

Abstract
Norcantharidin (NCTD), the demethylated form of Cantharidin, a reagent isolated from blister beetles, has been shown to be an anti‐tumor agent capable of inhibiting proliferation as well as inducing apoptosis in many cancer cell lines. However, little is known about the effect of NCTD in tumor angiogenesis. In this study, we demonstrated that NCTD inhibited vascular endothelial growth factor (VEGF)‐induced cell proliferation, migration, invasion, and capillary tube formation of primary human umbilical vein endothelial cells (HUVECs) in a dose‐dependent manner. Furthermore, we showed NCTD inhibited tumor growth and angiogenesis of colon cancer cells (LOVO) in vivo. We then mechanistically described that NCTD specifically abrogated the phosphorylation/activation of vascular endothelial growth factor receptor‐2 (VEGFR2)/MEK/ERK pathway kinases, with little effect on the phosphorylation of p38 MAPK and Akt, and on Cox‐2 expression. In summary, our results indicate that NCTD is a potential inhibitor of tumor angiogenesis by blocking VEGFR2/MEK/ERK signaling.
Tumor angiogenesis is considered as a key step in tumor growth, invasion, and metastasis, which is the formation of a blood vessel network that penetrates into a tumor to supply nutrients and oxygen and promotes tumor growth intensely.1, 2 Advances in this field are leading to novel treatments for many cancers.3 Angiogenesis or new blood vessel formation in most cancer types is initiated by cell proliferation and migration in response to chemotactic agents, such as vascular endothelial growth factor (VEGF), which is a potent pro‐angiogenic factor in tumor vasculature.4 Vascular endothelial growth factor expressed in endothelial cells exerts its biological effects by binding to its receptor tyrosine kinases such as vascular endothelial growth factor receptor 2 (VEGFR2), which is the major mediator of VEGF‐induced angiogenesis signaling molecules including signal transducers and activators of transcription (STATs),5, 6 mitogen‐activated protein kinases (MAPKs),7 focal adhesion kinase (FAK)8 and serine/threonine kinase (AKT).9
A variety of substances, particularly those present in dietary and medicinal plants, are proposed to have an inhibitory effect on angiogenesis.10 Cantharidin (exo‐2,3‐dimethyl‐7‐oxabicyclo‐hep‐tane‐2,3‐dicarb‐oxylic acidanhydride), a complex chemical isolated from blister beetles, has been shown to have anti‐tumor characteristics. Because of severe nephrotoxicity and inflammatory effects, cantharidin has been modified to a demethylated form, norcantharidin (NCTD), by usage of furan and maleic anhydride via the Diels–Alder reaction. Although NCTD has been reported to inhibit proliferation and induce apoptosis in many cancer cell lines,11 whether NCTD has any effect in tumor angiogenesis has remained elusive. Therefore, in this report, we examined if NCTD inhibited tumor angiogenesis by in vitro and in vivo studies.
Materials and Methods
Cell lines, cell culture and reagents
Norcantharidin (NCTD) was purchased from Sigma‐Aldrich (St. Louis, MO, USA). A 10‐mM solution of NCTD was prepared in sterile water, stored at −20°C and protected from light, and diluted to needed concentrations for studies. Primary HUVECs were a kind gift from Dr Dali Li (Institute of Biomedical Sciences and School of Life Sciences, East China Normal University in Shanghai). Human umbilical vascular endothelial cells were grown in endothelial cell medium (ECM), supplemented with 5% FBS, endothelial cell growth factor (ECGS) and P/S solution (10 000 U/mL penicillin, 10 000 μg/mL sreptomycin). Endothelial cell medium, FBS, ECGS and P/S solution were purchased from ScienCell (San Diego, CA, USA). The human colorectal cancer cell line (LOVO) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPM1640 medium supplemented with 10% FBS. VEGF165 was purchased from Fermentas (Shenzhen, China). Matrigel was purchased from BD Biosciences (San Jose, CA, USA), and mitomycin C was ordered from Roche (Shanghai, China). All cells were incubated at 37°C in an atmosphere of 5% CO2.
Cell viability assay
Human umbilical vascular endothelial cells and LOVO cell (2 × 104 cells/well) were treated without or with VEGF (4 ng/mL) and different concentrations of NCTD for 48 h. Cell viability was determined by MTT (Sigma‐Aldrich) with Bio‐Radmicroplate reader.
