

Abstract
Stalled replication forks undergo DNA double‐strand breaks (DSBs) under certain conditions. However, the precise mechanism underlying DSB induction and the cellular response to persistent replication fork stalling are not fully understood. Here we show that, in response to hydroxyurea exposure, DSBs are generated in an Artemis nuclease‐dependent manner following prolonged stalling with subsequent activation of the ataxia–telangiectasia mutated (ATM) signaling pathway. The kinase activity of the catalytic subunit of the DNA‐dependent protein kinase, a prerequisite for stimulation of the endonuclease activity of Artemis, is also required for DSB generation and subsequent ATM activation. Our findings indicate a novel function of Artemis as a molecular switch that converts stalled replication forks harboring single‐stranded gap DNA lesions into DSBs, thereby activating the ATM signaling pathway following prolonged replication fork stalling.
DNA replication is a crucial phase in cell proliferation and is always accompanied by the possibility of generating DNA irregularities. To prevent the disruption of genome integrity during replication by exogenous or endogenous stresses, replication fork progression is precisely regulated and monitored by the replication checkpoint.1 This machinery is one of the targets for cancer chemotherapy such as alkylating agents or inhibitors of ribonucleotide reductase, which cause an imbalance in the deoxynucleotide triphosphate pool. Stalled replication forks lead to the production of ssDNA lesions including ssDNA gaps, which in some cases are converted to DSBs, an event termed replication fork collapse, by a mechanism in which some nucleases play a key role. Double‐strand breaks (DSB) thus generated must be monitored and resolved by DNA damage response mechanisms to maintain genome integrity.
Ataxia–telangiectasia mutated (ATM) is mainly activated by DSBs and recruited to damage sites by the Mre11‐Rad50‐NBS1 complex.2 Ataxia‐telangiectasia mutated (ATM) exists as an inactive dimer and undergoes autophosphorylation, which triggers monomerization and activation.3 Another damage‐response protein, the ATR–ATR‐interacting protein complex, is principally activated by RPA‐coated ssDNA regions thatarise at stalled replication forks or during the processing of bulky lesions such as UV photoproducts and DSBs in S/G2 phases.4 Once ATM and ATR are activated by DNA lesions, they cooperatively stimulate DNA damage checkpoint pathways throughthe phosphorylation of numerous substrates, leading to cell cycle arrest, apoptosis, DNA repair, or cell senescence.5
The Artemis nuclease is mutated in individuals with RS‐SCID. In vitro, in the presence of ATP and DNA‐PK composed of DNA‐PKcs (officially known as protein kinase, DNA‐activated, catalytic polypeptide [PRKDC]) and the Ku70/80 heterodimer, Artemis acquires DNA endonuclease activity that specifically targets ssDNA–dsDNA junctions including 5′‐ or 3′‐ overhangs, hairpins, and gaps.6, 7Autophosphorylation of DNA‐PKcs at the ABCDE cluster (Thr2609, Ser2612, Thr2620, Ser2624, Thr2638, and Thr2647) is essential for Artemis endonuclease activity.6 Through its endonuclease activity, Artemis contributes to the repair of a fraction of DSBs (~10%) induced by ionizing radiation in vivo, suggesting that it processes the ends of DSBs that are refractory to repair by core non‐homologous end joining factors such as Ku70, Ku80, XRCC4, and DNA ligase IV.8
Here, we show that the ATR signaling pathway is activated at an early phase of replication fork stalling, and that extensive activation of the ATM signaling pathway is triggered by the generation of DSBs by Artemis nuclease following prolonged replication fork stalling. DNA‐PKcs kinase activity is also required for this DSB generation and subsequent ATM activation. Artemis‐deficient fibroblasts show resistance to HU. From these results, we propose that the Artemis/DNA‐PK machinery plays an essential role in the mechanism that responds to prolonged replication fork stalling by HU.
Materials and Methods
Cells
HeLa, U2OS, M059J, and M059K were obtained from ATCC (Manassas, VA, USA). Human telomerase reverse transcriptase immortalized human diploid fibroblasts (HDF2/326) were described previously. 9
Immunoblotting and immunofluorescence
Immunoblotting and immunofluorescence were carried out using standard methods. Details of the experimental procedure are also provided in Document S1.
Results
Activation of DNA damage response by HU
To obtain insights into the mechanism by which stalled DNA replication forks are converted to DSBs under certain conditions, we measured the phosphorylation of various ATM/ATR substrates to monitor the status of DNA damage checkpoint activation in response to HU. To assess the effect of HU treatment only in S phase, we synchronized HeLa cells at the G1‐S boundary with a double thymidine block, then released them into S phase for 1 h prior to HU exposure (Fig. S1a). As shown in Figure 1(a), Chk1 Ser345 and NBS1 Ser343 were strongly phosphorylated after a 2‐h exposure to HU. Phosphorylation of ATM at Ser1981 was weakly detectable at 2 h and significantly enhanced after 24 h continuous exposure to HU. Chk2 Thr68 was phosphorylated in parallel with ATM phosphorylation. The activation of the ATM signaling pathway after HU was confirmed by addition of the ATM specific inhibitor KU55933 that attenuated ATM phosphorylation at Ser1981 and SMC1 phosphorylation at Ser957 and Ser966 after HU exposure (Fig. 1b). In contrast, phosphorylation of Chk2, NBS1, and Chk1 was not inhibited by treatment with KU55933 (Fig. 1b, Fig. S1b). As Chk2 was phosphorylated even in ATM‐inhibited cells, Chk2 might be phosphorylated by ATR or DNA‐PKcs. This result indicates that ATM phosphorylation after HU exposure for 24 h is the result of autophosphorylation, and that ATM preferentially phosphorylates SMC1 after HU exposure.
