

Abstract
Since membrane type‐1 matrix metalloproteinase (MT1‐MMP) plays pivotal roles in tumor progression and metastasis and holds great promise as an early biomarker for malignant tumors, a method of evaluating MT1‐MMP expression levels would be valuable for molecular biological and clinical studies. Although we have previously developed a 99m Tc‐labeled anti‐MT1‐MMP monoclonal IgG (99m Tc‐MT1‐mAb) as an MT1‐MMP imaging probe by nuclear medical techniques for this purpose, slow pharmacokinetics were a problem due to its large molecular size. Thus, in this study, our aim was to develop miniaturized antibodies, a single chain antibody fragment (MT1‐scFv) and a dimer of two molecules of scFv (MT1‐diabody), as the basic structures of MT1‐MMP imaging probes followed by in vitro and in vivo evaluation with an 111In radiolabel. Phage display screening successfully provided MT1‐scFv and MT1‐diabody, which had sufficiently high affinity for MT1‐MMP (K D = 29.8 and 17.1 nM). Both 111In labeled miniaturized antibodies showed higher uptake in MT1‐MMP expressing HT1080 cells than in non‐expressing MCF7 cells. An in vivo biodistribution study showed rapid pharmacokinetics for both probes, which exhibited >20‐fold higher tumor to blood radioactivity ratios (T/B ratio), an index for in vivo imaging, than 99m Tc‐MT1‐mAb 6 h post‐administration, and significantly higher tumor accumulation in HT1080 than MCF7 cells. SPECT images showed heterogeneous distribution and ex vivo autoradiographic analysis revealed that the radioactivity distribution profiles in tumors corresponded to MT1‐MMP‐positive areas. These findings suggest that the newly developed miniaturized antibodies are promising probes for detection of MT1‐MMP in cancer cells.
Tumor metastasis is the most frequent cause of death for cancer patients. In order to metastasize, tumor cells must acquire the ability to break through the basement membrane and invade dense networks of interstitial ECM proteins.1 Matrix metalloproteinases (MMPs) are a family of enzymes responsible for degrading the various ECM components. While most MMPs are secreted as soluble zymogens, members of the subfamily of membrane‐type MMPs (MT‐MMPs) anchored to the cell membrane are suited for pericellular proteolysis.2, 3 MT1‐MMP is the major pericellular protease involved in processing triple helical collagen type I.4 In addition, MT1‐MMP activates MMP zymogens such as proMMP‐2 and proMMP‐13 that have significant involvement in tumor cell invasion and metastasis.5, 6 As MT1‐MMP has a close relationship with tumor malignancy and holds great promise as an early biomarker of malignant tumors,7, 8 in vivo monitoring and/or quantitation of MT1‐MMP expression could be valuable tools for molecular biological and clinical studies.
Recently, in the course of focusing on nuclear medical techniques for noninvasive quantitative evaluation of biological molecules deep within the body, we developed a 99mTc‐labeled anti‐MT1‐MMP monoclonal IgG (99mTc‐MT1‐mAb) as a radiolabeled probe for nuclear medical imaging of MT1‐MMP. Although this probe accumulated in the tumors of rodent models with a low effective dose,9 blood clearance was slow and the tumor to blood (T/B) ratio, an indicator of radiotracer availability for in vivo imaging, remained low up to 48 h post‐injection due to its high molecular weight (approximately 150 kDa), which led to a high systemic background radioactivity that prevented clear in vivo MT1‐MMP imaging. Improvement of this imaging probe was necessary for further applications.
Thus, we planned to develop two miniaturized structural variants (scFv, diabody) of the anti‐MT1‐MMP antibody to improve the kinetics for cancer imaging.10 scFv, single‐chain Fv, is a 30 kDa molecule composed of a variable region of the light chain (VL) and a variable region of the heavy chain (VH) joined via a peptide spacer sequence. As a monovalent fragment, scFv was expected to show extremely rapid kinetics due to its small size. On the other hand, the diabody, a dimer of scFv, was expected to show high sensitivity as an in vivo imaging agent11 due to its bivalency. Initially, scFv (MT1‐scFv) and diabody (MT1‐diabody) with affinity for MT1‐MMP were identified using phage display technology. Next, 111In‐labeled MT1‐scFv (111In‐MT1‐scFv) and MT1‐diabody (111In‐MT1‐diabody) were prepared and evaluated for MT1‐MMP imaging of cancers.
Materials and Methods
Preparation of MT1‐scFv and MT1‐diabody
We described the construction of phage display library in Data S1. The resulting library was subjected to five rounds of panning. After panning, ELISA assays were performed to select promising phages. Promising scFv (MT1‐scFv) and diabody (MT1‐diabody) were expressed (detail in Data S1) and purified by HisTALON Gravity Columns (Takara Bio, Tokyo, Japan), and then analyzed by 5–20% SDS‐PAGE. We also prepared anti‐polychlorobiphenyl scFv (NC‐scFv) as a negative control.