Flow cytometry analysis
Human umbilical vascular endothelial cells (1 × 106) were treated with different concentrations of NCTD for 48 h and then collected and analyzed for cell cycle distribution in a FACS flow cytometer (BD Sciences) with cell cycle Kit (BD Pharmingen).
Migration assay
Human umbilical vascular endothelial cells grew into full confluence in 6‐well plates and then were incubated with 10 μg/mL mitomycin C for 2 h to inactivate HUVECs proliferation. Cells were then scratched by pipette tip and washed with PBS. Endothelial cell medium supplemented with 0.5% FBS was added into well with or without 4 ng/mL VEGF and different concentrations of NCTD. Cell images were taken after 10 h of incubation at 37°C, 5% CO2. The migrated cells were calculated against untreated wells at 100%. Three independent experiments were preformed for statistics.
Transwell migration assay
The transwell (Corning, New York, NY, USA) were coated with 50 μL 1:8 diluted Matrigel (Growth factor reduced; BD Biosciences) for 30 min in a cell incubator. The bottom chambers of the transwell were filled with ECM with 0.5% FBS supplemented with 4 ng/mL VEGF and the top chambers were seeded with mytomycin C inactivated 4 × 104 cells/well HUVECs in 100 μL ECM (0.5% FBS) plus different concentrations of NCTD. After leaving overnight, the cells on the top surface of the membrane (non‐migrated cells) were scraped with a cotton swab and the cells spreading on the bottom sides of the membrane (invasive cells) were fixed with cold 4% paraformaldehyde and stained with crystal Violet. Images were taken using OLYPUS inverted microscope and invasive cells were quantified by manual counting. The inhibitory percentage of invasive cells was quantified against untreated control wells.
Tube formation assay
Each well of prechilled 24‐well plates was coated with 50 μL Matrigel and incubated at 37°C for 45 min. Human umbilical vascular endothelial cells (4 × 104 cells) were planted in 1 mL ECM (supplemented with 0.5% FBS and 4 ng/mL VEGF) with various concentrations of NCTD. After 6 h of incubation, endothelial cell tubular structure formation was quantified by calculating the tube length a high power fields (200×) with OLYMPUS inverted microscope, and the inhibitory percentage was quantified against expressed using untreated wells.
Xenograft mouse model
Xenograft mouse model was used to evaluate the effect of NCTD on tumor growth as described by Yi et al.12 The 5‐week‐old to 6‐week‐old SCID male mice (SLAC Laboratory Animal, Shanghai, China) weighing approximately 20 g was divided into five groups with five animals for each group: (1) control group, (2) 5‐FU group, and (3) NCTD treated groups with different dosage. LOVO cells, 2 × 106 cells per mouse, were s.c. injected into the mice. After the tumors had established (about 100 mm3), NCTD, dissolved in sterile PBS, was administered by intraperitoneal injection at 0.5‐, 1‐ and 2 mg/kg body weight, respectively. Nearly 5‐FU, dissolved in sterile PBS, was administered by intraperitoneal injection at 20 mg/kg per 2 days. Control animal was only given 0.2 mL sterile water as a vehicle control. The mouse bodyweight and tumor sizes were recorded every day and the tumor sizes were determined by Vernier caliper measurements and calculated as 1/2 × length × width × height. After 15 days, mice were killed. Animals use in the present study was approved by the Ethics Committee of Shanghai University of Traditional Chinese Medicine within which the work was undertaken and the present study conforms to the provisions of the Declaration of Helsinki in 1995.
Histology and immunohistochemistry
The organs and tumor were removed and fixed with formalin and embedded with paraffin. CD34 and Ki67 were performed on the 5‐μm sections with according antibody (Cell Signaling Technology, Beverly, MA, USA). Images were taken with Olympus IX71 microscope. The number of blood vessels and the percent of positive Ki67 cells were counted.
Western blotting
To determine the effects of NCTD on phosphorylation of VEGFR signaling pathway, HUVECs were first starved with 0.1% FBS medium for 12–14 h. After washout with new fresh culture medium, cells were pretreated with or without 50 ng/mL VEGF and different concentrations of NCTD for 30 min before the phosphorylation status of VEGFR signaling molecules was determined. The whole cell extracts were prepared by lysing cells with RIPA buffer supplemented with proteinase inhibitors. Antibodies used for different Western blot analyses, are anti‐phospho‐VEGF Receptor 2 (Tyr1175), phospho‐MEK1/2 (Ser221) mAb and phospho‐p44/42 MAPK (Erk1/2; Thr202/Tyr204; Cell Signaling Technology).