Figure 1.
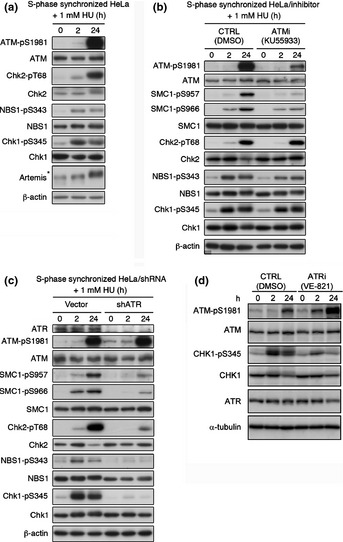
Ataxia–telangiectasia and RAD3‐related (ATR) and ataxia–telangiectasia mutated (ATM) pathways are differentially activated by exposure to hydroxyurea (HU). (a) S‐phase synchronized HeLa cells were treated with 1 mM HU 1 h after release from double thymidine block. Cell extracts from the indicated time points were immunoblotted with the indicated antibodies. *Hypersphosphorylated form of Artemis. (b) Synchronized HeLa cells were treated with 1 mM HU 1 h after release from double thymidine blockwith or without ATM‐specific inhibitor KU55933 (10 μM). (c) HeLa cells were transfected with shATR‐expressing construct or mock vector. The cells were synchronized 24 h after transfection. One hour after release from double thymidine block, cells were treated with 1 mM HU for 2 or 24 h. (d) Synchronized HeLa cells were treated with 1 mM HU 1 h after release from double thymidine blockwith or without ATR‐specific inhibitor VE‐821 (10 μM). CTRL, control.
Previously, phosphorylation of ATM after replication fork stalling has been reported to be dependent on ATR activation.10 Hence, we investigated whether DNA damage signaling caused by prolonged HU exposure was ATR‐dependent using ATR knockdown cells produced with an ATR‐specific shRNA11 (Fig. 1c) or siRNA to target a distinct ATR sequence (Fig. S1c). Increased ATM autophosphorylation was observed after ATR knockdown even in the absence of HU, presumably due to an increase in DSBs or genomic instability caused by ATR depletion.12 After 2 h exposure to HU, a slightly elevated level of ATM phosphorylation was observed in both cells. After 24 h exposure to HU, ATM was strongly autophosphorylated even in ATR knockdown cells. The ATR knockdown strikingly reduced Chk1 and NBS1 phosphorylation, and partly attenuated Chk2 and SMC1 phosphorylation after both 2 h and 24 h exposure to HU. Although NBS1, Chk2, and SMC1 are well‐known ATM targets, NBS1, Chk2, and SMC1 were also phosphorylated in an ATR‐dependent manner after HU treatment (Fig. 1c, Fig. S1d). Treatment with the ATR inhibitor VE‐821 also represented the siRNA‐dependent ATR knockdown experiment (Fig. 1d). These data are consistent with previous reports showing that Chk1, NBS1, and SMC1 are phosphorylated in an ATR‐dependent manner after HU or UV exposure13, 14 and that Chk2 can be phosphorylated by ATR in vitro.15 From these results, we concluded that ATM is activated in an ATR‐independent manner after prolonged exposure to HU.
Long continuous HU exposure induces DSB
To investigate the precise mechanisms underlying the activation of the ATM signaling pathway after prolonged HU exposure, the concomitant formation of foci of γH2AX, which is indicative of stalled replication forks and DNA DSBs,16, 17 and RPA2, a hallmark of ssDNA lesions, was investigated by immunofluorescence microscopy. γH2AX and RPA2 foci were detectable after a 2‐h exposure to HU, although these signals were relatively smaller and weaker than those observed after 24 h exposure. At the 2 h time point, most γH2AX foci colocalized with RPA2 foci (Fig. 2a,b,Fig. S2a). After 24 h HU treatment, γH2AX and RPA2 foci were more intense and granular. Interestingly, although both foci were detectable, most γH2AX foci no longer colocalized with RPA2 foci at this time point (Fig. 2a,b, Fig. S2a,b). After HU treatment, pS1981 phosphorylated ATM formed foci colocalizing with γH2AX in HeLa cells (Fig. S3a). After HU treatment for 24 h, pS1981 phosphorylated ATM was detected as clearly larger foci, and some of these were independent from RPA2 foci in wild‐type derived control fibroblasts (WT fibroblasts). However, ATM‐pS1981 foci, consisting of weakly stained fine granules, colocalized with RPA2 foci in Artemis‐mutated RS‐SCID derived AV2/326 cells (Artemis‐deficient fibroblasts) (Fig. S3b). These results are compatible with the interpretation that short‐term HU exposure causes replication fork arrest and prolonged HU exposure induces DSB, consistent with a previous report indicating the existence of DSB after prolonged HU treatment.18 To detect DSBs directly, DNA fragmentations after HU exposure were investigated by PFGE. After more than 12 h of HU exposure, increasing amounts of DSBs were clearly generated (Fig. 2c,d). Collectively, observations from Figures 1 and 2 support a two‐step model for the activation of DNA damage checkpoints in response to HU‐induced replication fork stalling: the primary activation of the ATR signaling as an early phase response to stalled replication fork and the secondary activation of the ATM signaling pathway as a late response to DSB.