Radiolabeling
MT1‐scFv, MT1‐diabody, and NC‐scFv in 50 mM NaHCO3 (pH 8.6) were mixed with 20 times equivalents of p‐SCN‐Bn‐DTPA (10 mg/mL; Macrocyclics, Dallas, TX, USA) for 1 h at room temperature (rt). Unreacted p‐SCN‐Bn‐DTPA was removed by size exclusion with an Amicon Ultra‐4 Centrifugal Filter Unit (10k) (Millipore, Billerica, MA, USA) concomitant with solvent exchange in 0.1 M acetate buffer (pH 6.0). Next, 111InCl3 (37 kBq/μg protein) was added and incubated for 1 h at rt. 111In‐labelled MT1‐scFv, MT1‐diabody, and NC‐scFv (111In‐MT1‐scFv, 111In‐MT1‐diabody and 111In‐NC‐scFv) were purified into molecular weight (MW)‐dependent fractions (250 μL × 60 fractions) by size exclusion chromatography on Superdex 75 10/300 GL (GE Healthcare, Tokyo, Japan) eluting with PBS(−) followed by radioactivity counting with a NaI well‐type scintillation counter (1470 WIZARD; PerkinElmer, Yokohama, Japan).
Immunoreactivity analysis
Surface plasmon resonance was analyzed with the ProteOn XPR36 system (Bio‐rad Laboratories, Tokyo, Japan). MT1‐MMP protein (ag6062; Proteintech Group, Chicago, IL, USA) or hinge region peptide conjugated with BSA (described in detail in Data S1) was coupled onto a GLM chip. MT1‐scFv, MT1‐diabody (unconjugated or DTPA‐conjugated) and NC‐scFv solutions, prepared as threefold serial dilutions (0, 7.7–625 nM), were injected onto the chip. The resulting sensorgrams were fitted by the simplest 1:1 interaction model using ProteOn analysis software to obtain the corresponding association and dissociation rate constants. Other determinations (antigen‐immobilized ELISA, cell ELISA, and indirect competitive ELISA) are described in Data S1.
Cellular uptake study
HT1080 human fibrosarcoma cells and MCF7 human breast adenocarcinoma cells, supplied by ATCC, were cultured in DMEM (Nissui Pharmaceutical, Tokyo, Japan) with 10% FBS at 37°C in a humidified atmosphere containing 5% CO2. HT1080 or MCF7 (5 × 105 cells) in FBS‐free‐DMEM were added to eppendorf tubes and were incubated with 111In‐MT1‐scFv, 111In‐MT1‐diabody or 111In‐NC‐scFv. After 1 or 3 h, cells were washed with PBS(−), lysed with 0.2 M NaOH, and then counted for radioactivity. Protein quantitation was performed by the BCA protein assay (Thermo Fisher Scientific, Yokohama, Japan).
Preparation of tumor‐bearing mice
Female Balb/c nu‐nu mice (5 weeks old; Japan SLC, Hamamatsu, Japan) and female SCID mice (5 weeks old; CLEA, Tokyo, Japan) were housed under a 12/12 h light/dark cycle and were given free access to food and water. Animal experiments were conducted in accordance with institutional guidelines and were approved by the Kyoto University Animal Care Committee. HT1080 cells were suspended in PBS(−) followed by subcutaneous inoculation into the right hind legs of Balb/c and SCID mice (5 × 106 cells/100 μL) and MCF7 cells (2 × 107 cells/100 μL) were injected into the left hind legs.
In vivo study
Animals were divided into groups (n = 3–4) for time points with approximately equal distribution of tumor sizes on the day before the study. Animals were fasted for 6 h before administration of the radiopharmaceutical. At time points 15 min, 1, 3, 6, and 24 h after intravenous administration of 111In‐MT1‐scFv or 111In‐MT1‐diabody (18.5 kBq/100 μL saline), mice were killed. The blood, heart, lung, liver, kidney, stomach, intestine, spleen, pancreas, muscle, and tumor tissues were excised, weighed, and counted for radioactivity. HT1080 and MCF7 cells co‐implantation model SCID mice were killed at 1 or 24 h for 111In‐MT1‐scFv and 3 or 24 h for 111In‐MT1‐diabody (18.5 kBq/100 μL saline), and tumor tissues were excised.
Ex vivo autoradiography and immunohistochemistry
Tumor‐bearing mice were killed 3 h after intravenous administration of 111In‐MT1‐scFv or 111In‐NC‐scFv and 24 h of the 111In‐MT1‐diabody (740 kBq/mouse). The tumors were removed and immediately frozen. After freezing, 10‐μm thick sections of the tumor were prepared with a cryomicrotome (CM1900; Leica Microsystems, Tokyo, Japan) and exposed to imaging plates (BAS‐SR; Fuji Photo Film, Tokyo, Japan) for 10 days. Autoradiograms of these sections were obtained with a BAS5000 scanner (Fuji Photo Film). Adjacent sections of the autoradiographic study were subjected to immunostaining against MT1‐MMP (described in details in Data S1).
SPECT/CT imaging study
111In‐MT1‐scFv and 111In‐MT1‐diabody (13.0 MBq) were injected into HT1080 tumor‐bearing mice. The mice were anesthetized by isoflurane and SPECT and CT images were obtained using the U‐SPECT‐II system (MILabs, Utrecht, the Netherlands) with 1.0‐mm pinhole collimators (SPECT conditions; 30 min × 1 frame, CT conditions; accurate full angle mode in 65 kV/615 μA) 3 h after injection of the 111In‐MT1‐scFv or 111In‐MT1‐diabody. SPECT images were reconstructed by the OSEM method (one subset, 40 iterations) with a 1.6‐mm Gaussian filter with 111In‐MT1‐scFv and a 0.9 mm filter for 111In‐MT1‐diabody.
