

Abstract
The tumor suppressor adenomatous polyposis coli (APC) is mutated in familial adenomatous polyposis and in many sporadic colorectal tumors. Adenomatous polyposis coli is known to negatively regulate Wnt signaling by inducing the degradation of β‐catenin. Adenomatous polyposis coli also interacts with the guanine nucleotide exchange factors Asef and Asef2 and stimulates their activity, thereby regulating cell adhesion and migration. Here we show that in confluent, non‐motile MDCK II cells, Asef/Asef2 are colocalized with APC at the sites of cell–cell adhesion at the apical and junctional levels. In contrast, in colorectal tumor cells containing mutated APC, significant amounts of Asef/Asef2 and the truncated mutant APCs are localized mainly in the cytoplasm. These results suggest that localization of the Asef/Asef2‐APC complex at the sites of cell–cell contact is critical for the regulation of cell adhesion, and that the aberrant subcellular localization of these complexes in colorectal tumor cells may contribute to the cell's aberrant adhesive and migratory properties.
The majority of somatic adenomatous polyposis coli (APC) mutations in colon cancers are found in its central region, called the mutation cluster region (MCR), and these typically result in the generation of truncated gene products.1 Normally, APC protein interacts with β‐catenin and induces its degradation.2 However, mutation of APC in colorectal tumor cells results in the accumulation of β‐catenin, which interacts with the T cell factor/lymphoid enhancer binding factor (TCF/LEF) family of transcription factors and activates transcription of target genes. Adenomatous polyposis coli also associates with the lateral plasma membrane in an actin‐dependent manner3 and has been shown to play a role in the maintenance of E‐cadherin based cellular adhesion.4, 5 Furthermore, APC interacts with Asef and the closely related Asef2, which are Cdc42‐ and Rac‐specific guanine nucleotide exchange factors (GEFs), and activates their GEF activity, thereby promoting reorganization of the actin cytoskeletal network and cell migration.6, 7 Asef was also found to inhibit E‐cadherin‐mediated cell–cell adhesion when small colonies of MDCK cells were examined. By contrast, in confluent non‐motile MDCK II cells, Asef increases E‐cadherin levels at the sites of cell‐cell adhesion.8
In the present study, we compared the subcellular localization of Asef/Asef2 in non‐motile versus motile cells. We also examined the relationship between the subcellular localization of Asef/Asef2 and the mutational state of APC in colorectal tumor cells. We show that Asef/Asef2 are colocalized with APC at the sites of cell–cell adhesion in non‐motile MDCK II cells and that they accumulate in lamellipodia in hepatocyte growth factor (HGF)‐treated motile cells. Moreover, we demonstrate that levels of cytoplasmic Asef and APC are higher in colorectal tumor cells containing a mutated APC compared to cells containing wild‐type APC. Hence, the altered subcellular localization of the Asef/Asef2‐APC complex may contribute to the aberrant adhesive and migratory properties of colorectal tumor cells.
Materials and Methods
Antibodies
Rabbit polyclonal antibody (pAb) to Asef/Asef2 was prepared by immunizing rabbits with a peptide containing amino acids 73–126 of Asef (amino acids 98–150 of Asef2). Mouse monoclonal antibody (mAb) to APC was generated as described previously.6 Rat mAb to E‐cadherin (ECCD‐2) was obtained from Calbiochem. Mouse mAb against α‐tubulin and integrin β4 were from Oncogene Research and BD Transduction Laboratories, respectively.
Cell culture
Cells were cultured as shown in Table S1. Polarized cells were grown on polycarbonate filters (Transwell, Costar, Cambridge, MA, USA) with a 0.4 μm pore diam and 24 mm filter diameter. Cells were plated at an initial density of 5.5 × 105/cm2 (2.5 × 106/filter) and cultured in large dishes with polypropylene filter supports. The cells were incubated for 6 days before fixation and processing for immunofluorescence.