Figure 2.
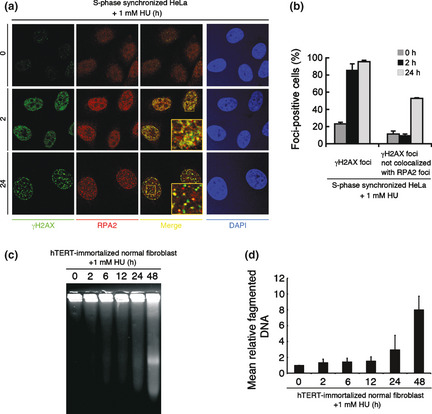
Short‐term hydroxyurea (HU) exposure causes replication fork arrest and 24 h continuous HU exposure induces double‐strand breaks (DSB). (a) HeLa cells were synchronized at S phase and treated with 1 mM HU for 2 h or 24 h. Cells were fixed and stained with anti‐phosphorylated histone H2AX (γH2AX) (Ser139) and anti‐replication protein A2(RPA2) antibodies. (b) More than 100 cells were counted, and the percentage showing >10 γH2AX foci or >4 γH2AX foci not colocalized with RPA2 foci was determined. Data represent the mean ± SEM from three independent experiments. (c) Human telomerase reverse transcriptase (hTERT)‐immortalized normal human fibroblasts (HDF2/326) were treated with 1 mM HU for indicated times. Cells were analyzed by pulse field gel electrophoresis (PFGE). (d) Mean relative fragmented DNA (1 = the fraction of DNA released from the gel plug in untreated fibroblasts) from (c) was calculated, and data are shown in the bar graph. Data represent the mean ± SEM from two independent experiments.
Artemis‐dependent DSBs activate ATM signaling
Because the relationship between prolonged replication fork stalling and DSB generation has not been precisely clarified, we sought to determine the key factor for DSB formation under these conditions. The recruitment of endonuclease(s) to the ssDNA–dsDNA junction, a structure that arises in replication fork stalling, is likely to lead to the generation of DSBs.16, 19 Since the Artemis nuclease is known to process ssDNA–dsDNA junctions in vitro,6, 7 we hypothesized that processing of stalled replication forks by the Artemis nuclease leads to generation of DSBs after prolonged HU exposure. To test this possibility, we investigated HU‐induced DSBs using WT and Artemis‐deficient hTERT‐immortalized fibroblasts derived from RS‐SCID patients. Pulse field gel electrophoresis (PFGE) showed a lower level of dose‐dependent generation of DSBs in Artemis‐deficient fibroblasts compared to WT fibroblasts after HU exposure (Fig. 3). Camptothecin treatment was used as a positive control for replication‐associated DSB,11, 20 showing no significant difference of DSBs in cells with or without Artemis (Fig. S4a,b). Identical results were obtained with the neutral comet assay, which detects DSBs at the single‐cell level, confirming our PFGE data (Fig. S4c,d). These data suggest that DSB generation after prolonged HU exposure is Artemis‐dependent.
Figure 3.
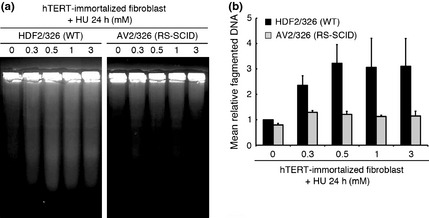
Double‐strand breaks (DSB) generation after replication fork stalling is Artemis‐dependent. (a) Human telomerase reverse transcriptase (hTERT)‐immortalized normal (HDF2/326;WT) and Artemis‐deficient (AV2/326; radiosensitive severe combined immunodeficiency [RS‐SCID]) human fibroblasts were treated with the indicated doses of hydroxyurea (HU) for 24 h. Cells were analyzed by pulse field gel electrophoresis (PFGE). (b) Mean relative fragmented DNA (1 = the fraction of DNA released from the gel plug in untreated fibroblasts) from (a) was calculated, and data are shown in the bar graph. Data represent the mean ± SEM from two independent experiments.