Statistics
Data are presented as means ± SD. The Mann–Whitney U‐test was used to evaluate the significance of differences in the cellular uptake and in vivo studies. Differences at the 95% confidence level (P < 0.05) were considered significant.
Results
In vitro characterization
Sodium dodecyl sulfate‐PAGE analysis showed the MT1‐scFv and MT1‐diabody as single bands (Fig. 1a). Enzyme linked immunosorbent assay revealed that both modified antibodies (MT1‐scFv and MT1‐diabody) had affinity and specificity for the antigen (MT1‐MMP hinge region) peptide as well as affinity for MT1‐MMP expressing cancer cells (Table S1). ProteOn revealed K D dissociation constants for MT1‐scFv and MT1‐diabody of 29.8 ± 4.1 and 17.1 ± 4.0 nM, respectively, against immobilized MT1‐MMP protein. MT1‐scFv and MT1‐diabody also showed high affinity for the hinge region peptide of MT1‐MMP (TKMPPQPRTTSRPSVPDKPKN) (K D = 51.1 ± 30.3, 0.27 ± 0.21 nM, respectively), but did not bind to a scrambled peptide (RKPRQPTSPTKPMVSNPTPDK). After conjugation with the bifunctional chelating agent (p‐SCN‐Bn‐DTPA), the K D of MT1‐scFv and MT1‐diabody were determined to be 38.5 ± 1.3 and 26.5 ± 2.9 nM, respectively. NC‐scFv did not show affinity for MT1‐MMP.
Figure 1.
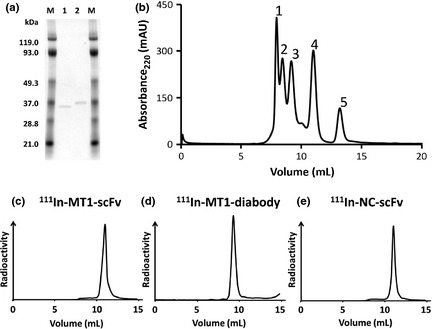
(a) Sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) of MT1‐scFv (1) and MT1‐diabody (2). (b) Chromatograms of MW‐marker; 1 (7.93 mL), glutamate dehydrogenase 290 kDa: 2 (8.43 mL), lactate dehydrogenase 142 kDa: 3 (9.15 mL), enolase 67 kDa: 4 (10.98 mL), myokinase 32 kDa: 5 (13.18 mL), cytochrome C 12.4 kDa. (c) Chromatogram of 111In‐MT1‐scFv (11.0 mL). (d) Chromatogram of 111In‐MT1‐diabody (9.25 mL). (e) Chromatogram of 111In‐NC‐scFv (11.0 mL).
Radiolabeling
The MT1‐scFv, MT1‐diabody and NC‐scFv were efficiently labeled with 111In in radiochemical yields and radiochemical purities evaluated as >95%. From size‐exclusion chromatography analysis using a Superdex 75 10/300 GL, retention volumes (mL) of the major peaks of 111In‐MT1‐scFv and 111In‐NC‐scFv (11.0 mL) were almost exactly corresponded to 32 kDa myokinase (10.98 mL) (Fig. 1b,c,e). 111In‐MT1‐diabody (9.25 mL) almost exactly corresponded to 67 kDa enolase (9.15 mL) (Fig. 1b,d).
Cellular uptake study
MT1‐MMP expressing HT1080 and MT1‐MMP non‐expressing MCF7 cells were incubated with 111In‐MT1‐scFv, 111In‐MT1‐diabody and 111In‐NC‐scFv followed by radioactivity measurement of the cells (Fig. 2). Radioactivities of HT1080 cells treated with 111In‐MT1‐scFv and 111In‐MT1‐diabody were significantly higher than those of MCF7 cells. HT1080 cells treated with 111In‐NC‐scFv showed lower radioactivity, not significantly different from MCF7 cells.
Figure 2.
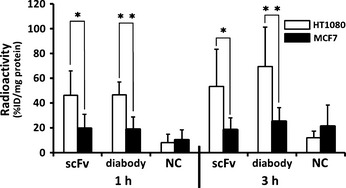
The radioactivity of HT1080 cells and MCF‐7 cells after incubation for 1 and 3 h with 111In‐MT1‐scFv (scFv), 111In‐MT1‐diabody (diabody) and 111In‐NC‐scFv (NC). Data are expressed as radioactivity per cell protein (mg) (mean ± standard deviation [SD]). Comparison between HT1080 and MCF‐7 cell groups was performed with the Mann–Whitney U‐test (*P < 0.005, **P < 0.0001 versus MCF‐7).
