221) | 57 (270) |
| GR041 | 5‐FU (16 h) | 26 (543) | 39 (556) |
| GR042 | Mitomycin C (16 h) | 25 (404) | 57 (605) |
| GR043 | Vorinostat (16 h) | 297 (1543) | 229 (1444) |
| GR044 | Bortezomib (16 h) | 118 (596) | 97 (904) |
| GR045 | Vorinostat (16 h) | 462 (2266) | 465 (2106) |
| GR046 | Bortezomib (16 h) | 307 (1551) | 373 (1893) |
| GR047 | Gemcitabine (16 h) | 15 (339) | 8 (186) |
| GR048 | Oxaliplatin (16 h) | 7 (167) | 53 (410) |
| GR049 | Bleomycin (16 h) | 3 (23) | 7 (29) |
| GR050 | Neocarzinostatin (16 h) | 13 (255) | 9 (180) |
| GR051 | Methotrexate (16 h) | 67 (692) | 53 (507) |
| GR052 | 6‐Mercaptopurine (16 h) | 35 (388) | 9 (273) |
| GR053 | PP242 (16 h) | 192 (1206) | 106 (924) |
| GR054 | Etoposide (16 h) | 33 (456) | 31 (405) |
| GR055 | Pemetrexed (16 h) | 24 (400) | 8 (186) |
†Number of probe sets such that the treatment‐to‐control ratio is more than 3 or <1/3 and the larger signal intensity of treatment or control is 300 is shown. ‡Results when the threshold values are changed (2 or 1/2 for ratio, 100 for signal intensity). 17‐AAG, 17‐N‐allylamino‐17‐demethoxygeldanamycin; 5‐FU, 5‐fluorouracil.
Hierarchical clustering and C‐map analysis
With the data that we obtained, we first used hierarchical cluster analysis. The analysis revealed that the drugs were divided into two major clusters, one containing conventional genotoxic drugs and the other including tubulin binding agents (paclitaxel, docetaxel, and vincristine), proteasome inhibitors (bortezomib and MG‐132) and Hsp90 inhibitors (geldanamycin and 17‐AAG) (Fig. 1). Moreover, we found that drugs with similar mechanisms of action were clustered together. As shown in Figure 1, DNA topoisomerase I inhibitors (camptothecin and SN‐38), HDAC inhibitors (vorinostat and trichostatin A), mammalian target of rapamycin (mTOR) inhibitors (everolimus, temsirolimus, and PP242), the tubulin binding agents (paclitaxel, docetaxel, and vincristine), the proteasome inhibitors (bortezomib and MG‐132), the Hsp90 inhibitors (geldanamycin and 17‐AAG), and ER stress inducers (thapsigargin, 2‐deoxyglucose, tunicamycin, and A23187) each formed a mechanism‐specific cluster. Further analysis revealed that each cluster of compounds modulates a cluster‐specific signature gene set (Table S1). However, the 16‐h treatment data tended to cluster together. Next we carried out the C‐map analyses for further validation. This analysis used a collection of genome‐wide transcriptional expression data from cells treated with chemical compounds and is useful in finding functional connections between compounds. When we used our gene signatures for HDAC inhibitors (trichostatin A and vorinostat) as “queries”, we were able to obtain output data that contained compounds with the same mode of action (Table 3). Similarly, when we entered the signature of the proteasome inhibitors, our output results contained proteasome inhibitors, MG‐262 and MG‐132, as hit compounds (Table S2).
Figure 1.