Statistical analysis
The data are presented as mean ± SE, and statistical comparisons between groups were performed using t‐test. P‐value ≤ 0.05 was considered statistically significant.
Results
Inhibition of HUVEC and LOVO proliferation by NCTD
Norcantharidin derives from cantharidin by demethylation and has a molecular weight of 168.15 g/mol (Fig. 1a). We first tested the in vitro anti‐proliferative effect of NCTD, on primary HUVECs using a standard proliferation assay (MTT). Our results showed that NCTD significantly reduced VEGF‐induced cell proliferation at an IC50 of 18.2 μM, whereas it inhibited VEGF‐free cell viability at a much higher concentrations, an IC50 of 30 μM (Fig. 1b,c). These data suggest that NCTD preferentially be an anti‐angiogenic reagent. Next we demonstrated that NCTD can also have inhibitory effects on non‐angiogenic, human colorectal cancer LOVO cells with an IC50 of 80 μM (Fig 1d), again suggesting that NCTD is more effective at inhibiting angiogenesis. Further, we examine whether NCTD affected cell cycle progression and apoptosis and our results revealed that it induced a G0/G1 phase block, 76.06% treated vs. 51.39% control and a decrease of cells in G2/M phase, from 33.95% to 12.57% (Table 1).
Figure 1.
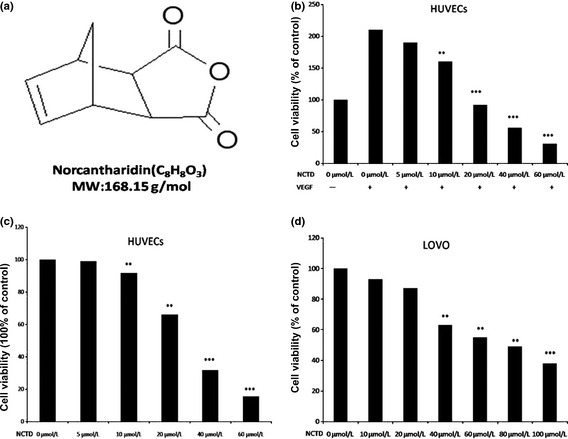
Norcantharidin (NCTD) inhibits cell viability in both human umbilical vein endothelial cells (HUVECs) and LOVO cells. (a) The chemical structure of NCTD with a molecular weight 186.15 g. (b) Norcantharidin inhibits vascular endothelial growth factor (VEGF)‐induced cell viability in dose‐dependent manner. HUVECs (2 × 104 cells/well) were starved with 0.1% fetal bovine serum (FBS) medium and then treated with or without VEGF (4 ng/mL) and different concentrations of NCTD for 24 h. Cell viability was quantified by MTT assay (*P < 0.05, **P < 0.01, ***P < 0.001 versus VEGF alone). (c) Effects of NCTD on HUVEC viability under normal culture condition. Human umbilical vein endothelial cells (2 × 104 cells/well) were treated with different concentrations of NCTD for 24 h (*P < 0.05, **P < 0.01, ***P < 0.001 versus control). (d) Norcantharidin inhibits colorectal cancer cell (LOVO) viability (the treatment is the same as C). Column, mean from three different experiments with six duplicates; bar, SE.
Table 1.
Norcantharidin (NCTD) arrests cell cycle progression in human umbilical vein endothelial cells (HUVECs)
| Population (%) | Norcantharidin (μmol/L) | ||||
|---|---|---|---|---|---|
| 0 | 5 | 10 | 20 | 40 | |
| G0/G1 phase | 51.39 ± 1.99 | 60.39 ± 3.95 | 65.44 ± 2.71 | 70.39 ± 2.38 | 76.06 ± 2.55 |
| G2/M phase | 33.95 ± 3.07 | 25.17 ± 1.49 | 20.61 ± 2.03 | 14.78 ± 1.56 | 12.57 ± 1.21 |
NCTD inhibits VEGF‐induced migration, invasion, and capillary structure formation of endothelial cells
Cell migration is critical for an endothelial cell to form blood vessels in angiogenesis. We then determined the effects of NCTD on the chemotactic motility of HUVECs using wound‐healing migration and transwell cell invasion assays. As shown in Figure 2(a,b), NCTD significantly inhibited HUVEC migration and invasion at 5 μM. Angiogenesis is a very complex process, in which tube formation of endothelial cells is a key step.(26) We then used two‐dimensional Matrigel assay to examine the effects of NCTD on the tubular structure formation by HUVECs. When HUVECs were placed on Matrigel, robust tube‐like structures were formed with the presence of VEGF (Fig. 2c). The ability of endothelial cells to form tubular structures was assessed in the presence or absence of NCTD by calculating the length of tubes with an inverted photomicroscope. As shown in Figure 2(c), we displayed that 10 μM NCTD inhibited 50% tube formation of HUVECs and 40 μM NCTD completely abrogated the process. These results demonstrated that NCTD blocked VEGF‐induced in vitro angiogenesis by inhibiting cell migration, invasion, and tube formation.