In vivo study
Biodistributions of 111In‐MT1‐scFv and 111In‐MT1‐diabody in tumor‐bearing mice are shown in Tables 1 and 2, respectively. Both probes showed rapid blood clearance with 0.75 ± 0.14 and 0.10 ± 0.01%ID/g at 1 and 24 h after injection of 111In‐MT1‐scFv, and 3.26 ± 0.37 and 0.09 ± 0.03%ID/g at 1 and 24 h after injection of 111In‐MT1‐diabody. Radioactivities for T/B ratios of 111In‐MT1‐scFv and 111In‐MT1‐diabody are shown in Figure 3. The T/B ratio 1 h post‐administration of 111In‐MT1‐scFv was 2.23 ± 0.38, which was greater than fivefold higher than 111In‐MT1‐diabody. The T/B ratio of the 111In‐MT1‐diabody increased up to 6 h post‐administration before reaching a plateau (6.15 ± 1.92 at 6 h, 5.86 ± 3.88 at 24 h). Tumor accumulation of both probes with HT1080 and MCF7 tumor cells in co‐implantation model SCID mice are shown in Figure 4. 111In‐MT1‐scFv gave a significantly higher accumulation with HT1080 (2.60 ± 0.19 at 1 h, 2.78 ± 0.24 at 24 h) than MCF7 cells (2.20 ± 0.15 at 1 h, 2.19 ± 0.43 at 24 h). The 111In‐MT1‐diabody had also significantly higher accumulation with HT1080 (2.25 ± 0.39 at 3 h, 2.06 ± 0.12 at 24 h) than MCF7 cells (1.80 ± 0.12 at 3 h, 1.79 ± 0.15 at 24 h).
Table 1.
Biodistribution of radioactivity after injection of 111In‐MT1‐scFv in tumor‐bearing mice†
| Time after injection (h) | |||||
|---|---|---|---|---|---|
| 0.25 | 1 | 3 | 6 | 24 | |
| Blood | 3.36 ± 0.53 | 0.75 ± 0.14 | 0.38 ± 0.07 | 0.26 ± 0.03 | 0.10 ± 0.01 |
| Heart | 0.82 ± 0.36 | 1.33 ± 0.27 | 0.71 ± 0.19 | 0.70 ± 0.07 | 0.49 ± 0.08 |
| Lung | 6.36 ± 1.58 | 1.72 ± 0.32 | 0.80 ± 0.26 | 0.62 ± 0.26 | 0.41 ± 0.17 |
| Liver | 25.87 ± 3.03 | 23.41 ± 2.38 | 21.24 ± 7.83 | 23.29 ± 0.91 | 14.80 ± 1.38 |
| Kidney | 249.93 ± 15.08 | 237.53 ± 23.96 | 220.34 ± 48.95 | 225.90 ± 8.71 | 182.00 ± 8.53 |
| Stomach‡ | 0.34 ± 0.07 | 0.20 ± 0.03 | 0.14 ± 0.02 | 0.19 ± 0.00 | 0.14 ± 0.04 |
| Intestine | 1.51 ± 0.11 | 0.85 ± 0.26 | 1.59 ± 0.62 | 1.48 ± 0.22 | 0.73 ± 0.11 |
| Pancreas | 1.66 ± 0.45 | 1.19 ± 0.22 | 0.85 ± 0.19 | 0.90 ± 0.03 | 0.64 ± 0.17 |
| Spleen | 16.90 ± 2.66 | 7.87 ± 1.71 | 6.00 ± 2.76 | 6.14 ± 1.16 | 5.77 ± 0.37 |
| Muscle | 0.96 ± 0.59 | 0.71 ± 0.04 | 0.65 ± 0.12 | 0.58 ± 0.19 | 0.45 ± 0.08 |
| Tumor | 0.71 ± 0.50 | 1.68 ± 0.51 | 1.35 ± 0.20 | 1.14 ± 0.22 | 0.64 ± 0.17 |
†Tissue radioactivity is expressed as % injected dose per gram. ‡Expressed as % injected dose.
Table 2.
Biodistribution of radioactivity after injection of 111In‐MT1‐diabody in tumor bearing mice†
| Time after injection (h) | |||||
|---|---|---|---|---|---|
| 0.25 | 1 | 3 | 6 | 24 | |
| Blood | 7.30 ± 1.18 | 3.26 ± 0.37 | 1.64 ± 0.12 | 0.21 ± 0.04 | 0.09 ± 0.03 |
| Heart | 1.78 ± 0.42 | 1.80 ± 0.32 | 1.34 ± 0.15 | 0.82 ± 0.06 | 0.59 ± 0.13 |
| Lung | 4.34 ± 0.55 | 2.04 ± 0.34 | 1.51 ± 0.24 | 0.40 ± 0.2 | 0.61 ± 0.10 |
| Liver | 47.00 ± 4.61 | 39.68 ± 3.56 | 40.63 ± 3.13 | 26.75 ± 1.64 | 15.96 ± 3.03 |
| Kidney | 65.98 ± 11.65 | 108.2 ± 15.05 | 131.98 ± 8.44 | 97.98 ± 10.66 | 241.74 ± 28.36 |
| Stomach‡ | 0.71 ± 0.06 | 1.27 ± 0.32 | 0.98 ± 0.33 | 1.29 ± 1.03 | 0.49 ± 0.22 |
| Intestine | 0.56 ± 0.12 | 1.14 ± 0.17 | 2.21 ± 0.13 | 2.14 ± 0.91 | 0.85 ± 0.17 |
| Pancreas | 0.52 ± 0.08 | 0.47 ± 0.07 | 0.56 ± 0.12 | 0.57 ± 0.07 | 0.83 ± 0.06 |
| Spleen | 13.03 ± 2.43 | 8.77 ± 1.47 | 8.34 ± 0.65 | 6.31 ± 0.36 | 6.50 ± 1.33 |
| Muscle | 0.29 ± 0.12 | 0.35 ± 0.08 | 0.29 ± 0.09 | 0.47 ± 0.53 | 0.94 ± 0.58 |
| Tumor | 1.14 ± 0.18 | 1.35 ± 0.33 | 1.52 ± 0.13 | 1.37 ± 0.73 | 0.47 ± 0.24 |
†Tissue radioactivity is expressed as % injected dose per gram. ‡Expressed as % injected dose.