We also investigated whether the extent of DSB formation induced by HU exposure correlates with the level of DNA damage checkpoint activation. Interestingly, Artemis‐deficient fibroblasts treated with HU for 24 h showed attenuated activation of ATM signaling compared to WT fibroblasts (Fig. 4a). To consolidate this finding, we also investigated the effect of Artemis knockdown in HeLa cells using two independent shRNA constructs. These transfectants showed cell cycle kinetics similar to control shRNA‐transfected cells (Fig. S5). Artemis knockdown HeLa cells thus generated showed attenuated activation of the ATM signaling pathway after 24 h HU exposure; in contrast, Chk1 phosphorylation levels were unchanged (Fig. 4b). RPA2 was hyperphosphorylated after only 24 h of treatment with 1 mM HU in Artemis‐competent cells, as previously described.11 Interestingly, hyperphosphorylation of RPA2 was attenuated in Artemis‐deficient fibroblasts. After DNA damage, ATM, ATR, and DNA‐PK dependent phosphorylation of RPA2 plays a key role in replication checkpoint activation. It has been reported hyperphosphorylated RPA2 associates with ssDNA and recombinase protein Rad51 in response to replication arrest by HU treatment and is critical for Rad51 recruitment and homologous recombination‐mediated repair.21Artemis‐deficient cells may also show homologous recombination‐mediated DNA repair defect (Fig 4c,d).
Figure 4.
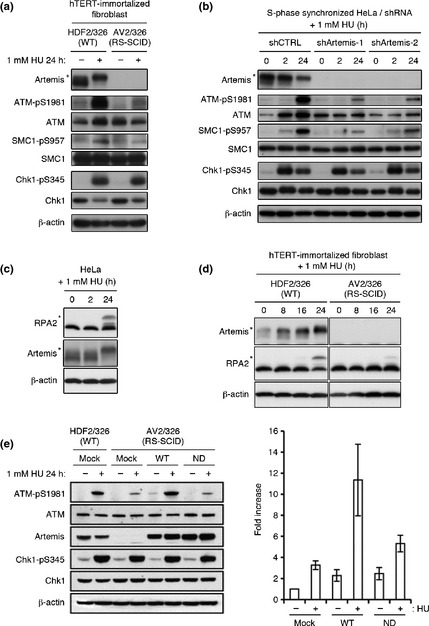
Activation of the ataxia–telangiectasia mutated (ATM) signaling pathway is Artemis‐dependent. (a) HDF2/326 and AV2/326 (radiosensitive severe combined immunodeficiency [RS‐SCID] human fibroblasts were treated with 1 mM hydroxyurea (HU) for 24 h. Cell extracts were immunoblotted with the indicated antibodies. (b) HeLa cells were transfected with a control (shCTRL) or two types of shArtemis‐expressing construct. Cells were synchronized 24 h after transfection. One hour after release, the cells were treated with 1 mM HU for 2 h or 24 h, and cell extracts were immunoblotted. (c) HeLa cells were treated with 1 mU HU and RPA2 phosphorylation status was determined by Western blotting. (d) Western blot analysis of RPA2 phosphorylation status in HDF2/326 and AV2/326 human fibroblasts. (e) HDF2/326 and AV2/326 human fibroblasts were infected with mock or Artemis WT or H254A nuclease dead mutant (ND). Cells were treated with 1 mM HU for 24 h and subjected to Western blot analysis. Right graph indicates relative ATM phosphorylation standardized to 1 as the base level of mock transduced Artemis‐deficient fibroblasts before HU treatment. Data represent the mean ± SEM from three independent experiments. hTERT, human telomerase reverse transcriptase. *Hypersphosphorylated form of Artemis or RPA2.
To address the functional importance of Artemis nuclease activity, we introduced WT or nuclease dead construct (H254A; histidine to alanine substitution on amino acid 254) of Artemis into Artemis‐deficient fibroblasts. As is shown in Figure 4(e), the Artemis nuclease‐dead mutant did not promote efficient activation of ATM signaling after HU treatment. This was in contrast to WT transfectant which efficiently restored ATM activation. Chk1 phosphorylation was indistinguishable between the cells expressing WT and nuclease‐dead mutant Artemis. These results are in accord with the interpretation that the nuclease activity of Artemis plays a critical role in the generation of DSBs when replication fork stalling is prolonged by long exposure to HU, and that DSBs, thus generated, lead to activation of the ATM‐dependent DNA damage response pathway.
DNA‐dependent protein kinase (DNA‐PK) is activated by replication fork stalling
In vitro, Artemis endonuclease activity is controlled by DNA‐PKcs autophosphorylation at the ABCDE cluster.6 Thus, we investigated whether the kinase activity of DNA‐PKcs is involved in Artemis‐dependent DSB formation and subsequent ATM activation following prolonged HU exposure. To gain direct evidence for the activation of DNA‐PKcs, we monitored its autophosphorylation at Ser2056 and/or Thr2609, which have been reported to be essential for DNA‐PKcs function.22, 23 A low level of DNA‐PKcs Ser2056 phosphorylation was detected after 2 h of HU exposure, which increased after 24 h HU exposure (Fig. 5a, Fig. S6a). Immunofluorescence examination using a phospho‐specific antibody against Thr2609 of DNA‐PKcs showed similar results in S‐phase synchronized HeLa cells, in which we observed small foci of phospho‐DNA‐PKcs after a 2 h HU exposure and an increased number and intensity of foci after 24 h HU exposure. These phospho‐DNA‐PKcs foci colocalized with RPA2 foci following 2 h exposure to HU and remained colocalized even after 24 h exposure (Fig. 5b, Fig. S6b), indicating that DNA‐PKcs is activated on stalled replication forks.
Figure 5.