Figure 2.
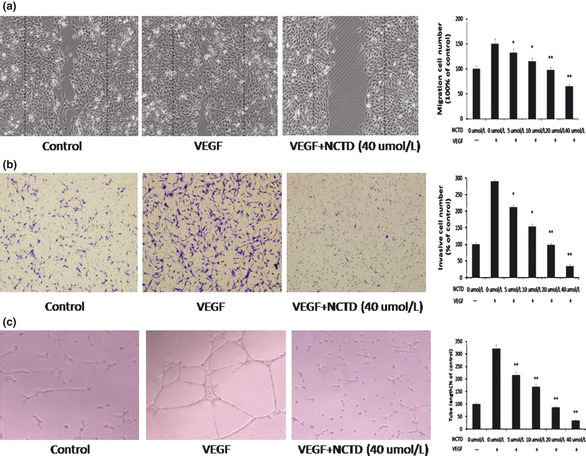
Norcantharidin (NCTD) inhibits vascular endothelial growth factor (VEGF)‐induced migration, invasion, and tubular structure formation of endothelial cells. (a) Norcantharidin inhibits human umbilical vein endothelial cell (HUVEC) migration. Human umbilical vein endothelial cells were allowed to grow into full confluence in six‐well plates, and inactivated with 10 μg/mL mitomycin C for 2 h. Cells were wounded with pipette and treated with or without 4 ng/mL VEGF and different concentration of NCTD in endothelial cell medium supplemented with 0.5% fetal bovine serum (FBS). After incubation, the migrated cells were quantified by manual counting. (b) Norcantharidin inhibits the invasion of HUVEC. Human umbilical vein endothelial cells were seeded in the upper chamber of transwell and treated with different concentrations of NCTD. The bottom chamber was filled with ECGM supplemented with VEGF. After about 8–10 h, the invasive HUVECs passed through the membrane and were quantified by counting the cells that migrated onto the membrane. (c) Norcantharidin inhibits VEGF‐induced tube formation of HUVECs. Human umbilical vein endothelial cells were placed in the 24‐well plates coated with Matrigel at the density of 4 × 104 cells/well. After 4–6 h, cells were fixed and tubular structure was quantified by manual counting of high power fields (200×). Column, mean from three different experiments with duplicates; bar, SE (*P < 0.05, **P < 0.01 versus VEGF alone).
NCTD inhibits tumor angiogenesis and tumor growth in vivo
Tumor angiogenesis is the rate‐limiting step in tumor progression, because it provides oxygen, nutrients for tumor growth, invasion and metastasis.13 We tested NCTD's in vivo effect in a xenograft human colorectal cancer model. Tumor‐bearing mice had been treated by NCTD or 5‐FU for 15 days. As shown in Figure 3(a), tumors in control animals grew rapidly (from 196.31 ± 31.14 to 1486.23 ± 148.62 mm3), whereas tumors in NCTD‐treated animals grew much slower (from 127.67 ± 20.11 to 645.71 ± 63.58 mm3). Accordingly, the average tumor weight in NCTD‐treated mice was significantly lower compared with that in the control group (Fig. 3c), suggesting that NCTD strongly inhibited tumor growth. To confirm that NCTD inhibited tumor angiogenesis, we analyzed the tumor sections for CD34, a marker for blood vessels. The results indicated that the average number of blood vessels in control animals was 23.47 ± 3.47/high power field (HPF), compared to that in NCTD treated mice, 8.14 ± 1.19/HPF (Fig. 3c), signifying that NCTD significantly inhibited in vivo tumor angiogenesis. Furthermore, our results showed NCTD repressed the proliferation of tumor cells in vivo as evidenced by Ki67 immunostaining of the tumor tissues. The average percent positive Ki67 cells in control animals was 37.41 ± 2.12%, whereas the average percent positive Ki67 cells in animals treated with NCTD at dosage of 0.5‐, 1‐ and 2‐mg/kg per day were 30.08 ± 3.35%, 22.24 ± 3.97%, and 19.72 ± 1.54%, respectively; the average percent positive Ki67 cells in 5‐Fu treated animals was 13.11 ± 3.04% (Fig. 4a). Mice treated with NCTD did not have altered body weight and the blood vessels in non‐cancerous tissues remained normal (Figs 3b,4b), suggesting no toxicity of the compound at the tested concentration.
