

Abstract
Arsenic trioxide (ATO) is one of the most potent drugs in cancer chemotherapy, and is highly effective in treating both newly diagnosed and relapse patients with acute promyelocytic leukemia (APL). Despite a number of reports regarding the molecular mechanisms by which ATO promotes anti‐tumor or pro‐apoptotic activity in hematological and other solid malignancies, the effects of ATO on immune responses remain poorly understood. To further understand and clarify the effects of ATO on immune responses, we sought to examine whether ATO affects the production of nitric oxide (NO) in a lipopolysaccharide (LPS)‐stimulated mouse macrophage cell line, RAW 264.7. Arsenic trioxide was found to prevent NO production in a dose‐dependent manner. Arsenic trioxide significantly inhibited the increase in inducible nitric oxide synthase (iNOS) at both the mRNA and protein levels. Furthermore, our analyses revealed that the inhibitory effect of ATO on iNOS expression was ascribed to the prevention of IRF3 phosphorylation, interferon (IFN)‐β expression, and STAT1 phosphorylation, but not the prevention of the MyD88‐dependent pathway. Taken together, our results indicate that ATO prevents NO production by inhibiting the TIR‐domain‐containing adaptor protein inducing IFN‐β (TRIF)‐dependent pathway, thus highlighting an anti‐inflammatory property of ATO in innate immunity.
Arsenic, an ancient drug used in traditional Chinese medicine, has attracted worldwide interest because it shows substantial anticancer activity in patients with acute promyelocytic leukemia (APL).1, 2 Arsenic trioxide (ATO) has been shown to exert anti‐cancer activity against APL by directly binding to promyelocytic leukemia retinoic acid receptor α (PML‐RARα), an onco‐protein regarded to be crucial for the pathogenesis of APL, which leads to subsequent degradation of PML‐RARα protein.3, 4, 5 Of note, a number of studies have demonstrated the pro‐apoptotic activity of ATO. Indeed, ATO induced apoptosis by increasing CASPASE‐10 expression, which is also associated with histone H3 phosphoacetylation.6 Second, ATO suppressed the expression of anti‐apoptotic genes such as hTERT, MYC and C17, which are associated with Sp1 oxidation via production of reaction oxygen species.7 Finally, ATO induced apoptosis of ATRA‐treated NB4 cells associated with the inhibition of nuclear factor (NF)‐κB activation and the enhancement of JNK activation.8
Bobé P et al.9 reported the therapeutic effects of ATO on the severe autoimmune disorders manifested in MRL/lpr mice, in which ATO significantly reduced lymphoproliferation, suppressed skin lesions, and reduced the serum concentration of immunoglobulins, Th1 cytokines, and NO. Thus, it is likely that ATO may play an important role in the regulation of inflammatory responses. However, the cellular and molecular mechanisms by which ATO regulates immune response remains poorly understood. Nitric oxide, a product of macrophages activated by cytokines, microbial compounds or both, is derived from the amino acid l‐arginine by the enzymatic activity of inducible nitric oxide synthase (iNOS). It functions as a tumoricidal and anti‐microbial molecule both in vitro and in vivo.10, 11, 12, 13, 14, 15 Nitric oxide also plays a diverse role in the pathogenesis of osteoclastogenesis,16, 17 pleurisy,18 and asthma,19, 20 indicating that NO is one of the most versatile molecules in the immune system. Sodium arsenite (SA), which forms a trivalent molecule (As3+), was reported to inhibit LPS‐induced NO production in mouse macrophages.21 Therefore, it is of particular interest to examine whether ATO affects NO production.
In this study, we show the inhibitory effect of ATO on LPS‐induced NO production in the mouse macrophage cell line RAW 264.7. We also discuss the molecular mechanisms underlying the inhibitory effect of ATO on iNOS gene expression.
Materials and Methods
Reagents
RPMI‐1640, penicillin‐streptomycin solution, and sodium nitrite were purchased from Wako Pure Chemical Industries (Osaka, Japan). Arsenic trioxide, LPS, modified Griess reagent, and rabbit anti‐iNOS antibody were obtained from Sigma‐Aldrich (St. Louis, MO, USA). Rabbit anti‐mouse IFN‐β antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Fluorescein isothiocyanate‐labeled rat anti‐mouse CD14 IgG and phycoerythrin (PE)/Cy7‐labeled rat anti‐mouse CD284 (toll like receptor 4: TLR4)/myeloid differentiation protein 2 (MD2) complex antibody were obtained from Biolegend (San Diego, CA, USA). All other primary and secondary antibodies for Western blot analysis (WBA) were obtained from Cell Signaling Technology Japan, (Tokyo, Japan). Cell proliferation kit I (MTT assay) was purchased from Roche (Tokyo, Japan).
Cell culture
RAW 264.7 cells (RAW cells, mouse macrophage cell line) were maintained in RPMI‐1640 supplemented with 5% FBS and penicillin‐streptomycin at 37°C in 5% CO2 humidified air.
