

Abstract
AML1 / RUNX1 is a frequent target of chromosome translocations and mutations in myeloid and B‐cell leukemias, and upregulation of AML1 is also observed in some cases of T‐cell leukemias and lymphomas. This study shows that the incidence of thymic lymphoma in p53‐null mice is less frequent in the Aml1+/− than in the Aml1+/+ background. AML1 is upregulated in p53‐null mouse bone‐marrow cells and embryonic fibroblasts. In the steady state, p53 binds to and inhibits the distal AML1 promoter. When the cells are exposed to stresses, p53 is released from the distal AML1 promoter, resulting in upregulation of AML1. Overexpression of AML1 stimulates T‐lymphocyte proliferation. These results suggest that upregulation of AML1 induced by loss of p53 promotes lymphoid‐cell proliferation, thereby inducing lymphoma development.
AML1, also known as the RUNX1 transcription factor, is encoded by one of the most frequently mutated genes in acute leukemia. The AML1 gene is disrupted by translocations in leukemia.1, 2 AML1 is also inactivated by point mutations within its DNA‐binding domain in both acute myeloid leukemia (AML)3 and myelodysplastic syndromes (MDS).4 AML1 haploinsufficiency underlies familial platelet disorder, an inherited leukemia predisposition syndrome.5 On the other hand, amplification of the locus containing AML1 and overexpression of AML1 are observed in some cases of leukemias and lymphomas,6, 7, 8, 9, 10 suggesting that an increased dosage of normal AML1 may contribute to leukemogenesis.
The p53 tumor suppressor functions in the most important checkpoint‐control pathway that prevents human cancer.11 Although hematopoiesis in p53‐null mice proceeds apparently normally, many studies have implicated p53 in the proliferation, differentiation, apoptosis, and aging of hematopoietic cells.12, 13, 14, 15, 16 Moreover, p53 deletions and mutations are detected at high frequencies in blast crisis chronic myelogenous leukemia and acute leukemia, as well as in almost 50% of adult T‐cell leukemias.17, 18 In the present study, we aimed to elucidate the relationship between AML1 and p53 in T‐cell malignancy. Because the most frequent tumor to develop in p53‐null mice is T‐cell lymphoma,19 we studied the role of AML1 in cancer development using the p53‐null mouse as an animal model system of T‐cell acute lymphoblastic leukemia (T‐ALL).
Materials and Methods
Mice
Aml1 +/− mice (a gift from T. Okuda) were mated to p53 +/− mice (a gift from H. Koseki) to generate Aml1 +/− p53 +/− male mice. The mice were then mated to p53 +/− female mice to generate Aml1 +/+ p53 −/− or Aml1 +/− p53 −/− mice. For in vitro T‐cell proliferation experiments, Aml1 fl/fl mice20 were mated to tamoxifen‐inducible ERT2‐Cre knock‐in mice (Artemis Pharmaceuticals GmbH, Cologne, Germany)21 to generate Aml1 fl/fl ERT2‐Cre mice. Active Cre‐recombinase was induced by the addition of 50 nM 4‐hydroxytamoxifen into the culture medium. The background of the mice used in this study is C57BL/6. The mice were kept at the Division of Animal Laboratory (National Cancer Center, Tokyo, Japan), according to institutional guidelines.
Quantitative RT‐PCR
Total RNA extraction, reverse transcription, and quantitative PCR were performed using ISOGEN (Wako Chemical, Osaka, Japan); the High Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA); and TaqMan probes for RUNX1 (Hs00231079_m1), RUNX3 (Hs00231709_m1), TP53 (Hs01034249_m1), Runx1 (Mm00486762_m1), Runx2 (Mm0003003491_m1), Runx3 (Mm00490666_m1), Trp53 (Mm01731290_g1), Pre T‐cell antigen receptor alpha (Mm00478363_m1), Fas (Mm00433237), and 18S rRNA (Hs99999901_s1), purchased from (Applied Biosystems). Expression levels were normalized to those of 18S rRNA.
Chromatin immunoprecipitation
ChIP analysis was performed as described,22 using primary antibodies specific for p53 (sc‐6243 and DO1) purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) or antibodies for Histone H3 (ab1791), Histone H3 K4Me3 (ab8580), or Histone H3 K9Ac (ab4441) purchased from Abcam (Cambridge, MA, USA). Quantitative real‐time PCR was performed on precipitated DNA using primers designed by primer express software (Applied Biosystems). The values relative to input were determined using a standard curve, and the relative quantification method was as described in ABI User Bulletin #2. The ChIP primer sequences corresponding to the mouse Aml1 or human AML1 distal promoter regions are available upon request.