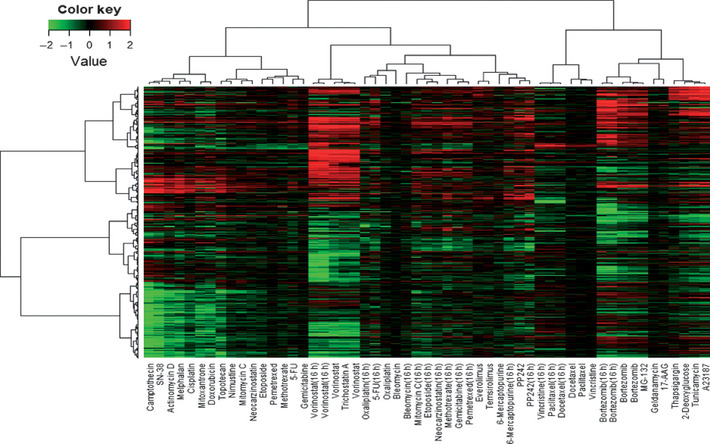
Hierarchical cluster analysis based on the collection of gene signatures of 35 anticancer compounds (55 treatment samples). In total, 3237 probe sets were used for clustering. The values in the heatmap are the logarithm of sample‐to‐control ratio of intensity values. Neither normalizing nor scaling was carried out. 17‐AAG, 17‐N‐allylamino‐17‐demethoxygeldanamycin; 5‐FU, 5‐fluorouracil. Green, downregulated genes; red, upregulated genes.
Table 3.
Results of the Connectivity map for HDAC inhibitors, showing list of ‘hit compounds’ as related compounds to the input gene signatures
| Rank | C‐map name | Dose | Cell | Score | Up | Down |
|---|---|---|---|---|---|---|
| (a) Trichostatin A (up, 164; down, 123) | ||||||
| 1 | Trichostatin A | 100 nM | MCF7 | 1.000 | 0.829 | −0.731 |
| 2 | Trichostatin A | 1 μM | MCF7 | 0.990 | 0.818 | −0.727 |
| 3 | Trichostatin A | 100 nM | MCF7 | 0.989 | 0.811 | −0.732 |
| 4 | Trichostatin A | 1 μM | MCF7 | 0.987 | 0.810 | −0.731 |
| 5 | Trichostatin A | 100 nM | MCF7 | 0.983 | 0.814 | −0.720 |
| 6 | Trichostatin A | 100 nM | MCF7 | 0.977 | 0.820 | −0.705 |
| 7 | Trichostatin A | 1 μM | MCF7 | 0.977 | 0.802 | −0.722 |
| 8 | Trichostatin A | 100 nM | MCF7 | 0.974 | 0.826 | −0.695 |
| 9 | Vorinostat | 10 μM | MCF7 | 0.972 | 0.802 | −0.716 |
| 10 | Trichostatin A | 100 nM | MCF7 | 0.970 | 0.807 | −0.707 |
| 11 | Trichostatin A | 100 nM | MCF7 | 0.970 | 0.806 | −0.708 |
| 12 | Trichostatin A | 100 nM | MCF7 | 0.970 | 0.792 | −0.722 |
| 13 | Trichostatin A | 100 nM | MCF7 | 0.969 | 0.821 | −0.691 |
| 14 | Vorinostat | 10 μM | MCF7 | 0.969 | 0.803 | −0.709 |
| 15 | Trichostatin A | 1 μM | MCF7 | 0.968 | 0.799 | −0.711 |
| 16 | Vorinostat | 10 μM | MCF7 | 0.967 | 0.783 | −0.727 |
| 17 | Trichostatin A | 100 nM | MCF7 | 0.967 | 0.770 | −0.738 |
| 18 | Trichostatin A | 1 μM | MCF7 | 0.965 | 0.784 | −0.723 |
| 19 | Trichostatin A | 100 nM | MCF7 | 0.965 | 0.803 | −0.703 |
| 20 | Trichostatin A | 100 nM | MCF7 | 0.963 | 0.787 | −0.716 |
| (b) Vorinostat (up, 157; down, 119) | ||||||
| 1 | Trichostatin A | 1 μM | MCF7 | 1.000 | 0.818 | −0.729 |
| 2 | Trichostatin A | 100 nM | MCF7 | 0.997 | 0.807 | −0.735 |
| 3 | Trichostatin A | 100 nM | MCF7 | 0.992 | 0.817 | −0.716 |
| 4 | Trichostatin A | 1 μM | MCF7 | 0.986 | 0.800 | −0.725 |
| 5 | Trichostatin A | 100 nM | MCF7 | 0.981 | 0.811 | −0.707 |
| 6 | Trichostatin A | 1 μM | MCF7 | 0.980 | 0.799 | −0.716 |
| 7 | Trichostatin A | 100 nM | MCF7 | 0.976 | 0.812 | −0.697 |
| 8 | Trichostatin A | 100 nM | MCF7 | 0.975 | 0.801 | −0.706 |
| 9 | Trichostatin A | 1 μM | MCF7 | 0.970 | 0.778 | −0.721 |
| 10 | Trichostatin A | 100 nM | MCF7 | 0.969 | 0.814 | −0.684 |
| 11 | Trichostatin A | 100 nM | MCF7 | 0.968 | 0.816 | −0.681 |