Measurement of nitric oxide
RAW cells were plated in a 96‐well culture plate (2.5 × 104 cells per well), and incubated for 24 h. Cells were pre‐incubated in a medium containing various concentrations of ATO (0.001, 0.01. 0.1, 0.5, 1, and 2 μM) for 90 min, and then stimulated with LPS (100 ng mL−1) for 24 h in the presence of ATO. The NO concentrations were determined using Griess reagent. Data represent the mean of triplicate determinations ± standard error (SE). To perform the cytotoxic evaluation of ATO on RAW cells, MTT assays were performed after the Griess assay according to the manufacturer's instructions. The percentage survival was determined by making comparisons with the untreated group.
Quantitative reverse transcription‐PCRs analysis
One million RAW cells were seeded in a 6‐well plate and incubated for 24 h. Cells were pre‐incubated in a medium containing 1 μM of ATO for 90 min, and then stimulated with LPS (100 ng mL−1) for the indicated times in the presence of ATO. Total RNA was extracted by using PureLink RNA purification Kit (Life Technologies Japan, Tokyo, Japan), and was reverse‐transcribed using cDNA reverse‐transcription kit (Life Technologies). cDNA was amplified by KOD FX Neo polymerase (Toyobo, Tokyo, Japan) with SYBR Green I (TaKaRa, Tokyo, Japan). Quantitative analysis was performed using the StepOne Real‐time PCR system (Life Technologies) according to the manufacturer's instructions. Glyceraldehyde‐3‐phosphate dehydrogenase was used as an internal standard. Primers used in this study are detailed in Table 1.
Table 1.
Primers applied for quantitative reverse transcription‐polymerase chain reaction
| Gene symbol | Sequences (5′‐) | Product size (bp) |
|---|---|---|
| iNOS | ||
| Sense | 5′‐ CTG CAG GTC TTT GAC GCT CG | 788 |
| Antisense | 5′‐ GTG GAA CAC AGG GGT GAT GC | |
| IFN‐β | ||
| Sense | 5′‐ GCT GAA TGG AAA GAT CAA CCT C | 216 |
| Antisense | 5′‐ TTC AGA AAC ACT GTC TGC TGG T | |
| GAPDH | ||
| Sense | 5′‐ GAT GAC ATC AAG AAG GTG GTG A | 199 |
| Antisense | 5′‐ TGC TGT AGC CGT ATT CAT TGT C | |
GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; IFN, interferon; iNOS, inducible nitric oxide synthase.
Flow cytometry analysis
RAW cells were stimulated with LPS as described above. Cells were scraped, washed, and re‐suspended in ice‐cold PBS containing 2% FBS, 1 mM EDTA, and 0.05% NaN3 (FACS buffer). Cells were then incubated with FITC‐labeled anti‐CD14 antibody and PE/Cy7‐labeled anti‐TLR4/MD2 antibody on ice for 1 h. After incubation, the cells were washed twice with FACS buffer. Cells were then re‐suspended in FACS buffer before being examined on a FACSCantoII (BD Bioscience, Tokyo, Japan) where 10 000 events were analyzed (determined by forward and side scatter). Data represent the mean fluorescence intensity of triplicate determinations ± SE.
Western blot analysis
RAW cells were stimulated with LPS as described above. Cells were washed with ice‐cold PBS and lysed in loading buffer, containing 125 mM of Tris (pH6.8), 4% sodium dodecyl sulfate (SDS), 10% β‐mercaptoethanol, 20% glycerol and 0.02% bromophenol blue. Western blot analysis was performed as described previously.23 Immune complexes were detected with ImmunoStar LD (Wako) by using LAS‐1000 image analyzer (FujiFilm, Tokyo, Japan). Band intensity was measured by using Image Gauge software (FujiFilm). The relative protein levels were calculated after normalization to an internal standard, β‐actin.
Luciferase reporter assay for NF‐κB activation
RAW cells (2 × 104 cells per well) were plated in 96‐well plate. On the following day, the cells were transiently transfected with 0.15 μg of pBIIX‐Luc reporter plasmid22 for measurement of NF‐κB activity and phRL‐TK vector for internal control by using TransIT‐2020 (Mirus Bio LLC, WI, USA). After 24 h, the cells were pre‐treated with 1 μM of ATO for 90 min, and then stimulated with LPS (100 ng mL−1) for 6 h in the presence of ATO. The cells were lysed and assayed for both firefly and Renilla luciferase activity by using the Dual Luciferase Assay system (Promega) and a Fluoreskan Ascent FL microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
RNA interference
RAW cells (7.5 × 104 cells per well) were plated in a 6‐well culture plate. On the following day, the cells were transfected by Lipofectamine RNAi/MAX (Life Technologies Japan, Tokyo, Japan) with 100 nM of STAT1 siRNA (STAT1 p84/p91 siRNA: sc‐44124) according to the manufacturer's protocol from Santa Cruz Biotechnology. An siGENOME RISC‐Free siRNA (Dharmacon‐Thermo Fisher Scientific, Tokyo, Japan) was used as a negative control.