Figure 3.
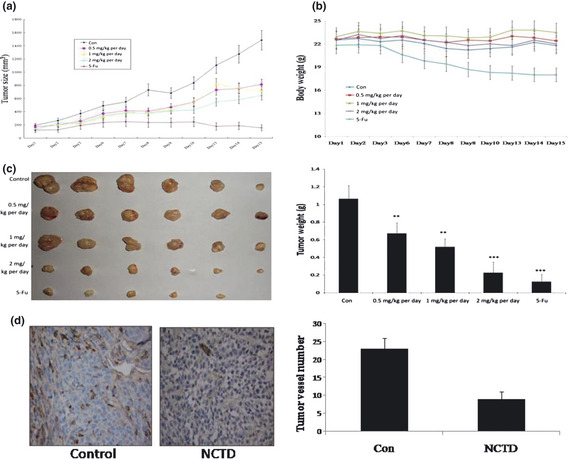
Norcantharidin (NCTD) inhibits tumor angiogenesis and tumor growth in vivo. (a) Norcantharidin inhibits solid cancer growth in xenograft colorectal cancer mouse model. Colorectal cancer cells (LOVO) were injected (2 × 106 per mouse) into the 5‐ to 6‐week‐old male nude mice. After the tumors had established (about 100 mm3), the mice were injected with or without NCTD at 0.5, 1 and 2 mg/kg per day, respectively. After 15 days, mice were killed, tumors were removed and photographed. Tumor sizes in control animals and NCTD treated animals were calculated and shown in (a). Tumors in control animals increased from 196.31 ± 31.14 to 1486.23 ± 148.62 mm3, whereas tumors in animals treated with NCTD at dose of 2 mg/kg per day increased from 127.67 ± 20.11 to 645.71 ± 63.58 mm3. (b) Treatment with NCTD has little effect on mouse body weight. No significant difference between animals treated with NCTD at dose of 2 mg/kg per day and the control animals. (c) Solid tumors in animals treated with NCTD at 2 mg/kg per day were significantly smaller than the control animals. The average weight of tumors from control animals was 1.0673 ± 0.189 g, whereas animals treated with NCTD at a dose of 2 mg/kg per day was 0.203 ± 0.116 g. (d) Effects of NCTD on tumor angiogenesis in xenograft mouse model. Solid tumors were fixed with 4% paraformaldehyde and embedded with paraffin. The 5‐μm sections were stained with anti‐mouse CD34 antibody and the blood vessel number was calculated. The average vessel number in control animals was 23.47 ± 3.47/high power field (HPF) (magnification, 200×), whereas the average blood vessel number in animals treated with NCTD at dose of 2 mg/kg per day was 8.14 ± 1.19/HPF. Column, mean; bar, SE (n = 7, *P < 0.05, **P < 0.01 versus control).
Figure 4.
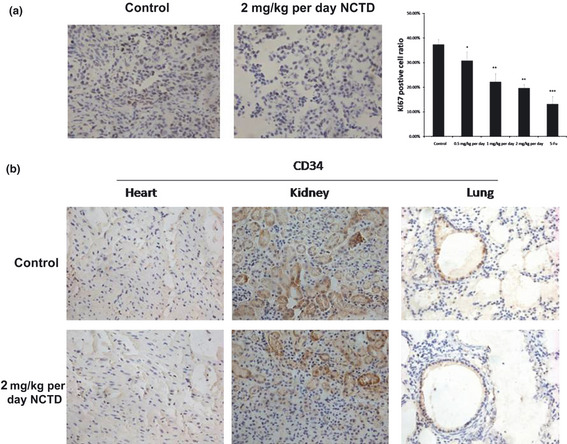
Effects of norcantharidin (NCTD) on Ki67 staining of tumor cells and CD34 staining of the blood vessels in non‐cancerous tissues in vivo. (a) Norcantharidin inhibits the proliferation of tumor cells in vivo measured by Ki67 immunostaining of the tumor tissues. The average percent positive Ki67 cells in control animals was 37.41 ± 2.12% (magnification, 200×), whereas the average percent positive Ki67 cells in animals treated with NCTD at dosage of 0.5, 1 and 2 mg/kg per day were 30.08 ± 3.3%, 22.24 ± 3.97%, and 19.72 ± 1.54%, respectively; percent positive Ki67 cells in 5‐Fu treated animals were 13.11 ± 3.04%. Column, mean; bar, SE (n = 6, *P < 0.05, **P < 0.01, ***P < 0.001 versus control). (b) Effects of NCTD on CD34 staining of the blood vessels in non‐cancerous tissues in vivo. The organs were fixed with 4% paraformaldehyde and embedded with paraffin. The 5‐μm sections were stained with anti‐mouse CD34 antibody. There was no difference of blood vessels in heart, kidney and lung between the control and the NCTD‐treat animals at dosage of 2 mg/kg per day.