| 12 | Trichostatin A | 100 nM | MCF7 | 0.967 | 0.767 | −0.729 |
| 13 | Vorinostat | 10 μM | MCF7 | 0.966 | 0.781 | −0.714 |
| 14 | Vorinostat | 10 μM | MCF7 | 0.964 | 0.803 | −0.688 |
| 15 | Vorinostat | 10 μM | MCF7 | 0.963 | 0.791 | −0.698 |
| 16 | Trichostatin A | 1 μM | MCF7 | 0.963 | 0.801 | −0.688 |
| 17 | Vorinostat | 10 μM | MCF7 | 0.963 | 0.789 | −0.700 |
| 18 | Trichostatin A | 1 μM | MCF7 | 0.963 | 0.784 | −0.705 |
| 19 | Trichostatin A | 100 nM | MCF7 | 0.962 | 0.795 | −0.693 |
| 20 | Vorinostat | 10 μM | MCF7 | 0.961 | 0.799 | −0.686 |
The number of up‐ and down‐signatures are shown in parenthesis.
To validate the applicability of our gene signature data to other types of cancer, we carried out an additional study on bortezomib. This agent is used for myeloma treatment. Therefore, we treated a human myeloma RPMI8226 cells with bortezomib and obtained gene expression data. Our clustering analysis revealed that the bortezomib signature data of RPMI8226 cells was clustered together with the proteasome inhibitors' data of HT‐29 cells. Similarly, when RPMI8226 cells were treated with SN‐38 or doxorubicin, the signature data were clustered with DNA damaging agents' data of HT‐29 cells. (Fig. S2). These results confirmed that our signature data are applicable to other cancer cells.
Collectively, these analyses showed that our gene expression data were reliable enough to analyze modes of action of the anticancer drugs.
Application of our database for analysis of the mode of action
Currently, there is no gene expression database open to the public that is specialized in anticancer agents. Based on the obtained gene expression data of anticancer agents, we developed our calculation program (connectivity scoring analysis) to compare gene signatures of test compounds to those of antitumor agents in our database for prediction of their likely modes of action. Basically, we adapted the algorithms of C‐map to our datasets. The C‐map contains data for a large number of compounds, but lacks information for several anticancer agents such as bortezomib. This makes it difficult to simply focus on a comparison of gene expression signatures for test compounds and standard anticancer agents. When we entered the signature gene set of the HDAC inhibitors as a “query” in our established system, we were able to obtain output data containing HDAC inhibitors, trichostatin A, or vorinostat (Table 4). Similarly, when we entered the signatures of the proteasome inhibitors, we obtained output data containing proteasome inhibitors (Table S3). These results indicate that our system can accurately predict the mode of action of an anticancer compound. Using this system, we were able to generate simpler results because our database specifically focuses on anticancer agents.
Table 4.