Results
Arsenic trioxide inhibits NO production in LPS‐stimulated RAW cells
We first examined whether ATO inhibits LPS‐induced NO production in RAW cells by using the Griess assay. As shown in Fig. 1(a), ATO treatment inhibited LPS‐induced NO production in a dose‐dependent manner.On the other hand, the ATO treatment alone at the same concentration range did not affect spontaneous NO production. To examine the viability of ATO‐treated RAW cells, MTT assays were performed 24 h after LPS stimulation. There was no significant difference in viability between the ATO‐treated and untreated RAW cells (data not shown), strongly suggesting that the inhibitory effect of ATO on the NO production was not due to its cytotoxicity. We also examined the effect of the treatment time of ATO on LPS‐induced NO production. The treatment of RAW cells with 1 μM of ATO for 10 min resulted in a marked reduction in LPS‐induced NO production (data not shown), indicating that the brief treatment with ATO was sufficient to inhibit NO production in LPS‐stimulated RAW cells.
Figure 1.
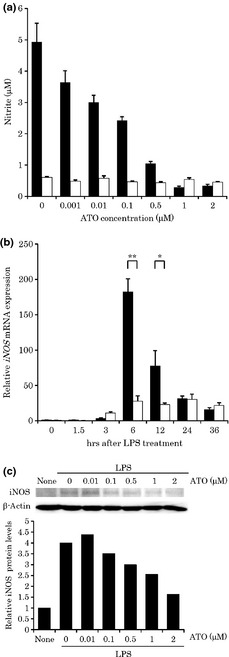
The effects of arsenic trioxide (ATO) treatment on nitric oxide (NO) production (a), inducible nitric oxide synthase (iNOS) mRNA expression (b), dose‐dependent inhibitory effect of ATO on iNOS protein expression (c) in lipopolysaccharide (LPS)‐stimulated RAW cells. (a) RAW cells were treated with ATO (1 μM) for 90 min, and then stimulated with (black bar) or without (white bar) LPS (100 ng mL−1) for 24 h in the presence of ATO (1 μM). The NO concentrations were measured using the Griess reaction (n = 6). (b) Expression of iNOS mRNA was examined using real‐time polymerase chain reaction (PCR). RAW cells were stimulated with LPS (100 ng mL−1) for the indicated times in the presence of ATO (1 μM). The relative iNOS mRNA expression level with the ATO treatment (white bar) compared to that without the treatment (black bar) is shown after normalization to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) mRNA expression. Data are expressed relative to the mRNA levels found in the control (at 0 h after LPS stimulation), which was arbitrarily defined as 1. An asterisk (**) or (*) indicates a significant difference with P‐value of less than 0.005 or 0.05, respectively (n = 3). (c) RAW cells were stimulated with LPS (100 ng mL−1) in the presence of various concentrations (0, 0.01, 0.1, 0.5, 1, and 2 μM) of ATO. Ten micrograms of the protein was prepared from RAW cells to detect the iNOS. After normalization to β‐actin protein, data were expressed relatively to the protein levels found in the untreated cells, which were arbitrarily defined as 1.
Arsenic trioxide inhibits the expression of iNOS mRNA and protein in LPS‐stimulated RAW cells
To further elucidate the molecular mechanism by which ATO inhibits the action of NO, we performed quantitative real‐time PCR analysis on LPS‐induced the iNOS mRNA expression. As shown in Fig. 1(b), the iNOS mRNA expression level was found to readily increase after LPS stimulation, and the maximum fold expression was detected 6 h after stimulation. ATO treatment significantly decreased LPS‐induced iNOS mRNA expression both at 6 h (P < 0.005) and 12 h (P < 0.05) compared with the cells not exposed to ATO. To confirm the inhibitory action of ATO on iNOS protein expression, WBA was performed. As shown in Fig. 1(c), the ATO treatment abrogated LPS‐induced iNOS protein expression in a dose‐dependent manner.
Arsenic trioxide does not alter the cell surface expression of CD14 or TLR4 in RAW cells
Lipopolysaccharide initially binds to CD14, a glycosylphosphatidylinositol (GPI)‐linked protein, which is expressed on the macrophage cell surface. Lipopolysaccharide is then transferred to MD‐2, which associates with and activates TLR4, thereby initiating intracellular signaling events.23 Therefore, we examined whether ATO affects the levels of CD14 and TLR4 expression. Fluorescence‐activated cell sorting analysis revealed no significant changes in the levels of CD14 or TLR4 expression (Table 2), suggesting that ATO may affect iNOS production by inhibiting intracellular signal transduction after LPS stimulation.
Table 2.