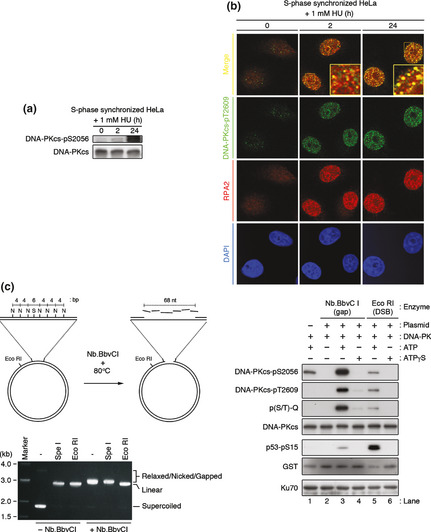
Activation of catalytic subunit of DNA‐dependent protein kinase (DNA‐PKcs) by stalled replication forks after treatment with hydroxyurea (HU). (a) S‐phase synchronized HeLa cells were treated with 1 mM HU 1 h after release from double thymidine block. Cell extracts from the indicated time points were immunoblotted with an anti‐phospho‐DNA‐PKcs (Ser2056) and generic DNA‐PKcs antibody. (b) Same as in (a), cells were stained with anti‐phospho‐DNA‐PKcs (Thr2609) and anti‐RPA2 antibodies. (c) Left panel, generation of a single‐stranded gap region on pG68 plasmid. Nb.BbvCI digestion generated a nick on the plasmid, and subsequent heat denaturation released DNA fragments. N, Nb.BbvCI site; S, SpeI site. Lower panel, restriction digestion analysis with indicated enzymes. Nb.BbvCI‐treated pG68 plasmid was resistant to SpeI digestion (different mobility from EcoRI digestion), indicating the existence of a single‐stranded DNA gap in the plasmid. The DNA‐PK holoenzyme is activated by a plasmid containing a single‐stranded gap DNA in vitro. The reaction was analyzed, followed by SDS‐PAGE and immunoblotting using indicated antibodies.
Because DNA‐PK has been shown to bind to dsDNA ends and is believed to require dsDNA ends for its activation,24, 25 our results in Figure 5(a,b) are difficult to interpret. However, several reports have suggested that DNA‐PKcs exerts kinase activity at ssDNA–dsDNA junctions in the absence of a dsDNA end in vitro.25, 26 The Ku heterodimer is also able to bind to ssDNA–dsDNA junctions in vitro.27 Therefore, we monitored autophosphorylation of DNA‐PKcs (Ser2056, Thr2609, and phospho‐(S/T)Q) and phosphorylation of p53 peptide (amino acids 1–100) as an indicator of kinase activity to investigate whether ssDNA gaps could activate DNA‐PK. To activate DNA‐PK, we used a pG68 plasmid carrying an array of seven Nb.BbvCI and one Eco RI sites.28 Nb.BbvCI or EcoRI treatment created a single‐stranded gap DNA region or dsDNA ends on the plasmid, respectively (Fig. 5c). These enzyme‐treated plasmids facilitated autophosphorylation of DNA‐PKcs only in the presence of ATP (Fig. 5c, lanes 3,5), as assays performed without ATP or with non‐hydrolysable ATPγS were unable to support DNA‐PKcs autophosphorylation (Fig. 5c, lanes 2,4,6). p53 peptide was preferentially phosphorylated in the presence of dsDNA end than single‐stranded gap DNA, which is in accordance with the results of previous studies that used oligonucleotide as activating DNA.25, 26, 27 Thus, our in vitro findings strongly support the interpretation that HU‐induced stalled replication forks activate DNA‐PKcs in cellulo. Therefore, we can conclude that DNA‐PKcs is activated on stalled replication forks (which are presumably single‐stranded gap DNA lesions harboring ssDNA–dsDNA junctions) by HU exposure.
Next, we addressed the relationship between activation of DNA‐PKcs and Artemis in cellulo. Autophosphorylation of DNA‐PKcs was detected in WT and Artemis‐deficient fibroblasts following exposure to HU (Fig. S6c). Immunofluorescence microscopy revealed that autophosphorylated DNA‐PKcs (Thr2609) foci were also formed in the absence of Artemis after HU exposure, with an almost equal percentage of foci‐positive cells in WT and Artemis‐deficient fibroblasts (Fig. S6d). These results suggest that the activation of DNA‐PKcs by HU treatment is not Artemis‐dependent.
Catalytic subunit of DNA‐dependent protein kinase (DNA‐PKcs) is required for HU‐induced DSB formation
Since the inhibition of DNA‐PKcs kinase activity affects the endonuclease activity of Artemis,6 we investigated whether suppression of DNA‐PKcs kinase activity by the specific inhibitor NU7026 affects DSB formation and subsequent ATM activation. Indeed, NU7026 attenuated DSB formation measured by PFGE (Fig. 6a,b) and neutral comet assay (Fig. S7a,b). Identical results were obtained with the DNA‐PKcs‐deficient human glioma cell line M059J and parental control M059K by neutral comet assay (Fig. S7c,d). NU7026 attenuated subsequent activation of ATM signaling after 24 h HU exposure (Fig. 6c). Taken together, these results suggest that DNA‐PKcs is initially autophosphorylated in response to single‐stranded gap DNA lesions at stalled replication forks, which leads to DSB induction mediated by Artemis nuclease and subsequent activation of the ATM signaling pathway.