Results of the connectivity scoring analysis using our database
| Rank | Compound name | Connectivity score | Up score | Down score |
|---|---|---|---|---|
| (a) Trichostatin A (up, 232; down, 181) | ||||
| 1 | Trichostatin A | 1.000 | 0.992 | −0.994 |
| 2 | Vorinostat | 0.992 | 0.987 | −0.984 |
| 3 | Vorinostat | 0.976 | 0.976 | −0.964 |
| 4 | Vorinostat (16 h) | 0.921 | 0.924 | −0.906 |
| 5 | Vorinostat (16 h) | 0.906 | 0.905 | −0.893 |
| 6 | PP242 | 0.672 | 0.693 | −0.642 |
| 7 | Doxorubicin | 0.667 | 0.552 | −0.773 |
| 8 | Etoposide (16 h) | 0.661 | 0.732 | −0.581 |
| 9 | Gemicitabine (16 h) | 0.658 | 0.717 | −0.591 |
| 10 | Neocarzinostatin (16 h) | 0.647 | 0.688 | −0.597 |
| (b) Vorinostat (up, 233; down, 173) | ||||
| 1 | Vorinostat | 1.000 | 0.992 | −0.994 |
| 2 | Trichostatin A | 0.988 | 0.981 | −0.982 |
| 3 | Vorinostat | 0.970 | 0.963 | −0.963 |
| 4 | Vorinostat (16 h) | 0.925 | 0.925 | −0.912 |
| 5 | Vorinostat (16 h) | 0.916 | 0.918 | −0.900 |
| 6 | PP242 | 0.684 | 0.698 | −0.660 |
| 7 | Etoposide (16 h) | 0.675 | 0.745 | −0.597 |
| 8 | Doxorubicin | 0.658 | 0.555 | −0.752 |
| 9 | Neocarzinostatin (16 h) | 0.658 | 0.715 | −0.591 |
| 10 | Gemicitabine (16 h) | 0.654 | 0.726 | −0.573 |
The number of up‐ and down‐signatures are shown in parentheses.
Recently, several reports have shown that ER stress is induced by a wide variety of chemotherapeutic agents.9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 However, to what extent ER stress is involved in the effect of these agents on cancer cells is still unclear. Because the proteasome inhibitors (bortezomib and MG‐132) and the Hsp90 inhibitors (geldanamycin and 17‐AAG), but not other agents, were closely clustered together with the four ER stress‐inducer agents (thapsigargin, 2‐deoxyglucose, tunicamycin, and A23187) (Fig. 1), we examined whether our connectivity scoring analysis could predict these agents as ER stress inducers. We first extracted the ER stress signature gene set, consisting of 58 probe sets whose expressions were all changed by treatment with the four ER stress‐inducing agents. Then we entered the ER signature gene set in our calculation program. As expected, we obtained a result containing the proteasome inhibitors (bortezomib and MG‐132) as “hit compounds” (Table 5). By contrast, the output result did not contain the Hsp90 inhibitors (geldanamycin and 17‐AAG). We examined expression changes of the ER stress genes by these agents in more detail. Our analysis revealed that the proteasome inhibitors strongly induced the ER stress‐related genes, but the Hsp90 inhibitors did not (Fig. 2). This finding is consistent with our connectivity scoring analysis. We further found that the proteasome inhibitors preferentially induced a subset of the ER stress‐related genes (class 1 genes in Fig. 2) and marginally induced the others (class 2 genes). We carried out the mapping of class 1 and 2 genes to the KEGG pathway of “protein processing in endoplasmic reticulum” and found the class 2 genes were mainly involved in the core protein processing machinery in the ER (Fig. S3). By contrast, most of the class 1 genes were not included in the core protein processing machinery in the ER, and many of them were known downstream effectors of main ER stress signaling pathways, including PERK‐eIF2alpha‐ATF4, ATF6, and IRE1‐XBP1.21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31 These results show that the proteasome inhibitors induce atypical ER stress. Thus, our database and its application program constitute an efficient tool for predicting the likely modes of action of anticancer agents.
Table 5.
Results of the connectivity scoring analysis using our database. Endoplasmic reticulum stress‐related genes (58 probe sets) were used as input queries
| Rank | Compound name | Connectivity score | Up score | Down score |
|---|---|---|---|---|
| 1 | Tunicamycin | 1.000 | 0.993 | −0.994 |
| 2 | A23187 | 0.999 | 0.989 | −0.997 |
| 3 | 2‐Deoxyglucose | 0.956 | 0.972 | −0.927 |
| 4 | Thapsigargin | 0.884 | 0.915 | −0.841 |
| 5 | Bortezomib | 0.757 | 0.766 | −0.738 |
| 6 | MG‐132 | 0.757 | 0.771 | −0.733 |
| 7 | Bortezomib | 0.746 | 0.770 | −0.712 |
| 8 | Everolimus | 0.711 | 0.790 | −0.622 |
| 9 | Temsirolimus | 0.704 | 0.734 | −0.664 |
| 10 | Bortezomib (16 h) | 0.700 | 0.729 | −0.661 |
Figure 2.