The cell surface expression of CD14 or TLR4 in RAW cells
| Mean fluorescence intensity | ||||
|---|---|---|---|---|
| CD14 | TLR4/MD2 | |||
| 12 h | 24 h | 12 h | 24 h | |
| Control | 58.0 ± 0.6 | 73 ± 1.5 | 48.3 ± 14 | 52.7 ± 0.3 |
| None | 5879 ± 47 | 5283 ± 27 | 1042 ± 22 | 909 ± 6 |
| ATO | 4953 ± 25 | 4229 ± 57 | 1037 ± 12 | 1132 ± 8 |
| LPS | 7631 ± 59 | 6669 ± 30 | 669 ± 12 | 1009 ± 14 |
| LPS+ATO | 8645 ± 27 | 7631 ± 93 | 500 ± 7 | 904 ± 24 |
In control group, IgG iso‐type matched control primary antibodies were used for analyses. ATO, arsenic trioxide; LPS, lipopolysaccharide.
Arsenic trioxide does not inhibit activation of NF‐κB or TNF‐α protein expression in LPS‐stimulated RAW cells
It has been reported that the activation of NF‐κB is involved in LPS‐induced NO production in RAW cells. Furthermore, sodium arsenite was reported to inhibit the activation of NF‐κB in LPS‐stimulated RAW cells by preventing the degradation of IκB‐α and IκB‐β.21 Therefore, we examined whether ATO affects LPS‐induced NF‐κB activation by using a luciferase reporter assay. As shown in Fig. 2(a), an increase in NF‐κB luciferase activity was detected 6 h after LPS stimulation. However, this activation was not significantly inhibited by ATO treatment. We also examined the effect of ATO on LPS‐induced phosphorylation of NF‐κB, IκBα by using WBA. As shown in Fig. 2(b), the phosphorylation of NF‐κB and IκB‐α was clearly detected 10 min after LPS stimulation. However, no significant change was observed in the phosphorylation levels with the ATO treatment. Next, we examined whether ATO affects the expression of LPS‐induced TNF‐α, a downstream molecule of NF‐κB, by using an enzyme‐linked immunosorbent assay (ELISA). As shown in Fig. 2(c), the TNF‐α protein was detected 6 h after LPS stimulation. In this setting, ATO had almost no effect on TNF‐α protein expression, further supporting the fact that the inhibitory effect of ATO on NO production was not due to the prevention of NF‐κB activation.
Figure 2.
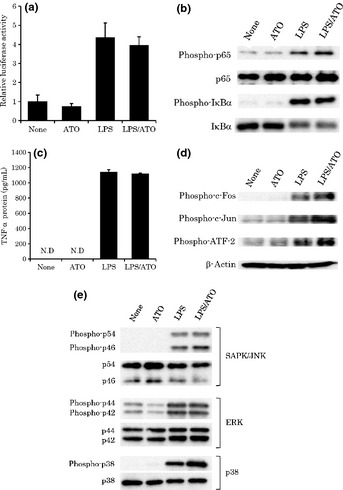
The effect of arsenic trioxide (ATO) treatment on nuclear factor (NF)‐κB activation (a), phosphorylation of NF‐κB and nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor α (b), tumor necrosis factor (TNF)‐α protein expression (c), phosphorylation of AP‐1, ATF‐2 (d), and mitogen‐activated protein kinases (MAPKs) (e) in lipopolysaccharide (LPS)‐stimulated RAW cells. (a) RAW cells were co‐transfected with 0.15 μg of pBIIX‐Luc vector for the NF‐κB firefly luciferase activity and phRL‐TK vector for the internal control Renilla luciferase activity. After 24 h, the cells were treated with or without ATO (1 μM) for 90 min, and then stimulated with LPS (100 ng mL−1) for 6 h in the presence of ATO. After normalization to Renilla luciferase activity, the fold‐change relative NF‐κB activity was expressed relative to that found in the untreated group, which was arbitrarily defined as 1 (n = 6). (b) RAW cells were stimulated with LPS (100 ng mL−1) for 10 min in the presence of ATO (1 μM). Five micrograms of the protein was prepared from RAW cells to detect the total or phosphorylated forms of p65 and IκB‐α. (c) z cells were stimulated with LPS (100 ng mL−1) for 6 h in the presence of ATO (1 μM). Tumor necrosis factor‐α protein expression was quantitatively determined using enzyme linked immunosorbent assay (ELISA) (n = 3). ND, not detected. (d) RAW cells were stimulated with LPS (100 ng mL−1) for 2 h in the presence of ATO (1 μM). Five micrograms of the protein was prepared from RAW cells to detect the phosphorylated forms of c‐Fos, c‐Jun, and ATF‐2. β‐actin protein was used as an internal control. (e) RAW cells were stimulated with LPS (100 ng mL−1) for 30 min in the presence of ATO (1 μM). Five micrograms of the protein was prepared from RAW cells to detect the total or phosphorylated forms of MAPKs.