Immunostaining
MDCK II cells were fixed with methanol‐acetone (1:1) at −20°C for 5 min (Figs 1, 2) or 3.7% formaldehyde in PBS (Fig. 1b). Fixed cells were double‐stained with rabbit pAb to Asef and rat mAb to E‐cadherin, or rabbit pAb to Asef and mouse mAb to APC, or rabbit pAb to Asef and trimetylrhodamine isothiocyanate (TRITC)‐conjugated phalloidin (Molecular Probes, Eugene, OR, USA) for 60 min at room temperature. Staining patterns obtained with these antibodies were visualized by incubation with fluorescein isothiocyanate (FITC)‐labeled anti‐rabbit IgG (for Asef), TRITC‐labeled anti‐rat IgG (for E‐cadherin) or TRITC‐labeled anti‐mouse IgG (for APC), respectively. The cells were photographed with a Carl Zeiss LSM510 (Carl Zeiss, Jena, Germany) laser scanning microscope.
Figure 1.
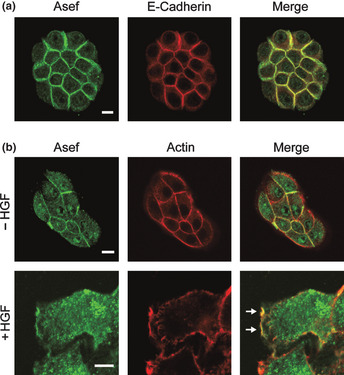
Subcellular localization of Asef/Asef2 in MDCK II cells. (a) Subcellular localization of Asef/Asef2 and E‐cadherin in small colonies of MDCK II cells. (b) Subcellular localization of Asef/Asef2 in MDCK II cells treated with HGF. After stimulation with HGF (20 ng/mL for 2 h), cells were stained for Asef/Asef2 and actin (trimetylrhodamine isothiocyanate [TRITC]‐phalloidin). Arrows indicate the regions of Asef/Asef2 accumulation in membrane ruffles. Bar, 10 μm.
Figure 2.
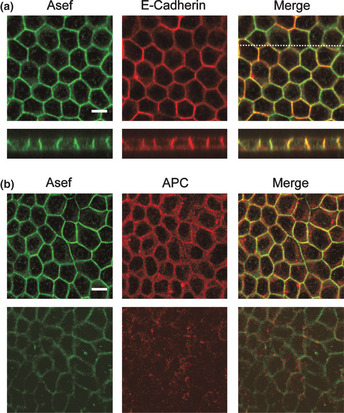
Subcellular localization of Asef/Asef2 and E‐cadherin in polarized MDCK II cells. Cells were stained with antibodies against the indicated proteins. (a, lower panels) z‐section along the line indicated in upper panel. (b, upper panels) Apical, junctional levels; (b, lower panels) basal levels. Bar, 10 μm.
Subcellular fractionation
Confluent cells grown on 10‐cm dishes were homogenized in 600 μL of lysis buffer (50 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM MgCl2, 2 mM EDTA, 10 μg/mL aprotinin and leupeptin, 1 mM phenylmethylsulfonyl fluoride, 500 μM sodium orthovanadate, 10 mM pyrophosphate, 10 mM NaF, and 1 mM dithiothreitol). The lysate was centrifuged at 500 g for 10 min to remove the nuclear fraction, and then the supernatant was recentrifuged at 120 000 g for 45 min (cytoplasmic fraction).
Immunoblotting
Immunoblotting was performed as described previously.7
Results
Subcellular localization of Asef/Asef2 in MDCK II cells
To examine the subcellular distribution of Asef and Asef2 in non‐motile cells, we stained small colonies of MDCK II cells with an antibody that recognizes both Asef and Asef2. We found that Asef/Asef2 were present predominantly at sites on the plasma membrane involved in cell–cell contact, and colocalized with E‐cadherin (Fig. 1a). Asef/Asef2 was not found in regions of the plasma membrane that were not in contact with other cells. We next examined the subcellular localization of Asef and Asef2 in motile cells using MDCK II cells treated with HGF, which induces cell scattering and migration. We found that Asef/Asef2 accumulated in membrane ruffles and lamellipodia in HGF‐treated MDCK II cells, as we reported previously9 (Fig. 1b). Thus, the subcellular localization of Asef and Asef2 are different between non‐motile and motile cells.