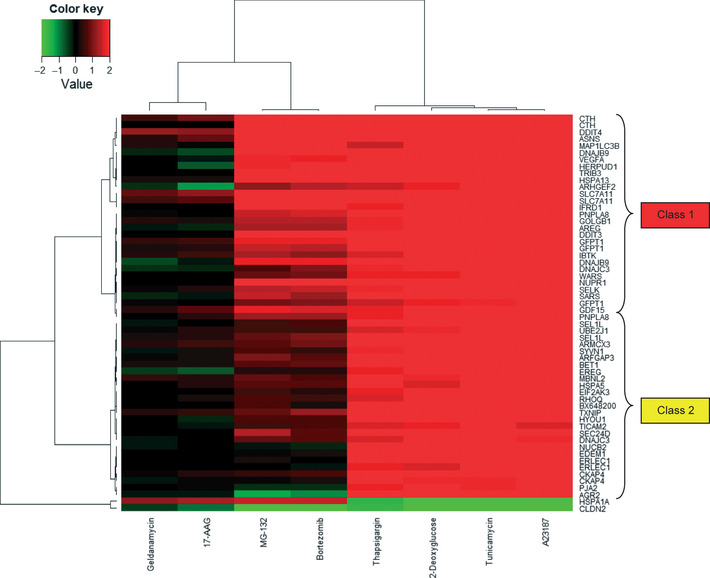
Heatmap of subcluster using endoplasmic reticulum stress‐related genes (58 probe sets), whose expressions were all changed by treatment with the four stress‐inducing agents. The row names are gene symbols of 58 probe sets, which were converted using the NetAffx database, NA32, supplied by Affymetrix. Two main clusters of upregulated genes were named “class 1” and “class 2” genes. 17‐AAG, 17‐N‐allylamino‐17‐demethoxygeldanamycin.
Discussion
Novel free‐access platform for evaluating antitumor agents
Several previous analyses, including the C‐map, have shown that genome‐wide gene expression analysis is effective in predicting modes of action of chemical compounds.3, 4, 32, 33, 34 In the present report, we describe the development of a comprehensive gene expression dataset specializing in the analysis of standard antitumor agents. This open‐to‐the‐public database is available for evaluating the likely mechanisms of action of new anticancer compounds.
In our clustering analysis, drugs with similar mechanisms of action such as genotoxic drugs, proteasome inhibitors, HDAC inhibitors, and ER stress inducers were clustered together (Fig. 1). These results strongly suggest that our gene expression data accurately reflect the mode of action of the agents. The KEGG pathway analysis confirmed that the ER stress‐related gene set was actually induced by the ER stressors (Table S4), which is consistent with our clustering results.
We acquired our gene expression data using human colon cancer HT‐29 cells, whereas the C‐map data mainly consisted of data that were obtained using human breast cancer MCF7 cells or human prostate cancer PC3 cells. It is noteworthy that our signature data of compounds in HT‐29 cells were closely related to those in the C‐map (Table 3). Moreover, we obtained gene expression data in human myeloma RPMI8226 cells and found that the data were closely related with the data obtained in HT‐29 cells (Fig. S2). These results indicate that our signature data could be applicable to data that are obtained in other types of cancer.