NCTD inhibits the VEGFR2\MEK\ERK signaling pathway in HUVECs
Vascular endothelial growth factor receptor 2 is the most biologically important receptor for VEGF. It regulates endothelial cell proliferation, migration, differentiation, tube formation and angiogenesis.14 To examine the underlying molecular mechanism of NCTD's inhibition on angiogenesis, we studied the effects of NCTD on the phosphorylation of VEGFR2/MEK/ERK signaling in HUVECs. Our results showed that NCTD at concentration of 40 μM significantly inhibited the phosphorylation of protein kinases involved in the activation of VEGFR2/MEK/ERK signaling pathway induced by VEGF at 10 min, including VEGFR2, MEK1/2, and pERK1/2 (Fig. 5a). We also studied a time course of VEGF's effect on HUVECs without or with the presence of NCTD (40 μM): 0, 5, 10, 30, 60, 120 min. The results showed that the NCTD got to inhibit the phosphorylation of VEGFR2/MEK/ERK signaling at 5 min and last for the duration of VEGF stimulation, which peaked at 10 min and went down to baseline (Fig. 5b,c). This inhibition by NCTD is specific because NCTD did not affect the phosphorylation of p38 MAPK, Akt and Cox‐2 expression level (Fig. 5d).
Figure 5.
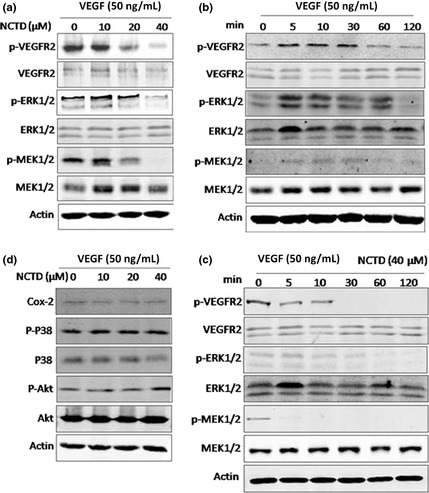
Norcantharidin (NCTD) inhibits vascular endothelial growth factor (VEGF)‐induced activation of the phosphorylation of VEGFR2/MEK/ERK signaling pathway. (a) Effects of NCTD on the phosphorylation and activation of VEGFR2/MEK/ERK pathway. Human umbilical vein endothelial cells (HUVECs) were pretreated with various concentrations of NCTD for 10 min before stimulation with 50 ng/mL VEGF for a certain time. After that, cells were washed with cold phosphate‐buffered saline (PBS) and lysed on the dish in RIPA buffer. Phosphorylation and activation of different protein kinases including pTyr1175VEGFR2, pSer217/221‐MEK1/2, and pThr202/Tyr204‐pERK1/2 were examined by specific antibodies. (b,c) A time course of VEGF's effect on HUVECs without or with the presence of NCTD (40 μM): 0, 5, 10, 30, 60, 120 min. (d) Norcantharidin could not affect the phosphorylation of pThr180/Tyr182‐p38 MAPK, pSer473‐Akt and Cox‐2 expression levels.