T‐cell proliferation assay
Number of living cells was determined by measuring ATP production using the Cell Titer Glo assay kit (Promega, Fitchburg, WI, USA) and a GLOMAX microplate luminometer (Promega).
Results
Dosage of Aml1/Runx1 is critical for development of T‐cell lymphoma in p53‐null mice
Loss of the p53 allele and amplification or overexpression of the AML1 gene is frequently observed in human lymphoblastic leukemia or lymphoma. Consistent with this, thymic lymphoma is the most frequent malignant disease in p53‐null mice.19 To analyze the effect of Aml1 gene dosage on the generation of lymphoma, we generated Aml1 +/− mice in a p53 −/− background. We were unable to produce Aml1 −/− p53 −/− mice because the Aml1 −/− mice died at embryonic days E11.5–E12.5, as reported.23 We then compared the frequency of spontaneously occurring tumors in Aml1 +/+ p53 −/− mice and Aml1 +/− p53 −/− mice. Within a period of 40 weeks after birth, the majority of malignant disease (89%) in Aml1 +/+ p53 −/− mice was T‐cell lymphoma. However, in Aml1 +/− p53 −/− mice, a wide range of malignancies was observed, with sarcoma being the major malignancy (26%) (Fig. 1a, Table S1); by contrast, only two cases of lymphoma (9%) were observed in Aml1 +/− p53 −/− mice. In those lymphomas, the levels of Aml1 mRNA were higher than those in normal T cells (Fig. 1b). Fluorescence‐activated cell sorting and RT‐PCR analyses revealed that lymphoma cells derived from Aml1 +/+ p53 −/− and Aml1 +/− p53 −/− mice were positive for T‐cell receptor, CD4 and CD8 (Fig. S1a,b). Event‐free survival was significantly different between the two genotypes (P = 0.0325) (Fig. 1c). These results suggest that expression levels of AML1 affect the incidence of malignancies in p53‐null mice.
Figure 1.
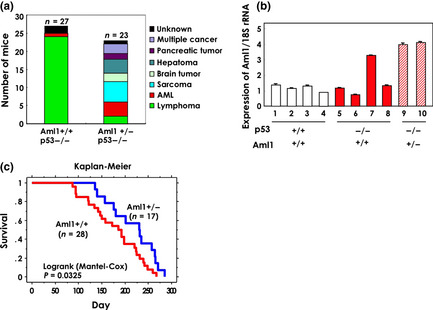
Aml1 haploinsufficiency in p53‐null mice prevents the development of T‐cell lymphoma. (a) Spontaneous onset of malignant transformation in p53‐null mice of Aml1+/+ and Aml1+/− genotypes. The majority of lymphomas developed in Aml1+/+ mice were T‐cell lymphomas (for details, see Table S1). (b) The level of Aml1 transcripts in T‐cell lymphoma. Total RNA of thymus from six mice with lymphoma (red bar, filled: Aml1+/+p53−/−, red bar, diagonal stripes: Aml1+/−p53−/−) and four normal controls (white) was isolated and quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) was performed as described in Experimental Procedures. The data shown are the means ± standard error of the mean (SEM). (c) Event‐free survival curves for p53−/−Aml1+/+ mice (n = 28) and p53−/−Aml1+/− mice (n = 17). Mice carrying sarcomas (n = 6) were not included in this analysis, because these mice were killed at the onset of disease. Statistical analysis was performed using the statview software.
AML1 is upregulated in cells with p53 aberrations
To determine whether the level of Aml1 transcripts correlates with the presence or absence of p53, we compared expression of Aml1 in various types of cells from littermate mice of the p53 +/− and p53 −/− genotypes. We observed increased expression of Aml1 in total bone‐marrow (BM) cells, c‐Kit+ BM cells, common lymphoid progenitors (CLPs), splenic T cells, and thymic T cells, as well as in p53 −/− mouse embryonic fibroblast (MEF) cells (Fig. 2a–d). Considering that the major function of p53 is regulation of stress‐response genes, we tested the effect of stress on the expression of Aml1 in BM cells from another littermate mouse. Aml1 is induced by ionizing radiation (IR) in mice with normal p53, but this induction was not observed in p53 −/− cells (Fig. 2e). Levels of Aml1 expression were increased by p53 shRNA treatment (Fig. S2). Together, these data strongly suggest that Aml1 is transcriptionally regulated by p53 both in the steady state and following genotoxic stress.