Figure 6.
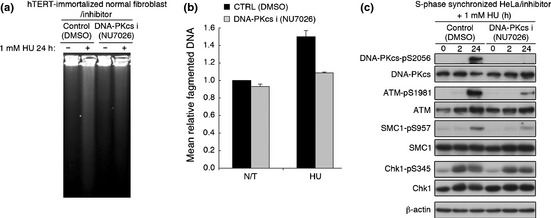
Double‐strand breaks (DSB) generation and ataxia‐telangiectasia mutatedactivation (ATM) activation following prolonged replication fork stalling are dependent on catalytic subunit of DNA‐dependent protein kinase (DNA‐PKcs) activity. (a) Normal human fibroblasts (HDF2/326) were pretreated with DMSO or 10 μM NU7026 for 1 h, and treated with 1 mM HU for 24 h in the presence or absence (DMSO) of NU7026 (10 μM). Cells were analyzed by pulse field gel electrophoresis (PFGE). (b) Mean relative fragmented DNA (1 = the average of the fraction of DNA released from the gel plug in untreated control) from (a) was calculated, and data are shown in the bar graph. Data represent the mean ± SEM from two independent experiments. (c) S‐phase synchronized HeLa cells were treated with 1 mM HU 1 h after release from double thymidine block with DMSO or NU7026 (10 μM). Cell extracts from the indicated time points were immunoblottedwith the indicated antibodies.
Discussion
We revealed that the Artemis/DNA‐PK machinery plays a critical role in generating DSBs after prolonged replication fork stalling by continuous HU exposure. Involvement of some nucleases has been speculated. Recently, the endonuclease Mus81 was shown to be involved in DSB formation after prolonged inhibition of DNA replication by HU and aphidicolin in mouse embryonic stem cells.19 Mus81 was also shown to interact with human Apollo/SNM1B, a member of the SNM1 nuclease family characterized by the presence of a metallo‐β‐lactamase domain, and they have been speculated to work cooperatively for DSB formation after replication stress.29 Because Artemis/SNM1C is also a member of the SNM1 nuclease family, we hypothesized that, like Apollo/SNM1B, Artemis/SMN1C also works cooperatively with Mus81. However, Artemis and Mus81 did not associate with each other before or after HU exposure (data not shown), suggesting that these proteins might function independently against different targets/substrates. For example, Artemis can cleave hairpin or bubble structures, but Mus81 does not process these structures.30
After replication fork stalling, secondary structures such as hairpins, stem‐loops/bubbles, or similar structures might be formed on ssDNA gaps.31, 32 There is a strong possibility that these secondary structures activate DNA‐PK, and are processed by Artemis on the stalled replication fork in cellulo. This is in accord with the idea that autophosphorylation and simultaneous conformational changes in DNA‐PK enhance cleavage of ssDNA–dsDNA junctions by Artemis.6, 33
Replication‐associated DSBs are thought to be one‐sided DSBs, and can cause chromosomal aberrations such as translocation, when these one‐sided DSBs are rejoined with incorrect partners. Indeed, replication‐associated DSBs are thought to be tumorigenic.34, 35 Thus, genome integrity during replication needs to be monitored by several fail‐safe mechanisms. Increase of DSB generation and apoptosis induction is one of the ways to avoid tumorigenesis in Artemis‐competent cells. In contrast, induction of apoptosis was impaired and cellular survival was increased with fewer DSBs in Artemis‐deficient fibroblasts after prolonged HU.36 Artemis‐deficient fibroblasts may have aberrant chromosomes in M‐phase after prolonged HU treatment, as is the case in Mus81‐knockout embryonic stem cells,19 but fail to be eradicated by cell death through apoptosis/mitotic catastrophe. These are in accord with the recent finding that apoptosis induction after massive DSB is Artemis‐dependent.37 Thus, further study is ongoing in order to assess whether genomic abnormalities increase in Artemis‐deficient fibroblasts surviving after HU‐induced replication stress.
In summary, our findings indicate a novel function of the DNA nuclease Artemis in resolving stalled DNA replication forks in cells in S phase, with potential implications for understanding carcinogenesis and therapeutic responses to DNA damaging drugs. Although stalled replication forks initially activate an ATR‐dependent DNA damage response, when stalling is prolonged, cells may trigger a second wave of DNA damage checkpoint response mediated by Artemis‐dependent DSB generation (Fig. 7). Thus, Artemis plays an essential role in the response to DNA damage caused by HU‐induced replication fork stalling. Subsequent activation of the ATM signaling pathway in response to DSB generation could allow two alternative cell fates through either induction of cell death or DSB‐dependent DNA repair/replication restart, thereby preventing the disruption of DNA integrity.
Figure 7.