Discussion
The formation of new blood vessels is a complex multistep process. Endothelial cells resting in the parent vessels are activated by an angiogenesis signal to allow endothelial cells to migrate, proliferate, and differentiate to give rise to capillary tubules. Any of these steps can be a potential targets for pharmacologic intervention.15 In this report, we found that NCTD, a synthetic demethylated analog of cantharidin, which has long been used as an anti‐cancer drug, inhibited VEGFR2/MEK/ERK signaling resulting in reduced tumor angiogenesis. It has been shown that NCTD attenuates the migration, adhesion and vascular network formation of HUVECs, expression levels of VEGF and VEGFR2 proteins and angiogenesis and the growth of human gallbladder carcinoma.16 Our current studies showed NCTD repressed VEGF‐mediated endothelial cell proliferation, migration, invasion and capillary formation through inhibiting the VEGFR2/MEK/ERK signaling cascade. Yu et al. also showed NCTD could inhibit the migration, adhesion and vascular network tube formation of HUVECs, expression levels of VEGF and VEGFR2 proteins and angiogenesis and the growth of human gallbladder carcinoma.16, 17, 18 But our research comprehensively focuses on the NCTD could repress VEGF‐mediated endothelial cell proliferation, migration, invasion and capillary structure formation through inhibiting the phosphorylation of VEGFR2. As a result, NCTD inhibited in vitro and in vivo growth of colorectal cancer cells. In a xenografts mouse model, we found that NCTD (2 mg/kg per day) did not affect the body weight of the mice, but exhibited significant inhibitory effects on solid tumor growth and tumor angiogenesis. In most cases, NCTD effectively induces cell growth inhibition of cultured tumor LOVO cells at concentrations over 80 μM. In contrast, NCTD sufficiently inhibited VEGF induced angiogenic responses only at or less than 20 μM in vitro and in vivo angiogenesis assays. Such concentration has little effect on endothelial cell viability. Because VEGF is a major inducer for the formation of tumor vasculature,19, 20 our results demonstrate that NCTD inhibits tumor growth in vivo via their anti‐angiogenic activity at a much lower concentration and much earlier than their cytotoxicity effects on tumor cells.
As we knew, VEGFR2 mediates the majority of the downstream effects of VEGF in angiogenesis, including microvascular permeability, endothelial cell proliferation, invasion, migration and survival.21 We found that NCTD inhibited VEGF‐induced phosphorylation of VEGFR2 in HUVECs, but did not affect the levels of total VEGFR2 proteins. The reduction of phosphorylated VEGFR2 might explain the observed decrease of VEGF‐induced phosphorylation of ERK1/2, the major downstream target of VEGFR2, following treatment with NCTD. The possible mechanism of how NTCD decreases the phosphorylation of VEGFR2 was that NCTD was directly binding to the kinase domain of VEGFR2. MEK–ERK signaling pathway has been implicated in endothelial cell proliferation,22 VEGF‐mediated survival,23, 24 and protection against receptor‐mediated apoptosis.(25) MEK–ERK signaling is thought to stimulate angiogenesis by promoting endothelial cell motility.(26) Although the mechanism of activation of MEK–ERK in endothelial cells by growth factors and integrins(27) and the differential regulation of Raf‐1 by different stimuli have been characterized,(28) it is not clear how MEK–ERK signaling mediates its effects during tumor angiogenesis in vivo. Our results illustrated that NCTD could significantly inhibit the activation of VEGFR2/MEK–ERK protein kinases in a concentration‐dependent manner with a modest, effective concentration of 40 μM.
In summary, this study shows that NCTD inhibits VEGF‐induced phosphorylation of VEGFR2, MEK and ERK both in vitro, inhibits tumor angiogenesis and is efficacious against a model of human colorectal cancer in nude mice. Taken together, our studies indicate that NCTD is a potential inhibitor of tumor angiogenesis by blocking VEGFR2/MEK/ERK signaling pathway.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work is partially supported by the specialized training plan of Putuo Hospital Shanghai University of Traditional Chinese Medicine (2011Y008). We thank Professor Dali Li (East China Normal University) for providing us the HUVECs used in our experiments.