Figure 2.
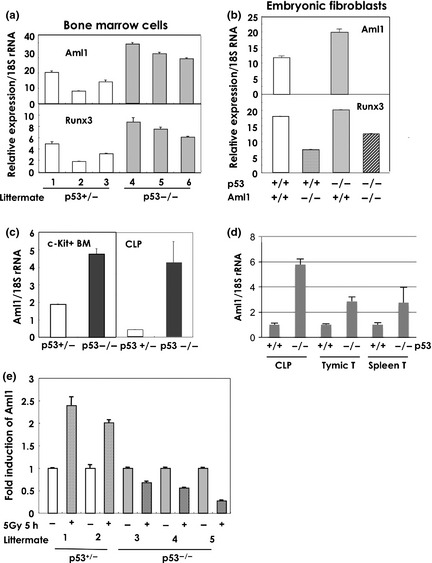
AML1 is overexpressed in cells with p53‐null mutations and is induced by stress. (a) Aml1 is overexpressed in bone‐marrow cells from p53‐null mice in the steady state. The level of Aml1 transcripts in bone marrow cells from littermate mice, generated by mating a p53+/− female and a p53−/− male mouse, was measured. Total RNA was prepared and subjected to quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) analysis of Aml1and Runx3. 1–3: p53+/− littermates, 4–6: p53−/− littermates. (b) Aml1 is overexpressed in mouse embryonic fibroblast (MEF) cells from p53‐null mice The level of Aml1 and Runx3 transcripts in the MEFs of the indicated phenotype was measured by qRT‐PCR using total RNA of cells at the time of passage 2 as described in Experimental Procedures. MEF cells were isolated from littermate E11.5 embryos that were generated by crossing Aml1+/− male and Aml1+/− female mice of p53 wild type background. The data shown are the means ± standard error of the mean (SEM). (c) Aml1 is overexpressed in bone marrow cells from p53‐null mice. The level of Aml1 transcripts, and control 18S rRNA, in c‐Kit+ bone marrow cells and in the bone marrow CLP (c‐Kitlow, Sca1+, IL‐7Ra+, Lin−) fraction of littermate mice, generated by mating a p53+/− female and a p53−/− male mouse. Total RNA was prepared and subjected to qRT‐PCR for assay of Aml1 and 18S rRNA. The data shown are the means ± SEM. (d) Aml1 is overexpressed in p53‐null cells. Total RNA was isolated from c‐Kit+ bone marrow (BM) cells, CLP, thymocytes, CD4+ splenic T cells, and MEFs, all of which were prepared from wild‐type and p53 −/− littermates. The level of Aml1 transcripts was analyzed. The data shown are means ± SEM. (e) Aml1 expression is also induced by IR in mouse cells. The level of Aml1 transcripts was measured in bone‐marrow cells from littermate mice, generated by mating a p53+/− female and a p53−/− male mouse. The cells were exposed to 5 Gy of IR and cultured for 5 h. Total RNA was prepared and subjected to semi‐quantitative RT‐PCR for analysis of Aml1. 1, 2: p53+/− littermates, 3–5: p53−/− littermates.