To obtain gene expression data that reflect the mode of action of agents, the exposure time of cells to drugs is an important issue. We basically chose a short exposure time (6 h) and, in most of the agents, significant gene expression changes were observed (Table 2) and the data were clustered in a mechanism of action‐dependent manner (Fig. 1). These results indicate that the exposure time would be basically suitable for many agents. By contrast, for some agents that did not show dramatic effects on gene expression in the 6‐h treatment, we tested a longer exposure time (16 h) (Table 1). However, we found that the 16‐h treatment data tended to cluster together, even though the agents had different modes of action (Fig. 1). These observations suggest that longer exposure time might not necessarily be better than shorter exposure time even though gene expression changes are increasingly dramatic. Thus, the drug treatment regime must be carefully chosen for gene signature acquisition and subsequent mechanism analysis.
Mining cryptic linkages and gaps between ER stress and related agents
As reported previously, ER stress is closely related with tumor microenvironment conditions as well as the effect of several antitumor agents.32 Therefore, we included well‐known ER stress‐inducing agents in our compound panel. We found that the tubulin binding agents, proteasome inhibitors, and Hsp90 inhibitors were clustered together with the ER stress inducers in a group different from classical genotoxic agents (Fig. 1). This observation indicates that these drugs could have unique modes of action.
It has been reported that proteasome inhibitors induce ER stress as well as suppressing nuclear factor‐κB activation by interfering with the degradation of I‐κB.12 In our clustering analysis, the protease inhibitors formed a cluster with the ER stress inducers (Fig. 1) and the analysis with our program predicted that the inhibitors induce ER stress (Table S3). Moreover, KEGG pathway analysis revealed that these agents modulate the expression of ER stress‐related genes (indicated as “protein processing in endoplasmic reticulum” in Table S4). These data support the notion that ER stress could play an essential role in the mode of action of proteasome inhibitors.
Our gene signature analysis further revealed that the proteasome inhibitors induce an atypical type of ER stress. In particular, we found that the inhibitors induce a subset of ER stress‐related genes (class 1 genes) while only marginally inducing the other genes (class 2 genes) (Figs. 2, S3). It is still unclear what causes this atypical gene expression pattern. Presumably there is a negative feedback mechanism that selectively suppresses class 2 gene expression. It is noteworthy that the analysis with our program was able to detect the mechanistic difference between the proteasome inhibitors and the well‐known ER stress‐inducing agents. Namely, when we entered the ER signature gene set in our program, the ER stress inducers ranked significantly higher than the proteasome inhibitors (Table 5). Thus, our program could potentially predict detailed mechanisms of action of anticancer drugs.
The Hsp90 inhibitors did not typically induce the ER stress‐related genes (Fig. 2, Table 5), although they were clustered with the ER stress inducers (Fig. 1). To determine the connection between the inhibitors and ER stress, we carried out our connectivity scoring analysis. We found that the signature of the Hsp90 inhibitors weakly correlated with those of some ER stress inducers, such as thapsigargin or tunicamycin (Table S5), suggesting that the inhibitors would not be typical ER stressors but could marginally induce ER stress. It was reported that some Hsp90 inhibitors induce ER stress, but the level of ER stress induction differs among the inhibitors.20 These data suggest that ER stress induction by Hsp90 inhibitors could depend both on compound types and on cell types.
As described above, we developed in‐house R programs for calculating scores for ranking gene expression changes, and for searching statistically significant pathways from the KEGG database. The program for searching pathways enables us to easily produce simple charts for each pathway analyzed and give graphical information of the location of genes in each pathway. The programs and database developed in this study will be made available on our website. Our database included some compounds that are not present in the C‐map database. Therefore, unanticipated characteristics of a novel compound might be obtained by using our database.
In summary, we have developed a publicly available gene expression database of standard anticancer agents as well as some related application programs. Our gene expression database is specialized in antitumor agents, and our datasets include some anticancer agents not contained in other databases, such as C‐map. To establish a more comprehensive database, we plan to add new antitumor agents and update our database. Thus, our database would be suitable for primary characterization of new candidate compounds in comparison with known anticancer agents. We have also acquired data concerning differential sensitivity of human cancer cell lines to anticancer agents and established a “sensitivity‐based” signature database.33, 34, 35, 36 Further trials are planned to develop an integrated database of antitumor agents that include both gene expression‐based and sensitivity‐based signatures. Our public database and related programs will be helpful for evaluating candidate compounds as novel antitumor agents.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- 17‐AAG
17‐N‐allylamino‐17‐demethoxygeldanamycin
- 5‐FU
5‐fluorouracil
- C‐map
Connectivity map
- ER
endoplasmic reticulum
- HDAC
histone deacetylase
- Hsp90
heat shock protein 90
- KEGG
Kyoto Encyclopedia of Genes and Genomes
Supporting information
Fig. S1. Dose–response curves of 35 anticancer drugs.