Arsenic trioxide does not inhibit the phosphorylation of MAPKs, including JNK/SAPK, Erk1/2, and p38, in LPS‐stimulated RAW cells
Lipopolysaccharide is known to activate a series of MAPKs, such as Erk1/2, p38, and JNK/SAPK, which leads to the activation of downstream transcriptional factors such as activator protein 1 (AP‐1) and activating transcription factor 2 (ATF‐2). These signaling pathways are dependent on MyD88, and known to be involved in the regulation of iNOS expression.25 Therefore, we examined the effects of ATO on LPS‐induced phosphorylation of AP‐1 subunits by using WBA. As shown in Fig. 2(d), the phosphorylation of c‐Fos and c‐Jun proteins was detected 2 h after LPS stimulation. However, no significant reduction in the phosphorylation of these transcriptional factors was observed with ATO treatment, whereas the phosphorylation of c‐Jun appeared to be partially enhanced. Similarly, the activation of ATF‐2 (phosphorylated ATF‐2), a trans‐acting element of CREB, was not prevented, suggesting that the inhibitory effect of ATO on NO production did not affect the activation of these transcriptional factors. Next, we examined the effect of ATO on the activation of the MAPKs, including Erk1/2, p38, and JNK/SAPK, the upstream molecules of AP‐1 and ATF‐2. As shown in Fig. 2(e), the phosphorylation of MAPKs was detected 30 min after LPS stimulation. However, no significant reduction was observed in the phosphorylation of these transcriptional factors with ATO treatment. Taken together, these results demonstrate that the inhibitory effect of ATO on iNOS expression is not due to the prevention of the MyD88‐dependent pathway.
Arsenic trioxide prevents the TRIF‐dependent pathway through the inhibition of IRF3 phosphorylation, IFN‐β expression and STAT1 activation
Lipopolysaccharide ‐induced interferon (IFN)‐α/β and STAT1 activation are critical determinants of iNOS gene expression.25 Interferon‐β expression, and subsequently, STAT1 phosphorylation are normally induced in LPS‐stimulated MyD88 deficient mouse macrophages, suggesting that the MyD88‐independent pathway may play an important role in iNOS gene expression. Therefore, we next sought to examine whether ATO affects the LPS‐induced TRIF‐dependent pathway. First, we examined the effect of ATO both on IFN‐β mRNA and protein level in LPS‐stimulated RAW cells. As shown in Fig. 3(a), the increase of IFN‐β mRNA level was detected 2 h after LPS stimulation; this increase was prevented by ATO treatment. To confirm the inhibitory action of ATO on IFN‐β protein expression, we performed WBA. As shown in Fig. 3(b), the ATO treatment reduced the IFN‐β protein expression in LPS‐stimulated RAW cells. To further elucidate the inhibitory effect of ATO on the TRIF‐dependent pathway, we examined the effect of ATO on LPS‐induced phosphorylation of IRF3 transcriptional factor. As shown in Fig. 3(c), the phosphorylation of IRF3 was successfully detected 2 h after LPS stimulation. The phosphorylation was clearly prevented by ATO. In addition, we also examined whether ATO affects the phosphorylation of STAT1α/β. As shown in Fig. 3(d), the phosphorylation of STAT1α/β was detected 4 h after LPS stimulation. Again, this phosphorylation was prevented by ATO. Finally, we performed siRNA experiments to investigate the iNOS protein expression and NO production under knock‐down of STAT1. As shown in Fig. 3(e and f), knockdown of STAT1 resulted in the significant reduction of iNOS protein and NO production (P < 0.005) in LPS‐stimulated RAW cells, suggesting that STAT1 can act as one of the key transcriptional activators of iNOS gene. Collectively, we found that the inhibitory effect of ATO on NO production was ascribed to the prevention of the TRIF‐dependent pathway in LPS‐stimulated RAW cells.
Figure 3.
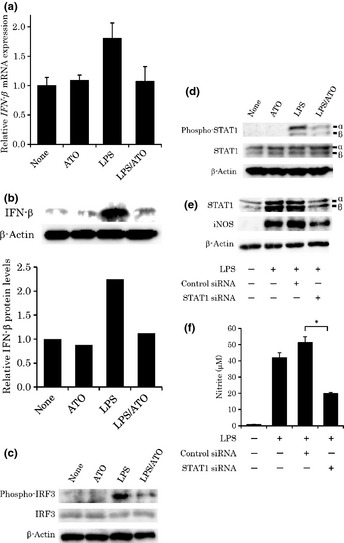
The effect of arsenic trioxide (ATO) on interferon (IFN)‐β mRNA expression (a), IFN‐β protein level (b), phosphorylation of IRF3 (c) and STAT1α/β (d), and the effect of STAT1 siRNA on STAT1 and inducible nitric oxide synthase (iNOS) protein levels (e), and nitric oxide (NO) production (f) in lipopolysaccharide (LPS)‐stimulated RAW cells. (a) RAW cells were stimulated with LPS (100 ng mL−1) for 2 h in the presence of ATO (1 μM). The IFN‐β mRNA expression levels are shown after normalization to GAPDH mRNA expression relative to those found in the untreated group, which were arbitrarily defined as 100% (n = 3). (b) RAW cells were stimulated with LPS (100 ng mL−1) for 2 h in the presence of ATO (1 μM). Twenty micrograms of the protein was prepared from RAW cells to detect the IFN‐β. After normalization to β‐actin protein, bar graph shows the expression relative to the protein levels in the untreated cells, which were arbitrarily defined as 1. (c) RAW cells were stimulated with LPS (100 ng mL−1) for 2 h in the presence of ATO (1 μM). Thirty micrograms of the protein was prepared from RAW cells to detect the phosphorylated forms of IRF3. β‐actin was used as an internal control. (d) RAW cells were stimulated with LPS (100 ng mL−1) for 4 h in the presence of ATO (1 μM). Ten micrograms of the protein was prepared from RAW cells to detect the total or the phosphorylated forms of STAT1α/β. (e) RAW cells were transfected with 100 nM of siRNA specific to mouse STAT1 or nonspecific control siRNA. After 48 h, RAW cells were stimulated with LPS (100 ng mL−1) for 24 h. Ten micrograms of the protein was prepared from RAW cells to detect the total STAT1 or iNOS. β‐actin was used as an internal control. (f) After 48 h of siRNA transfection, RAW cells were stimulated with LPS for 24 h. Nitric oxide concentrations were measured using the Griess reaction. An asterisk (*) indicates a significant difference with a P‐value of less than 0.005 (n = 6).