(Cancer Sci 2013; 104: 604–610)
References
- 1. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285: 1182–6. [DOI] [PubMed] [Google Scholar]
- 2. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000; 407: 249–57. [DOI] [PubMed] [Google Scholar]
- 3. Burstein HJ, Schwartz RS. Molecular origins of cancer. N Engl J Med 2008; 358: 527. [DOI] [PubMed] [Google Scholar]
- 4. Kerbel RS. Tumor angiogenesis. N Engl J Med 2008; 358: 2039–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen SH, Murphy DA, Lassoued W, Thurston G, Feldman MD, Lee WM. Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Biol Ther 2008; 7: 1994–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Z, Han ZC. STAT3: a critical transcription activator in angiogenesis. Med Res Rev 2008; 28: 185–200. [DOI] [PubMed] [Google Scholar]
- 7. Masson‐Gadais B, Houle F, Laferriere J, Huot J. Integrin alphavbeta3, requirement for VEGFR2‐mediated activation of SAPK2/p38 and for Hsp90‐dependent phosphorylation of focal adhesion kinase in endothelial cells activated by VEGF. Cell Stress Chaperones 2003; 8: 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor‐2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 2007; 19: 2003–12. [DOI] [PubMed] [Google Scholar]
- 9. Kitamura T, Asai N, Enomoto A et al Regulation of VEGF‐mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol 2008; 10: 329–37. [DOI] [PubMed] [Google Scholar]
- 10. Mojzis J, Varinska L, Mojzisova G, Kostova I, Mirossay L. Antiangiogenic effects of flavonoids and chalcones. Pharmacol Res 2008; 57: 259–65. [DOI] [PubMed] [Google Scholar]
- 11. Wang GS. Medical uses of mylabris in ancient China and recent studies. J Ethnopharmacol 1989; 26: 147–62. [DOI] [PubMed] [Google Scholar]
- 12. Yi T, Yi Z, Cho SG et al Gambogic acid inhibits angiogenesis and prostate tumor growth by suppressing vascular endothelial growth factor receptor 2 signaling. Cancer Res 2008; 68: 1843–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer 2005; 5: 423–35. [DOI] [PubMed] [Google Scholar]
- 14. Olsson AK, Dimberg A, Kreuger J, Claesson‐Welsh L. VEGF receptor signalling ‐ in control of vascular function. Nat Rev Mol Cell Biol 2006; 7: 359–71. [DOI] [PubMed] [Google Scholar]
- 15. Quesada AR, Munoz‐Chapuli R, Medina MA. Anti‐angiogenic drugs: from bench to clinical trials. Med Res Rev 2006; 26: 483–530. [DOI] [PubMed] [Google Scholar]
- 16. Yu T, Hou F, Liu M et al Norcantharidin anti‐angiogenesis activity possibly through an endothelial cell pathway in human colorectal cancer. Asian Pac J Cancer Prev 2012; 13: 499–503. [DOI] [PubMed] [Google Scholar]
- 17. Fan YZ, Fu JY, Zhao ZM, Chen CQ. Effect of norcantharidin on proliferation and invasion of human gallbladder carcinoma GBC‐SD cells. World J Gastroenterol 2005; 11: 2431–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang JT, Fan YZ, Chen CQ, Zhao ZM, Sun W. Norcantharidin: A potential antiangiogenic agent for gallbladder cancers in vitro and in vivo. Int J Oncol 2012; 40: 1501–14. [DOI] [PubMed] [Google Scholar]
- 19. Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature 2005; 438: 967–74. [DOI] [PubMed] [Google Scholar]
- 20. Carmeliet P. Angiogenesis in life, disease and medicine. Nature 2005; 438: 932–6. [DOI] [PubMed] [Google Scholar]
- 21. Lu N, Gao Y, Ling Y et al Wogonin suppresses tumor growth in vivo and VEGF‐induced angiogenesis through inhibiting tyrosine phosphorylation of VEGFR2. Life Sci 2008; 82: 956–63. [DOI] [PubMed] [Google Scholar]
- 22. Meadows KN, Bryant P, Pumiglia K. Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J Biol Chem 2001; 276: 49289–98. [DOI] [PubMed] [Google Scholar]
- 23. Berra E, Milanini J, Richard DE et al Signaling angiogenesis via p42/p44 MAP kinase and hypoxia. Biochem Pharmacol 2000; 60: 1171–8. [DOI] [PubMed] [Google Scholar]
- 24. Gupta K, Kshirsagar S, Li W et al VEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signaling. Exp Cell Res 1999; 247: 495–504. [DOI] [PubMed] [Google Scholar]
- 25. Alavi A, Hood JD, Frausto R, Stupack DG, Cheresh DA. Role of Raf in vascular protection from distinct apoptotic stimuli. Science 2003; 301: 94–6. [DOI] [PubMed] [Google Scholar]
- 26. Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF‐induced angiogenesis and vascular permeability. Mol Cell 1999; 4: 915–24. [DOI] [PubMed] [Google Scholar]
- 27. Eliceiri BP, Klemke R, Stromblad S, Cheresh DA. Integrin alphavbeta3 requirement for sustained mitogen‐activated protein kinase activity during angiogenesis. J Cell Biol 1998; 140: 1255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hood JD, Bednarski M, Frausto R et al Tumor regression by targeted gene delivery to the neovasculature. Science 2002; 296: 2404–7. [DOI] [PubMed] [Google Scholar]