Fig. S2. Hierarchical cluster analysis including RPMI8226 cell line data.
Fig. S3. KEGG pathway of “protein processing in endoplasmic reticulum”.
Table S1. Upregulated and downregulated probe sets in each cluster of compounds.
Table S2. Results of the Connectivity map for proteasome inhibitors.
Table S3. Results of the connectivity scoring analysis for proteasome inhibitors.
Table S4. Results of pathway analysis with the KEGG database.
Table S5. Results of the connectivity scoring analysis for Hsp90 inhibitors.
Doc S1. Supplementary methods.
Acknowledgments
The present study is supported by the Scientific Support Programs for Cancer Research project/Screening Committee of Anticancer Drugs (SCADS), Grant‐in‐Aid for Scientific Research on Innovative Areas from The Ministry of Education, Culture, Sports, Science and Technology, Japan.
(Cancer Sci, doi: 10.1111/cas.12071, 2012)
References
- 1. Sawyers C. Targeted cancer therapy. Nature 2004; 432: 294–7. [DOI] [PubMed] [Google Scholar]
- 2. Tsuruo T, Naito M, Tomida A et al Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci 2003; 94: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lamb J, Crawford ED, Peck D et al The connectivity map: using gene‐expression signatures to connect small molecules, genes, and disease. Science 2006; 313: 1929–35. [DOI] [PubMed] [Google Scholar]
- 4. Lamb J. The connectivity map: a new tool for biomedical research. Nat Rev Cancer 2007; 7: 54–60. [DOI] [PubMed] [Google Scholar]
- 5. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000; 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benjamini Y, Hochberg Y. Controling the false discovery rate: a pratical and powerful approach to multiple testing. J Roy Statist Soc B 1995; 57: 289–300. [Google Scholar]
- 7. Storey JD. The positive false discovery rate: a Bayesian interpretation and the q‐value. Ann Statist 2003; 31: 2013–35. [Google Scholar]
- 8. Storey JD, Tibshirani R. Statistical significance for genome‐wide experiments. Proc Natl Acad Sci U S A 2003; 100: 9440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS‐341 induces apoptosis through induction of endoplasmic reticulum stress‐reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol 2004; 24: 9695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nawrocki ST, Carew JS, Dunner K Jr et al Bortezomib inhibits PKR‐like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res 2005; 65: 11510–9. [DOI] [PubMed] [Google Scholar]
- 11. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006; 107: 4907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther 2011; 10: 2034–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davenport EL, Moore HE, Dunlop AS et al Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 2007; 110: 2641–9. [DOI] [PubMed] [Google Scholar]
- 14. De Raedt T, Walton Z, Yecies JL et al Exploiting cancer cell vulnerabilities to develop a combination therapy for ras‐driven tumors. Cancer Cell 2011; 20: 400–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mhaidat NM, Thorne R, Zhang XD, Hersey P. Involvement of endoplasmic reticulum stress in Docetaxel‐induced JNK‐dependent apoptosis of human melanoma. Apoptosis 2008; 13: 1505–12. [DOI] [PubMed] [Google Scholar]
- 16. Liao PC, Tan SK, Lieu CH, Jung HK. Involvement of endoplasmic reticulum in paclitaxel‐induced apoptosis. J Cell Biochem 2008; 104: 1509–23. [DOI] [PubMed] [Google Scholar]
- 17. Mozos A, Roué G, López‐Guillermo A et al The expression of the endoplasmic reticulum stress sensor BiP/GRP78 predicts response to chemotherapy and determines the efficacy of proteasome inhibitors in diffuse large b‐cell lymphoma. Am J Pathol 2011; 179: 2601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus‐independent apoptotic signaling. J Biol Chem 2003; 278: 9100–6. [DOI] [PubMed] [Google Scholar]