Discussion
In the present study, we demonstrate that ATO inhibits iNOS expression, leading to the reduction of NO production in LPS‐stimulated RAW mouse macrophage cells by inhibiting the TRIF‐dependent pathway. It is unlikely that the inhibition of iNOS expression by ATO is due to its cytotoxicity or its alteration of TLR4 or CD14 expression levels. Rather, this inhibition is due to the impairment of the LPS‐inducible intracellular signal transduction pathway. After LPS recognition, TLR4 initiates distinct signaling pathways depending on the adapters MyD88 and TRIF, leading to the activation of the transcription factors NF‐κB, AP‐1, ATF2, and IRF3 (Fig. 4). Activation of these transcription factors consequentially induces the production of pro‐inflammatory cytokines such as type I interferons.24 It is well known that iNOS expression is induced by the activation of the TLR4 signaling pathway.25, 27 It is also known that NF‐κB acts as a transcriptional factor for iNOS gene expression through the MyD88‐dependent pathway in LPS‐stimulated mouse macrophages.25, 28 Chakravortty et al. previously reported that sodium arsenite inhibits iNOS expression via prevention of NF‐κB activity.21 These studies prompted us to examine whether ATO affects the activation of NF‐κB. However, in this study, ATO did not exhibit any inhibitory effects on the transcriptional activity of NF‐κB. The transcriptional factors AP‐1 and ATF2, which are activated through MyD88 in the TLR4 signaling pathway, are known to play an important role in the transcriptional regulation of the iNOS gene.25 Therefore, we examined whether ATO affects the activation of AP‐1 and ATF‐2, and the activation of MAP kinases after LPS stimulation. Again, ATO did not show any inhibitory effects on the phosphorylation levels of AP‐1 and ATF‐2 or the phosphorylation levels of MAP kinases, including ERK1/2, p38, and JNK/SAPK. Thus, our results indicate that the inhibitory effect of ATO on iNOS expression is not mediated by the prevention of the activation of the MyD88‐dependent pathway.
Figure 4.
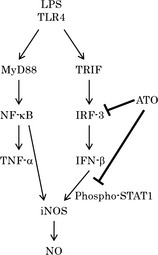
A possible mechanism by which arsenic trioxide (ATO) inhibits nitric oxide (NO) production in the lipopolysaccharide (LPS)/TLR4 signaling pathway. The induction of the iNOS mRNA after LPS stimulation is mediated by the MyD88‐dependent and/or TRIF‐dependent pathway. Nuclear factor (NF)‐κB is independently activated and acts as the transcriptional factor in inducing pro‐inflammatory cytokines, such as tumor necrosis factor (TNF)‐α, through the MyD88‐dependent pathway. In the TRIF‐dependent pathway, IRF3 is phosphorylated, forms homodimers, and acts as a transcriptional activator of interferon (IFN)‐β expression. Subsequently, IFN‐β induces the phosphorylation of STAT1α/β, which acts as a transcriptional activator of the iNOS gene expression. In this study, ATO was found to inhibit NO production in LPS‐stimulated RAW cells through the inhibition of the TRIF‐dependent pathway.