Colocalization of membrane‐associated Asef/Asef2 and APC in polarized MDCK II cells
We next examined the localization of Asef/Asef2 in MDCK II cells that had been grown on micropore filters to produce polarized, epithelial‐like monolayers. Confocal z‐sections of polarized MDCK II cells showed that Asef/Asef2 and E‐cadherin were localized predominantly at the lateral plasma membrane and were barely detectable at either the apical or basal membrane (Fig. 2a). We also examined whether Asef/Asef2 are colocalized with APC in polarized MDCK II cells. Adenomatous polyposis coli has been reported to exhibit two distinct peripheral locations in these cells: an actin‐dependent APC pool associated with the lateral plasma membrane and basal APC clusters that depend predominantly on microtubules.3 We found that Asef/Asef2 and APC were colocalized at the sites of cell–cell contact in polarized MDCK II cells, and were barely detectable in clusters of APC at the basal zones (Fig. 2b). These results suggest that the Asef /Asef2‐APC complex plays some role at the sites of cell–cell adhesion.
Subcellular distribution of Asef in colorectal tumor cells
It is known that the subcellular localization of truncated mutant APCs in colorectal tumor cells is different from that of wild‐type APC.3 We therefore examined the subcellular distribution of Asef in colorectal tumor cells expressing either wild‐type or truncated mutant APC. Colorectal tumor cell lines cultured to confluence were fractionated into cytoplasmic and membrane fractions and the amounts of Asef and APC in each fraction were measured by immunoblotting analysis. Consistent with previous reports,3 the levels of cytoplasmic APC in colorectal tumor cells harboring an APC mutation (SW480, SW620, HT29, Caco‐2 and LoVo cells) were significantly higher than those in MDCK II and colorectal tumor cells with wild‐type APC (HCT116 and SW48 cells) (Fig. 3). Furthermore, we found that the levels of Asef in the cytoplasm were also increased in colorectal tumor cells having APC mutations. In addition, we found that the total amounts of APC in colorectal tumor cells containing mutant APC were greater than those containing wild‐type APC mutations, except for LoVo cells. However, we found no relationship between the total expression levels of Asef and the mutational state of APC in colorectal tumor cells (Fig. 3c). These results suggest that a significant fraction of the Asef‐APC complex is not associated with the membrane, but rather is present in the cytoplasm in colorectal tumor cells having a mutated APC.
Figure 3.
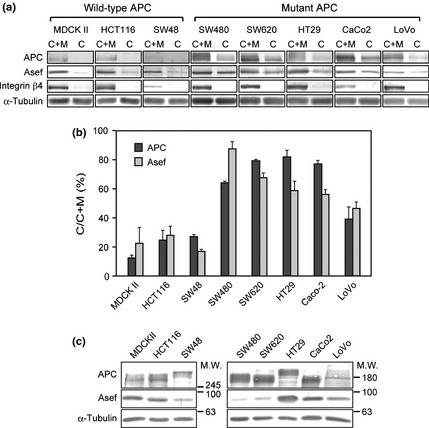
Subcellular distribution of Asef and adenomatous polyposis coli (APC) in colorectal tumor cells. (a) The cytoplasmic (C) and membrane fractions (M) prepared from MDCK II or colorectal tumor cells were subjected to immunoblotting with anti‐Asef/Asef2 and anti‐APC antibodies. The bands of Asef and APC are shown. (b) The ratios of the amounts of cytoplasmic Asef and APC (C) to the total amounts of Asef and APC (C + M) were estimated using NIH image (National Institutes of Health). Each bar represents the mean ± standard error of the mean (SEM) of two independent experiments. (c) The total amounts of APC and Asef/Asef2 in lysates (50 μg of protein) were also analyzed by immunoblotting. Integrin β4 was used as a marker for the membrane fraction. α‐tubulin was used as a loading control and a marker for the cytoplasmic fraction.