- 19. Baumeister P, Dong D, Fu Y, Lee AS. Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor‐induced apoptosis. Mol Cancer Ther 2009; 8: 1086–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol 2002; 22: 8506–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Whitney ML, Jefferson LS, Kimball SR. ATF4 is necessary and sufficient for ER stress‐induced upregulation of REDD1 expression. Biochem Biophys Res Commun 2009; 379: 451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ye J, Kumanova M, Hart LS et al The GCN2‐ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 2010; 29: 2082–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rzymski T, Milani M, Pike L et al Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010; 29: 4424–35. [DOI] [PubMed] [Google Scholar]
- 24. Lee AH, Iwakoshi NN, Glimcher LH. XBP‐1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003; 23: 7448–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma Y, Hendershot LM. Herp is dually regulated by both the endoplasmic reticulum stress‐specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J Biol Chem 2004; 279: 13792–9. [DOI] [PubMed] [Google Scholar]
- 26. Kokame K, Agarwala KL, Kato H, Miyata T. Herp, a new ubiquitin‐like membrane protein induced by endoplasmic reticulum stress. J Biol Chem 2000; 275: 32846–53. [DOI] [PubMed] [Google Scholar]
- 27. Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress‐inducible gene, is induced via ATF4‐CHOP pathway and is involved in cell death. EMBO J 2005; 24: 1243–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salazar M, Carracedo A, Salanueva IJ et al TRB3 links ER stress to autophagy in cannabinoid anti‐tumoral action. Autophagy 2009; 5: 1048–9. [DOI] [PubMed] [Google Scholar]
- 29. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004; 11: 381–9. [DOI] [PubMed] [Google Scholar]
- 30. Shchedrina VA, Everley RA, Zhang Y et al Selenoprotein K binds multiprotein complexes and is involved in the regulation of endoplasmic reticulum homeostasis. J Biol Chem 2011; 286: 42937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Du S, Zhou J, Jia Y, Huang K. SelK is a novel ER stress‐regulated protein and protects HepG2 cells from ER stress agent‐induced apoptosis. Arch Biochem Biophys 2010; 502: 137–43. [DOI] [PubMed] [Google Scholar]
- 32. Saito S, Furuno A, Sakurai J et al Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res 2009; 69: 4225–34. [DOI] [PubMed] [Google Scholar]
- 33. Dan S, Tsunoda T, Kitahara O et al An integrated database of chemosensitivity to 55 anticancer drugs and gene expression profiles of 39 human cancer cell lines. Cancer Res 2002; 62: 1139–47. [PubMed] [Google Scholar]
- 34. Dan S, Okamura M, Seki M et al Correlating phosphatidylinositol 3‐kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations. Cancer Res 2010; 70: 4982–94. [DOI] [PubMed] [Google Scholar]
- 35. Yaguchi S, Fukui Y, Koshimizu I et al Antitumor activity of ZSTK474, a new phosphatidylinositol 3‐kinase inhibitor. J Natl Cancer Inst 2006; 98: 545–56. [DOI] [PubMed] [Google Scholar]
- 36. Mashima T, Oh‐hara T, Sato S et al p53‐defective tumors with a functional apoptosome‐mediated pathway: a new therapeutic target. J Natl Cancer Inst 2005; 97: 765–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Dose–response curves of 35 anticancer drugs.
Fig. S2. Hierarchical cluster analysis including RPMI8226 cell line data.
Fig. S3. KEGG pathway of “protein processing in endoplasmic reticulum”.
Table S1. Upregulated and downregulated probe sets in each cluster of compounds.
Table S2. Results of the Connectivity map for proteasome inhibitors.
Table S3. Results of the connectivity scoring analysis for proteasome inhibitors.
Table S4. Results of pathway analysis with the KEGG database.
Table S5. Results of the connectivity scoring analysis for Hsp90 inhibitors.
Doc S1. Supplementary methods.