Apart from the MyD88‐dependent pathway, TLR4 triggers the TRIF‐dependent signaling pathway to induce IFN‐β (Fig. 4).24 In the TRIF‐dependent pathway, IRF3 is phosphorylated, forms homodimers, and acts as a transcriptional activator of IFN‐β expression.29 Finally, IFN‐β induces the phosphorylation of STAT1α/β, which acts as a crucial transcriptional factor of the iNOS gene in LPS‐stimulated mouse macrophages.26, 30 Meraz MA et al. and Ohmori Y et al. 31, 32 have independently reported that STAT1 knockout mice failed to produce NO from macrophages after LPS/IFN stimulation. Collectively, these reports prompted us to examine the involvement of the TRIF‐dependent pathway in the inhibitory effect of ATO on iNOS expression. Our results demonstrated that ATO treatment decreases both IFN‐β mRNA and protein expression, and decreases the phosphorylation levels of IRF3. Finally, knockdown of STAT1 resulted in the reduction of iNOS expression, strongly indicating that STAT1 can act as one of the key transcriptional activators of iNOS gene expression. Arsenic trioxide was found to decrease STAT1α/β phosphorylation. This decrease in STAT1 phosphorylation can be partly explained by the prevention of IRF3 activation by ATO. Since IRF3 activation is known to occur through its phosphorylation by IκB kinase ε, it is possible that ATO may prevent the activation of IRF3 by inhibiting this upstream kinase.
Arsenic trioxide has been shown to have pro‐apoptotic activity by affecting a number of signal transduction pathways in APL cells.3, 4, 5, 6, 7, 8, 33, 34 However, the relationship between NO production and apoptosis in APL cells remains poorly understood. In this study, we found that ATO prevents NO production in LPS‐stimulated RAW cells. Therefore, it will be interesting to examine whether ATO exerts pro‐apoptotic activity in APL cells by preventing NO production.
In conclusion, we demonstrate that ATO prevents NO production, at least in part, through inhibition of the TRIF‐dependent pathway. This finding raises the possibility that ATO can regulate TLR4‐inducible pro‐inflammatory gene expression and modulate the TLR4‐mediated innate immune systems. In this study, we have done the experiments by using only one cell line, RAW 264.7. Thus, it will be interesting to examine whether ATO prevents NO production in either mouse or human naive macrophages. Further studies are certainly warranted to contribute to the understanding of the molecular action of ATO in immune defense systems.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This study was supported in part by a grant from the Strategic Research Foundation Grant‐aided Project for Private Universities from Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT) (S1101027) to S. Karnan, H. Konishi and Y. Hosokawa.
(Cancer Sci, doi: 10.1111/cas.12053, 2012)
References
- 1. Zhu J, Chen Z, Lallemand‐Breitenbach V, de The H. How acute promyelocytic leukaemia revived arsenic. Nat Rev Cancer 2002; 2: 705–13. [DOI] [PubMed] [Google Scholar]
- 2. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008; 111: 2505–15. [DOI] [PubMed] [Google Scholar]
- 3. Zhang XW, Yan XJ, Zhou ZR et al Arsenic trioxide controls the fate of the PML‐RARalpha oncoprotein by directly binding PML. Science 2010; 328: 240–3. [DOI] [PubMed] [Google Scholar]
- 4. Lallemand‐Breitenbach V, Jeanne M, Benhenda S et al Arsenic degrades PML or PML‐RARalpha through a SUMO‐triggered RNF4/ubiquitin‐mediated pathway. Nat Cell Biol 2008; 10: 547–55. [DOI] [PubMed] [Google Scholar]
- 5. Tatham MH, Geoffroy MC, Shen L et al RNF4 is a poly‐SUMO‐specific E3 ubiquitin ligase required for arsenic‐induced PML degradation. Nat Cell Biol 2008; 10: 538–46. [DOI] [PubMed] [Google Scholar]
- 6. Li J, Chen P, Sinogeeva N et al Arsenic trioxide promotes histone H3 phosphoacetylation at the chromatin of CASPASE‐10 in acute promyelocytic leukemia cells. J Biol Chem 2002; 277: 49. [DOI] [PubMed] [Google Scholar]
- 7. Chou WC, Chen HY, Yu SL, Cheng L, Yang PC, Dang CV. Arsenic suppresses gene expression in promyelocytic leukemia cells partly through Sp1 oxidation. Blood 2005; 106: 304–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mathieu J, Besançon F. Arsenic trioxide represses NF‐kappaB activation and increases apoptosis in ATRA‐treated APL cells. Ann N Y Acad Sci 2006; 1090: 203–8. [DOI] [PubMed] [Google Scholar]
- 9. Bobe′ P, Bonardelle D, Benihoud K, Opolon P, Chelbi‐Alix MK. Arsenic trioxide: syndromes in MRL/lpr mice. Blood 2006; 108: 3967–75. [DOI] [PubMed] [Google Scholar]
- 10. Bogdan C. Nitric oxide and the immune response. Nat Immunol 2001; 2: 907–16. [DOI] [PubMed] [Google Scholar]
- 11. Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J 1992; 6: 3051–64. [PubMed] [Google Scholar]