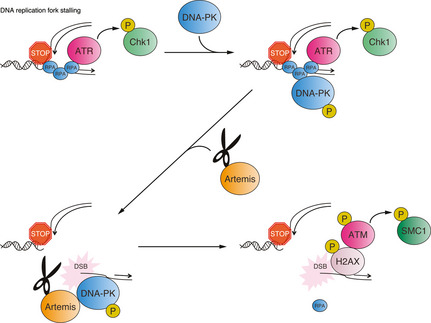
Model for Artemis‐mediated double‐strand break (DSB) formation and subsequent DNA damage checkpoint activation. ATM, ataxia–telangiectasia mutated; ATR, ataxia–telangiectasia and RAD3‐related; DNA‐PK, DNA‐dependent protein kinase;P, phosphorylation; RPA, replication protein A.
Disclosure Statement
The authors have no conflicts of interest.
Abbreviations
- ATM
ataxia–telangiectasia mutated
- ATR
ataxia–telangiectasia and RAD3‐related
- DNA‐PK
DNA‐dependent protein kinase
- DNA‐PKcs
catalytic subunit of DNA‐dependent protein kinase
- dsDNA
double‐stranded DNA
- DSB
double‐strand break
- γH2AX
phosphorylated histone H2AX
- hTERT
human telomerase reverse transcriptase
- HU
hydroxyurea
- PFGE
pulse field gel electrophoresis
- RPA
replication protein A
- RS‐SCID
radiosensitive severe combined immunodeficiency
- shRNA
short hairpin RNA
- siATR
short interfering RNA against ATR
- siRNA
short interfering RNA
- ssDNA
single‐stranded DNA
Supporting information
Doc. S1. Supplementary experimental procedure.
Fig. S1. Cell cycle distribution of S‐phase synchronized HeLa cells, quantification of Figure 1(b); ATM activation in HeLa cells transfected with siATR, quantification of Fig. 1(b) .
Fig. S2. Magnified image of γH2AX and RPA2 foci and signal intensity chromatographs.
Fig. S3. RPA2 and phosphorylated ATM colocalization after hydroxyurea treatment, analyzed by immunofluorescence.
Fig. S4. Camptothecin (CPT) induces double‐strand breaks.
Fig. S5. Cell cycle profiles of shRNA transfected HeLa cells.
Fig. S6. Quantitation of immunofluorescence data shown in Figure 5(b). Magnified image of phospho‐DNA‐PKcs (T2609) and RPA2 fociand signal intensity chromatographs. Artemis‐independent activation of DNA‐PKcs analyzed by Western blotting and immunofluorescence.
Fig. S7. Generation of DSBs dependent on DNA‐PKcs analyzed by comet assay.
Acknowledgments
We thank Dr. Leonard Wu (University of Oxford, Oxford, UK) for providing the pG68 and pG48 plasmids, and Dr. Minoru Takata (Kyoto University, Kyoto, Japan) for providing the Artemis expression plasmid. We also thank N. Terada and M. Sato for characterizing Artemis‐deficient fibroblasts by genomic PCR, S. Nakada, A. Shibata, R. Sakasai, and Y. Ichijima for inspiring discussions, and the members of the H.T. and S.M. laboratories for helpful discussions. This work was supported by a Grant‐in‐Aid from the Ministry of Education, Science, and Culture (Japan) to S.M. and by a Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labor and Welfare (Japan) to S.M. and M.T.
(Cancer Sci, doi: 10.1111/cas.12144, 2013)
References
- 1. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9: 616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA‐PKcs to sites of DNA damage. Nature 2005; 434: 605–11. [DOI] [PubMed] [Google Scholar]
- 3. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421: 499–506. [DOI] [PubMed] [Google Scholar]
- 4. Jazayeri A, Falck J, Lukas C, et al ATM‐ and cell cycle‐dependent regulation of ATR in response to DNA double‐strand breaks. Nat Cell Biol 2006; 8: 37–45. [DOI] [PubMed] [Google Scholar]
- 5. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 2008; 9: 297–308. [DOI] [PubMed] [Google Scholar]
- 6. Goodarzi AA, Yu Y, Riballo E, et al DNA‐PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J 2006; 25: 3880–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma Y, Schwarz K, Lieber MR. The Artemis: DNA‐PKcs endonuclease cleaves DNA loops, flaps, and gaps. DNA Repair (Amst) 2005; 4: 845–51. [DOI] [PubMed] [Google Scholar]
- 8. Riballo E, Kuhne M, Rief N, et al A pathway of double‐strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma‐H2AX foci. Mol Cell 2004; 16: 715–24. [DOI] [PubMed] [Google Scholar]
- 9. Kobayashi N, Agematsu K, Sugita K, et al Novel Artemis gene mutations of radiosensitive severe combined immunodeficiency in Japanese families. Hum Genet 2003; 112: 348–52. [DOI] [PubMed] [Google Scholar]
- 10. Stiff T, Walker SA, Cerosaletti K, et al ATR‐dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J 2006; 25: 5775–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakasai R, Shinohe K, Ichijima Y, et al Differential involvement of phosphatidylinositol 3‐kinase‐related protein kinases in hyperphosphorylation of replication protein A2 in response to replication‐mediated DNA double‐strand breaks. Genes Cells 2006; 11: 237–46. [DOI] [PubMed] [Google Scholar]
- 12. Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14: 397–402. [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao H, Piwnica‐Worms H. ATR‐mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21: 4129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu S, Bekker‐Jensen S, Mailand N, Lukas C, Bartek J, Lukas J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol 2006; 26: 6056–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia‐mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA 2000; 97: 10389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takahashi A, Ohnishi T. Does gammaH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett 2005; 229: 171–9. [DOI] [PubMed] [Google Scholar]