Figure 3.
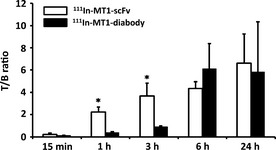
Tumor to blood radioactivity ratio (T/B) of 111In‐MT1‐scFv and 111In‐MT1‐diabody. Comparisons between 111In‐MT1‐scFv and 111In‐MT1‐diabody were performed with the Mann–Whitney U‐test (*P < 0.05 versus 111In‐MT1‐diabody).
Figure 4.
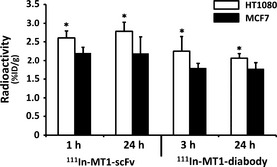
Tumor accumulations of 111In‐MT1‐scFv (1, 24 h) and 111In‐MT1‐diabody (3, 24 h) in HT1080 and MCF7 tumors. Comparisons of accumulations between HT1080 and MCF7 were performed with the Mann–Whitney U‐test (*P < 0.05 versus MCF7).
SPECT/CT imaging study
SPECT images obtained 3 h after administration of the 111In‐MT1‐scFv and 111In‐MT1‐diabody showed heterogeneous distribution of radioactivity. Transverse, sagittal and coronal images are shown in Figure 5.
Figure 5.
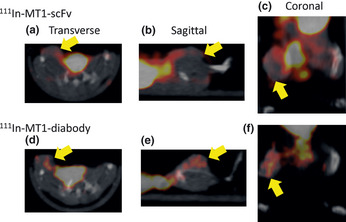
Transverse, sagittal and coronal images of tumor‐bearing mice 3 h after administration with 111In‐MT1‐scFv (a–c) and 111In‐MT1‐diabody (d–f). Yellow arrows indicate tumors.
Ex vivo autoradiography and immunohistochemistry
Autoradiograms showed that the distributions of 111In‐MT1‐scFv, 111In‐MT1‐diabody and 111In‐NC‐scFv in tumors were heterogeneous (Fig. 6b,d,f). Immunohistochemistry indicated the presence of MT1‐MMP areas in tumors (Fig. 6a,c,e). The accumulation profiles of 111In‐MT1‐scFv and 111In‐MT1‐diabody tended to correspond to MT1‐MMP positive areas, but 111In‐NC‐scFv didn't.
Figure 6.
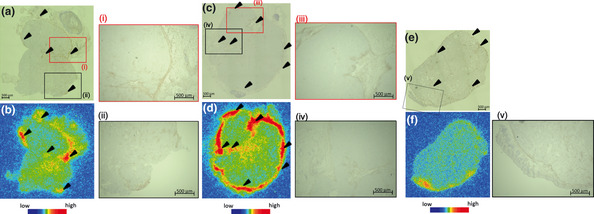
Representative images of MT1‐MMP immunostainings and autoradiograms of 111In‐MT1‐scFv (a,b), 111In‐MT1‐diabody (c,d), and 111In‐NC‐scFv (e,f). Black arrowheads indicate areas of MT1‐MMP expression.
Discussion
The results of affinity analysis indicated that both probes had sufficiently high affinity for MT1‐MMP imaging and that both were successfully radiolabeled with 111In. Biodistribution studies in tumor‐bearing mice demonstrated that 111In‐MT1‐scFv and 111In‐MT1‐diabody had rapid blood clearance. The T/B ratio, an important index for in vivo imaging, of the 111In‐MT1‐scFv and 111In‐MT1‐diabody was about 20‐fold higher than the previously reported whole body antibody probe (99mTc‐MT1‐mAb9) 6 h post‐administration, suggesting that the newly developed 111In labeled miniaturized antibodies have potential for clinical evaluation. In addition, the rapid pharmacokinetics of these agents may allow adoption of 99mTc, the most useful generator‐produced radioisotope with a short (6 h) half‐life, as the signal moiety instead of 111In.