- 12. Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA 2000; 97: 8841–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bogdan C, Röllinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol 2000; 12: 64–76. [DOI] [PubMed] [Google Scholar]
- 14. Xie K, Dong Z, Fidler IJ. Activation of nitric oxide gene for inhibition of cancer metastasis. J Leukoc Biol 1996; 59: 797–803. [DOI] [PubMed] [Google Scholar]
- 15. Pervin S, Singh R, Chaudhuri G. Nitric oxide‐induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA‐MB‐231): potential role of cyclin D1. Proc Natl Acad Sci USA 2001; 98: 3583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shibata K, Yoshimura Y, Kikuiri T et al Effect of the release from mechanical stress on osteoclastogenesis in RAW264.7 cells. Int J Mol Med 2011; 28: 73–9. [DOI] [PubMed] [Google Scholar]
- 17. Zheng H, Yu X, Collin‐Osdoby P, Osdoby P. RANKL stimulates inducible nitric‐oxide synthase expression and nitric oxide production in developing osteoclasts. An autocrine negative feedback mechanism triggered by RANKL‐induced interferon‐beta via NF‐kappaB that restrains osteoclastogenesis and bone resorption. J Biol Chem 2006; 81: 15. 809–20. [DOI] [PubMed] [Google Scholar]
- 18. Impellizzeri D, Di Paola R, Esposito E et al CGS 21680, an agonist of the adenosine (A2A) receptor, decreases acute lung inflammation. Eur J Pharmacol 2011; 668: 305–16. [DOI] [PubMed] [Google Scholar]
- 19. Hamid Q, Springall DR, Riveros‐Moreno V et al Induction of nitric oxide synthase in asthma. Lancet 1993; 342: 1510–13. [DOI] [PubMed] [Google Scholar]
- 20. Sordillo JE, Webb T, Kwan D, Kamel J, Hoffman E, Milton DK. Allergen exposure modifies the relation of sensitization to fraction of exhaled nitric oxide levels in children at risk for allergy and asthma. J Allergy Clin Immunol 2011; 127: 1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chakravortty D, Kato Y, Sugiyama T et al The inhibitory action of sodium arsenite on lipopolysaccharide‐induced nitric oxide production in RAW 267.4 macrophage cells: a role of Raf‐1 in lipopolysaccharide signaling. J Immunol 2001; 166: 2011–17. [DOI] [PubMed] [Google Scholar]
- 22. Hosokawa Y, Suzuki H, Nakagawa M, Lee TH, Seto M. API2‐MALT1 fusion protein induces transcriptional activation of the API2 gene through NF‐kappaB binding elements: evidence for a positive feed‐back loop pathway resulting in unremitting NF‐kappaB activation. Biochem Biophys Res Commun 2005; 334: 51–60. [DOI] [PubMed] [Google Scholar]
- 23. Ota A, Yamamoto M, Hori T et al Upregulation of plasma CCL8 in mouse model of graft‐vs‐host disease. Exp Hematol 2009; 37: 525–31. [DOI] [PubMed] [Google Scholar]
- 24. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124: 783–801. [DOI] [PubMed] [Google Scholar]
- 25. Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol 2004; 500: 255–66. [DOI] [PubMed] [Google Scholar]
- 26. Toshchakov V, Jones BW, Perera PY et al TLR4, but not TLR2, mediates IFN‐beta‐induced STAT1alpha/beta‐dependent gene expression in macrophages. Nat Immunol 2002; 3: 392–8. [DOI] [PubMed] [Google Scholar]
- 27. Lowenstein CJ, Snyder SH. Nitric oxide, a novel biologic messenger. Cell 1992; 70: 705. [DOI] [PubMed] [Google Scholar]
- 28. Goldring CE, Reveneau S, Algarté M, Jeannin JF. In vivo footprinting of the mouse inducible nitric oxide synthase gene: inducible protein occupation of numerous sites including Oct and NF‐IL6. Nucleic Acids Res 1996; 24: 1682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamamoto M, Sato S, Hemmi H et al Role of adaptor TRIF in the MyD88‐independent toll‐like receptor signaling pathway. Science 2003; 301: 640–3. [DOI] [PubMed] [Google Scholar]
- 30. Gao JJ, Filla MB, Fultz MJ, Vogel SN, Russell SW, Murphy WJ. Autocrine/paracrine IFNα/β mediates the lipopolysaccharide‐induced activation of transcription factor Stat1α in mouse macrophages: pivotal role of Stat1α in induction of the inducible nitric oxide synthase gene. J Immunol 1998; 161: 4803–10. [PubMed] [Google Scholar]
- 31. Meraz MA, White JM, Sheehan KC et al Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK‐STAT signaling pathway. Cell 1996; 84: 431–42. [DOI] [PubMed] [Google Scholar]
- 32. Ohmori Y, Hamilton TA. Requirement for STAT1 in LPS‐induced gene expression in macrophages. J Leukoc Biol 2001; 69: 598–604. [PubMed] [Google Scholar]
- 33. Chen GQ, Zhu J, Shi XG et al In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl‐2 expression and modulation of PML‐RAR alpha/PML proteins. Blood 1996; 88: 1052–61. [PubMed] [Google Scholar]
- 34. Shao W, Fanelli M, Ferrara FF et al Arsenic trioxide as an inducer of apoptosis and loss of PML/RAR alpha protein in acute promyelocytic leukemia cells. J Natl Cancer Inst 1998; 90: 124–33. [DOI] [PubMed] [Google Scholar]