- 17. Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR‐dependent manner in response to replicational stress. J Biol Chem 2001; 276: 47759–62. [DOI] [PubMed] [Google Scholar]
- 18. Robison JG, Lu L, Dixon K, Bissler JJ. DNA lesion‐specific co‐localization of the Mre11/Rad50/Nbs1 (MRN) complex and replication protein A (RPA) to repair foci. J Biol Chem 2005; 280: 12927–34. [DOI] [PubMed] [Google Scholar]
- 19. Hanada K, Budzowska M, Davies SL, et al The structure‐specific endonuclease Mus81 contributes to replication restart by generating double‐strand DNA breaks. Nat Struct Mol Biol 2007; 14: 1096–104. [DOI] [PubMed] [Google Scholar]
- 20. Furuta T, Takemura H, Liao ZY, et al Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication‐dependent DNA double‐strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem 2003; 278: 20303–12. [DOI] [PubMed] [Google Scholar]
- 21. Shi W, Feng Z, Zhang J, et al The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis 2010; 31: 994–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan DW, Chen BP, Prithivirajsingh S, et al Autophosphorylation of the DNA‐dependent protein kinase catalytic subunit is required for rejoining of DNA double‐strand breaks. Genes Dev 2002; 16: 2333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen BP, Chan DW, Kobayashi J, et al Cell cycle dependence of DNA‐dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem 2005; 280: 14709–15. [DOI] [PubMed] [Google Scholar]
- 24. Hammarsten O, Chu G. DNA‐dependent protein kinase: DNA binding and activation in the absence of Ku. Proc Natl Acad Sci USA 1998; 95: 525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martensson S, Hammarsten O. DNA‐dependent protein kinase catalytic subunit. Structural requirements for kinase activation by DNA ends. J Biol Chem 2002; 277: 3020–9. [DOI] [PubMed] [Google Scholar]
- 26. Morozov VE, Falzon M, Anderson CW, Kuff EL. DNA‐dependent protein kinase is activated by nicks and larger single‐stranded gaps. J Biol Chem 1994; 269: 16684–8. [PubMed] [Google Scholar]
- 27. Falzon M, Fewell JW, Kuff EL. EBP‐80, a transcription factor closely resembling the human autoantigen Ku, recognizes single‐ to double‐strand transitions in DNA. J Biol Chem 1993; 268: 10546–52. [PubMed] [Google Scholar]
- 28. Ralf C, Hickson ID, Wu L. The Bloom's syndrome helicase can promote the regression of a model replication fork. J Biol Chem 2006; 281: 22839–46. [DOI] [PubMed] [Google Scholar]
- 29. Bae JB, Mukhopadhyay SS, Liu L, et al Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross‐links. Oncogene 2008; 27: 5045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ciccia A, McDonald N, West SC. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem 2008; 77: 259–87. [DOI] [PubMed] [Google Scholar]
- 31. Burrow AA, Marullo A, Holder LR, Wang YH. Secondary structure formation and DNA instability at fragile site FRA16B. Nucleic Acids Res 2010; 38: 2865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA 2008; 105: 9936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ding Q, Reddy YV, Wang W, et al Autophosphorylation of the catalytic subunit of the DNA‐dependent protein kinase is required for efficient end processing during DNA double‐strand break repair. Mol Cell Biol 2003; 23: 5836–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bartkova J, Horejsi Z, Koed K, et al DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 2005; 434: 864–70. [DOI] [PubMed] [Google Scholar]
- 35. Gorgoulis VG, Vassiliou LV, Karakaidos P, et al Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434: 907–13. [DOI] [PubMed] [Google Scholar]
- 36. Unno J. Artemis‐dependent cell death by prolonged replication fork stalling. Jpn J Pediatr Hematol. 2011; 25: 86–90. [Google Scholar]
- 37. Abe T, Ishiai M, Hosono Y, et al KU70/80, DNA‐PKcs, and Artemis are essential for the rapid induction of apoptosis after massive DSB formation. Cell Signal 2008; 20: 1978–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Doc. S1. Supplementary experimental procedure.
Fig. S1. Cell cycle distribution of S‐phase synchronized HeLa cells, quantification of Figure 1(b); ATM activation in HeLa cells transfected with siATR, quantification of Fig. 1(b) .
Fig. S2. Magnified image of γH2AX and RPA2 foci and signal intensity chromatographs.
Fig. S3. RPA2 and phosphorylated ATM colocalization after hydroxyurea treatment, analyzed by immunofluorescence.
Fig. S4. Camptothecin (CPT) induces double‐strand breaks.
Fig. S5. Cell cycle profiles of shRNA transfected HeLa cells.
Fig. S6. Quantitation of immunofluorescence data shown in Figure 5(b). Magnified image of phospho‐DNA‐PKcs (T2609) and RPA2 fociand signal intensity chromatographs. Artemis‐independent activation of DNA‐PKcs analyzed by Western blotting and immunofluorescence.
Fig. S7. Generation of DSBs dependent on DNA‐PKcs analyzed by comet assay.