MT1‐MMP comprises several domains that include propeptide, catalytic, hinge region, hemopexin, and transmembrane domains and a cytoplasmic tail.12 Recently, several research groups have revealed non‐proteolytic roles for MT1‐MMP in tumor malignancy.13, 14 Therefore, in this study, we proposed to evaluate not proteolytic activity, but the expression level of MT1‐MMP, in which case the extracellular domains of MT1‐MMP could be targeted. However, the propeptide and the catalytic domain are eliminated from the cell membrane by an autocatalytic processing.15, 16 The hemopexin domain is involved in various interactions.17, 18 Therefore, other proteins may interfere with binding to the hemopexin domain. Thus, we decided to target the hinge region of MT1‐MMP. Promising clones (including MT1‐scFv and MT1‐diabody) were first obtained using phage‐display technology. Next, expressed and purified clones were evaluated for their affinity to immobilized antigen by ELISA. Enzyme linked immunosorbent assay revealed the MT1‐scFv and MT1‐diabody had higher affinity for MT1‐MMP hinge region than other clones. ProteOn analysis revealed the MT1‐diabody had better affinity than MT1‐scFv, which may result from its bivalency. Furthermore, ProteOn revealed both probes specifically recognized the hinge region sequence, which does not share homology with other MMPs. Thus, MT1‐scFv and MT1‐diabody are believed to be free from cross‐reactivity with other MMPs. Radiolabeling with 111In was successfully performed with high radiochemical yield and radiochemical purity (>95%), which meant the conjugates could be used for in vitro and in vivo experiments without additional purification. Furthermore, ProteOn revealed the affinity of both probes for MT1‐MMP was not affected by conjugation with p‐SCN‐Bn‐DTPA.
To evaluate immunoreactivity for MT1‐MMP on cancer cells, a cellular uptake study was performed using HT1080 as MT1‐MMP positive cells and MCF7 cells as MT1‐MMP negative cells in a Western blot analysis.19 We used 111In‐MT1‐scFv and 111In‐MT1‐diabody, in addition, 111In‐NC‐scFv was used to evaluate nonspecific uptake into tumor cells. Although 111In‐NC‐scFv showed low accumulation in both HT1080 and MCF7 cells, the 111In‐MT1‐scFv and 111In‐MT1‐diabody showed significantly high accumulation in HT1080 indicating both had affinity for MT1‐MMP in cells and retained their immunoreactivity for MT1‐MMP after conjugation with p‐SCN‐Bn‐DTPA and radiolabeling with 111In.
Biodistribution studies of 111In‐MT1‐scFv and 111In‐MT1‐diabody in tumor‐bearing mice indicated their rapid blood clearance, especially in the 1–3 h period after injection, with faster blood clearance of 111In‐MT1‐scFv than the 111In‐diabody because of its smaller size. High radioactivity in the kidneys indicated renal excretion of the probes. The T/B ratio 1 h after injection of 111In‐MT1‐scFv was more than 5‐ and 30‐fold higher than the 111In‐MT1‐diabody and our previously reported 99mTc‐MT1‐mAb, respectively, indicating that the scFv format would be useful for imaging cancers shortly after being injected. The T/B ratios of 111In‐MT1‐scFv and 111In‐MT1‐diabody after 6 h were similar; however, tumor accumulation of 111In‐MT1‐diabody was higher than 111In‐MT1‐scFv and had lower accumulation in the kidneys, suggesting 111In‐MT1‐diabody would be more suitable for high sensitivity cancer imaging and/or a lower injection dose than 111In‐MT1‐scFv. Thus, both agents could shorten the time for cancer imaging compared to the previous mAb imaging agent.
The SPECT/CT imaging study indicated both probes showed heterogeneous distribution in tumors. We found that the 111In‐MT1‐diabody gave clearer tumor imaging than the 111In‐MT1‐scFv due to higher accumulation in tumors. In the coronal image of 111In‐MT1‐scFv, we found relatively high radioactivity, which was not derived from tissues around the tumor. We believe this radioactivity was caused by excreted urine.
Furthermore, we confirmed probe accumulation into HT1080 and MCF7 tumor cells in co‐implantation model SCID mice. Both 111In‐MT1‐scFv and 111In‐MT1‐diabody showed significantly higher accumulation into HT1080 cells. Finally, ex vivo autoradiography and immunohistochemistry were examined to confirm the accumulation of 111In‐MT1‐scFv and 111In‐MT1‐diabody in HT1080 tumors according to MT1‐MMP expression. Consequently, these radioactivity distribution profiles corresponded to MT1‐MMP‐positive areas. These data indicate both probes maintained an affinity for MT1‐MMP in vivo, and the accumulation in tumors did reflect MT1‐MMP expression.
Many research groups have been developing in vivo imaging probes for MT1‐MMP. In fact, some fluorogenic probes activated by or binding with MT1‐MMP have successfully imaged tumors.20, 21, 22 However, these probes were insufficient for quantitative analysis of MT1‐MMP in vivo. In this regard, radiolabeled probes were expected to achieve more accurate quantitation; therefore, several radiolabeled probes for MT1‐MMP have been developed including the 99mTc‐MT1‐mAb9 and 123I‐labelled TIMP‐2.23 However, 99mTc‐MT1‐mAb displayed a relatively low T/B ratio because of the large molecular size of the mAb, and tumor accumulation of 123I‐labelled TIMP‐2 was inhibited by endogenous TIMP‐2. Furthermore, TIMP‐2 also had affinity for MMP‐2. This broad‐spectrum among MMPs is problematic in terms of MT1‐MMP quantitation. To improve the T/B ratio, we previously reported using a pre‐targeting method.24 While this modification improved the T/B ratio, the method required multiple administrations, which would be undesirable for clinical use. In this study, the newly developed 111In‐MT1‐scFv and 111In‐MT1‐diabody showed specific affinity for MT1‐MMP hinge region and gave high T/B ratios. The specificity of these agents for MT1‐MMP and their good contrast would improve quantitation of MT1‐MMP expression levels and could contribute to molecular biological and clinical studies. MT1‐MMP is thought to be a promising predictor for recurrence risk in early stage breast cancer.25 Although accumulation of 111In‐MT1‐scFv and 111In‐MT1‐diabody in abdominal tissues like kidney, liver, spleen and intestine for excretion would hamper imaging tumors, we expect both probes to be used for breast cancer imaging, which is less affected by the background radioactivity of surrounding tissues because breast cancers are typically located from these other tissues.