Discussion
In the present study, we showed that Asef is colocalized with APC at the sites of cell–cell contact in polarized non‐motile MDCK II cells. We previously showed that expression of Asef causes an increase in the amounts of E‐cadherin and actin filaments present at the sites of cell–cell contact.8 Furthermore, it has been shown that APC is involved in the maintenance of E‐cadherin‐mediated cellular adhesion.4, 5 Thus, our findings raise the possibility that APC‐induced activation of Asef at the sites of cell–cell contact in non‐motile cells may be important for the regulation of E‐cadherin‐mediated cell adhesion. On the other hand, Asef is colocalized with APC in the lamellipodia in motile MDCK II cells treated with HGF. We speculate that the different subcellular localizations of Asef may determine whether Asef stimulates E‐cadherin‐mediated cell–cell adhesion or cell migration.
We previously reported that the preckstrin homology (PH) domain of Asef binds to PtdIns(3–5)P3 and targets Asef to the cell–cell adhesion sites in MDCK II cells.8 However, we found that the levels of plasma membrane‐associated Asef in colorectal tumor cells containing a mutant APC were significantly lower than those in colorectal tumor cells containing wild‐type APC. Truncated mutant APCs present in colorectal tumor cells retain the armadillo repeat domain and are still able to interact with Asef via this domain.6 Thus, the cytoplasmic localization of Asef in colorectal tumor cells containing mutant APC may be directed by the truncated APC. Our results suggest that the aberrant subcellular localization of the Asef/Asef2‐APC complex in colorectal tumor cells may contribute to their aberrant adhesive and migratory properties.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Table S1. Cell culture medium.
Acknowledgments
This work was supported by the Research Program of Innovative Cell Biology by Innovative Technology (Integrated Systems Analysis of Cellular Oncogenic Signaling Networks), Grants‐in‐Aid for Scientific Research on Innovative Areas (Integrative Research on Cancer Microenvironment Network, and Regulation of Polarity Signaling during Morphogenesis, Remodeling, and Breakdown of Epithelial Tubule Structure), Takeda Science Foundation, Kobayashi Foundation for Cancer Research and in part by Global COE Program (Integrative Life Science Based on the Study of Biosignaling Mechanisms), MEXT, Japan.
(Cancer Sci 2013; 104: 1135–1138)
References
- 1. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet 2001; 10: 721–33. [DOI] [PubMed] [Google Scholar]
- 2. Clevers H, Nusse R. Wnt/β‐catenin signaling and disease. Cell 2012; 149: 1192–205. [DOI] [PubMed] [Google Scholar]
- 3. Rosin‐Arbesfeld R, Ihrke G, Bienz M. Actin‐dependent membrane association of the APC tumour suppressor in polarized mammalian epithelial cells. EMBO J 2001; 20: 5929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carothers AM, Melstrom KA Jr, Mueller JD, Weyant MJ, Bertagnolli MM. Progressive changes in adherens junction structure during intestinal adenoma formation in Apc mutant mice. J Biol Chem 2001; 276: 39. [DOI] [PubMed] [Google Scholar]
- 5. Townsley FM, Bienz M. Actin‐dependent membrane association of a Drosophila epithelial APC protein and its effect on junctional Armadillo. Curr Biol 2000; 10: 1339–48. [DOI] [PubMed] [Google Scholar]
- 6. Kawasaki Y, Senda T, Ishidate T et al Asef, a link between the tumor suppressor APC and G‐protein signaling. Science 2000; 289: 1194–7. [DOI] [PubMed] [Google Scholar]
- 7. Kawasaki Y, Sato R, Akiyama T. Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat Cell Biol 2003; 5: 211–15. [DOI] [PubMed] [Google Scholar]
- 8. Muroya K, Kawasaki Y, Hayashi T, Ohwada S, Akiyama T. PH domain‐mediated membrane targeting of Asef. Biochem Biophys Res Commun 2007; 355: 85–8. [DOI] [PubMed] [Google Scholar]
- 9. Kawasaki Y, Tsuji S, Sagara M, Echizen K, Shibata Y, Akiyama T. Adenomatous polyposis coli and asef function downstream of hepatocyte growth factor and phosphatidylinositol 3‐Kinase. J Biol Chem 2009; 284: 22436–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Cell culture medium.