For further applications involving imaging of other cancers, non‐specific accumulation in other tissues should be decreased. For instance, imaging for pancreatic cancers would be hampered by non‐specific accumulation in the liver. One of the possible reasons for non‐specific accumulation may be the cationic character of both probes (pI of MT1‐scFv: 9.54, MT1‐diabody: 9.40). Because cationic probes typically have increased tissue retention, non‐specific accumulation may be reduced by anionization, such as converting positively charged groups into negatively charged groups, while maintaining the immunoreactivity of the probes.26 High accumulation in the kidneys could be overcome by radiolabeling with a renal enzyme‐cleavable linkage.27
Increasing the accumulation in tumors is a straightforward strategy for clearly imaging cancers. To increase the accumulation of the probes in tumors, we believe affinity for MT1‐MMP is an important factor. Since the diabody can bind to antigen in a bivalent manner, we expected the MT1‐diabody to have a higher affinity for MT1‐MMP than MT1‐scFv. Consequently, the MT1‐diabody showed an extremely high affinity against the hinge region peptide (K D = 0.27 nM), but it showed mediocre affinity for MT1‐MMP protein. We postulated that the recognition site of MT1‐diabody might be blocked to some degree by the secondary structure of MT1‐MMP. We believe converting MT1‐scFv into the diabody form by shortening the linker would be an effective alternative method. The converted MT1‐scFv could acquire higher affinity through bivalent binding.28 We consider higher specific radioactivity could also be effective. In this study, we administered 111In‐MT1‐scFv and 111In‐MT1‐diabody (18.5 kBq/0.5 μg protein) to mice. Although these conditions were similar to previously reported conditions from other researchers,29, 30 about 1000‐fold unlabeled MT1‐scFv and MT1‐diabody used as precursors were also administered with 111In‐MT1‐scFv and 111In‐MT1‐diabody and might block binding sites on MT1‐MMP to some extent. Therefore, decreasing the amount of unlabeled precursor, which could be achieved by refining the reaction conditions, may lead to increased specific accumulation in tumors. These attempts would lead to increased specific accumulation and allow a more precise evaluation of MT1‐MMP expression in tumors.
As this research constitutes a preliminary study of miniaturized antibodies for imaging MT1‐MMP, there are a number of further modifications that could improve the characteristics of these imaging agents.
In conclusion, we have developed novel 111In labeled MT1‐MMP imaging probes based on scFv and diabody (MT1‐scFv and MT1‐diabody) for sensitive cancer imaging within a short time after administration. Compared with the previously developed whole antibody probe, faster blood clearance and sufficiently higher T/B ratios during the early period after administration with the new agents were achieved. This study shows the strategy of using antibody fragments is effective for improving the kinetics of imaging MT1‐MMP in cancer cells, and both novel probes have the potential for further applications.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Table S1. Data from the antigen‐immobilized ELISA, indirect competitive ELISA, cell ELISA.
Data S1. Materials and Methods.
Acknowledgments
This study was partly supported by Grants‐in‐Aid for Scientific Research (22791189 and 23113509) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Part of this study was supported by the New Energy and Industrial Technology Development Organization (NEDO), Japan.
(Cancer Sci 2013; 104: 495–501)
References
- 1. Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol 2005; 17: 559–64. [DOI] [PubMed] [Google Scholar]
- 2. Page‐McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 2007; 8: 221–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones JL, Glynn P, Walker RA. Expression of MMP‐2 and MMP‐9, their inhibitors, and the activator MT1‐MMP in primary breast carcinomas. J Pathol 1999; 189: 161–8. [DOI] [PubMed] [Google Scholar]
- 4. Ohuchi E, Imai K, Fujii Y, Sato H, Seiki M, Okada Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem 1997; 272: 2446–51. [DOI] [PubMed] [Google Scholar]
- 5. Knäuper V, Will H, López‐Otin C et al Cellular mechanisms for human procollagenase‐3 (MMP‐13) activation. Evidence that MT1‐MMP (MMP‐14) and gelatinase a (MMP‐2) are able to generate active enzyme. J Biol Chem 1996; 271: 17124–31. [DOI] [PubMed] [Google Scholar]
- 6. Itoh Y, Seiki M. MT1‐MMP: a potent modifier of pericellular microenvironment. J Cell Physiol 2006; 206: 1–8. [DOI] [PubMed] [Google Scholar]
- 7. Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol 2002; 3: 207–14. [DOI] [PubMed] [Google Scholar]
- 8. Crispi S, Calogero RA, Santini M et al Global gene expression profiling of human pleural mesotheliomas: identification of matrix metalloproteinase 14 (MMP‐14) as potential tumour target. PLoS ONE 2009; 4: e7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Temma T, Sano K, Kuge Y et al Development of a radiolabeled probe for detecting membrane type‐1 matrix metalloproteinase on malignant tumors. Biol Pharm Bull 2009; 32: 1272–7. [DOI] [PubMed] [Google Scholar]
- 10. Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol 2005; 23: 1126–36. [DOI] [PubMed] [Google Scholar]
- 11. Sundaresan G, Yazaki PJ, Shively JE et al 124I‐labeled engineered anti‐CEA minibodies and diabodies allow high‐contrast, antigen‐specific small‐animal PET imaging of xenografts in athymic mice. J Nucl Med 2003; 44: 1962–9. [PMC free article] [PubMed] [Google Scholar]
- 12. Nagase H, Woessner JF Jr. Matrix metalloproteinases. J Biol Chem 1999; 274: 21491–4. [DOI] [PubMed] [Google Scholar]
- 13. D'Alessio S, Ferrari G, Cinnante K et al Tissue inhibitor of metalloproteinases‐2 binding to membrane‐type 1 matrix metalloproteinase induces MAPK activation and cell growth by a non‐proteolytic mechanism. J Biol Chem 2008; 283: 87–99. [DOI] [PubMed] [Google Scholar]
- 14. Remacle AG, Golubkov VS, Shiryaev SA et al Novel MT1‐MMP small‐molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res 2012; 72: 2339–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yana I, Weiss SJ. Regulation of membrane type‐1 matrix metalloproteinase activation by proprotein convertases. Mol Biol Cell 2000; 11: 2387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toth M, Hernandez‐Barrantes S, Osenkowski P et al Complex pattern of membrane type 1 matrix metalloproteinase shedding. Regulation by autocatalytic cells surface inactivation of active enzyme. J Biol Chem 2002; 277: 26340–50. [DOI] [PubMed] [Google Scholar]
- 17. Itoh Y, Ito N, Nagase H, Evans RD, Bird SA, Seiki M. Cell surface collagenolysis requires homodimerization of the membrane‐bound collagenase MT1‐MMP. Mol Biol Cell 2006; 17: 5390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mori H, Tomari T, Koshikawa N et al CD44 directs membrane‐type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin‐like domain. EMBO J 2002; 21: 3949–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Atkinson JM, Falconer RA, Edwards DR et al Development of a novel tumor‐targeted vascular disrupting agent activated by membrane‐type matrix metalloproteinases. Cancer Res 2010; 70: 6902–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimizu Y, Temma T, Sano K, Ono M, Saji H. Development of membrane type‐1 matrix metalloproteinase‐specific activatable fluorescent probe for malignant tumor detection. Cancer Sci 2011; 102: 1897–903. [DOI] [PubMed] [Google Scholar]
- 21. Zhu L, Wang H, Wang L et al High‐affinity peptide against MT1‐MMP for in vivo tumor imaging. J Control Release 2011; 150: 248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu L, Zhang F, Ma Y et al In vivo optical imaging of membrane‐type matrix metalloproteinase (MT‐MMP) activity. Mol Pharm 2011; 8: 2331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Steenkiste M, Oltenfreiter R, Frankenne F et al Membrane type 1 matrix metalloproteinase detection in tumors, using the iodinated endogenous [123I]‐tissue inhibitor 2 of metalloproteinases as imaging agent. Cancer Biother Radiopharm 2010; 25: 511–20. [DOI] [PubMed] [Google Scholar]
- 24. Sano K, Temma T, Kuge Y et al Radioimmunodetection of membrane type‐1 matrix metalloproteinase relevant to tumor malignancy with a pre‐targeting method. Biol Pharm Bull 2010; 33: 1589–95. [DOI] [PubMed] [Google Scholar]
- 25. Ogura S, Ohdaira T, Hozumi Y, Omoto Y, Nagai H. Metastasis‐related factors expressed in pT1 pN0 breast cancer: assessment of recurrence risk. J Surg Oncol 2007; 96: 46–53. [DOI] [PubMed] [Google Scholar]
- 26. Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem 2010; 21: 2153–63. [DOI] [PubMed] [Google Scholar]
- 27. Uehara T, Koike M, Nakata H et al Design, synthesis, and evaluation of [188Re]organorhenium‐labeled antibody fragments with renal enzyme‐cleavable linkage for low renal radioactivity levels. Bioconjug Chem 2007; 18: 190–8. [DOI] [PubMed] [Google Scholar]
- 28. Adams GP, Schier R, McCall AM et al Prolonged in vivo tumour retention of a human diabody targeting the extracellular domain of human HER2/neu. Br J Cancer 1998; 77: 1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li L, Yazaki PJ, Anderson AL et al Improved biodistribution and radioimmunoimaging with poly(ethylene glycol)‐DOTA‐conjugated anti‐CEA diabody. Bioconjug Chem 2006; 17: 68–76. [DOI] [PubMed] [Google Scholar]
- 30. Lub‐de Hooge MN, Kosterink JG, Perik PJ et al Preclinical characterization of 111In‐DTPA‐Trastuzumab. Br J Pharmacol 2004; 143: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Data from the antigen‐immobilized ELISA, indirect competitive ELISA, cell ELISA.
Data S1. Materials and Methods.