AML1 is regulated by p53
There are two promoters in the AML1 gene.24 The distal promoter is more active than the proximal promoter in hematopoietic stem cells and developing T cells.25 To determine whether p53 binds to the distal promoter region of Aml1, we performed chromatin immunoprecipitation (ChIP) using c‐Kit+ cells from mouse BM. For this purpose, we used PCR primers corresponding to regions ranging from −10 kb upstream to +4 kb downstream of the Aml1 transcription start site (Fig. 3b). When c‐Kit+ cells were cultured in vitro, the expression of Aml1 was induced (Fig. 3a). Binding of p53 to the Aml1 promoter could be detected in ChIP analysis before, but not after, cultivation (Fig. 3b). ChIP analysis of histone H3 trimethylated at lysine 4 (H3K4Me3) and histone H3 acetylated at lysine 9 (H3K9Ac), both of which are hallmarks of active gene transcription, indicated that activating modifications of chromatin increased in proportion to the Aml1 mRNA level. H3 trimethylated at lysine 9 (H3K9Me3), which is a repressive mark, was not altered by the cultivation. These results indicated that, at steady state immediately following the isolation of c‐Kit+ cells from BM, p53 was preferentially bound close to and downstream of the transcription start site. However, after a 3‐day culture of c‐Kit+ cells, significant binding of p53 was no longer detected at any locus tested (Fig. 3b), even though the expression of p53 was high at this time. At steady state, the Aml1 distal promoter region appeared to be inactive, as demonstrated by the relatively lower levels of H3K4Me3 and H3K9Ac occupancy revealed by ChIP analysis. Conversely, levels of H3K4Me3 and H3K9Ac increased at the downstream region of the Aml1 promoter (primers no. 3–5) after a 3‐day culture (Fig. 3b and Fig. S3). Occupancy of the Aml1 promoter by p53 was also decreased following irradiation (Fig. 3c). The results of these ChIP experiments indicate that the activity of the Aml1 promoter is inhibited by binding of p53 under normal conditions, and that this inhibition was abolished under conditions of stress. In a reporter assay, the Aml1 promoter was inhibited by wild‐type p53 and activated by the p53 R175H mutant, which is defective in binding to DNA (Fig. 4). These results indicate that p53 inhibits transcription of the Aml1 gene. The p53 R175H mutant may function in a dominant‐negative manner.
Figure 3.
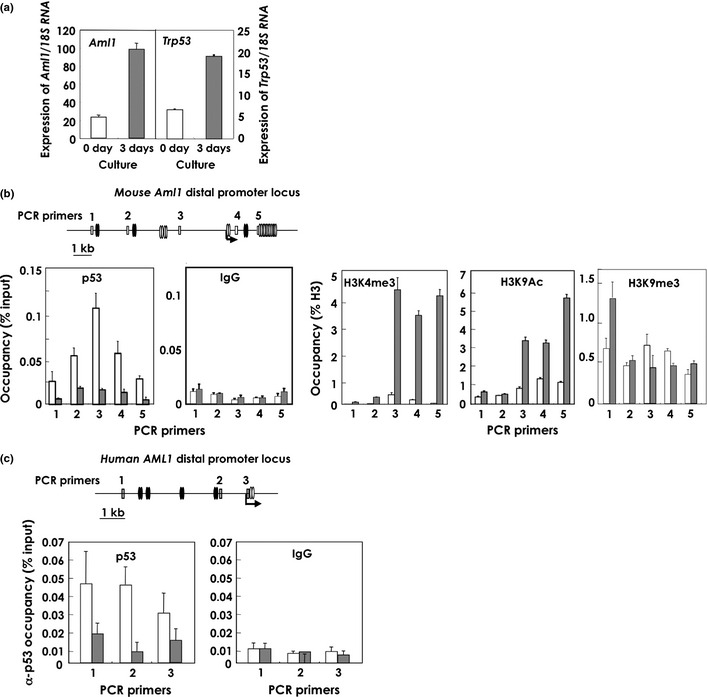
p53 binds to the AML1 promoter of hematopoietic cells at steady state, and the binding is reduced after exposure to stresses. (a) Quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) of the Aml1 and Trp53 transcripts in mouse bone‐marrow c‐Kit+ cells before (0 day) or after (3 day) 3‐day in vitro culture with cytokines (SCF and IL‐3). The data shown are means ± standard error of the mean (SEM). (b) Upper panel: Mouse Aml1 distal promoter regions contain potential AML1‐ and p53‐binding motifs. The primers designed for ChIP assay of the mouse Aml1 distal promoter region are indicated by numbers. The arrow indicates the transcription start site. Oval doublets represent two separate DNA motifs within 20 bp (black: p53‐binding candidate motifs, white: AML1‐binding motifs). Lower panel: ChIP assays for the detection of p53 binding and chromatin modification (methylation: H3K4me3; acetylation: H3K9Ac), as indicated in the figures. White bars: uncultured mouse bone‐marrow c‐Kit+ cells; gray bars: c‐Kit+ cells after a 3‐day culture. ChIP primers, corresponding to bases within the DP2 region of the Aml1 promoter as shown in a, were used. The data shown are means ± SEM. (c) Upper panel: Primers designed for the ChIP assay of the human AML1 distal promoter region are indicated as numbers. Lower panel: ChIP assay for detection of anti‐p53 antibody binding. The data shown are means ± SEM. White bars: MOLT4 cells without IR treatment; gray bars: MOLT4 cells 2 h after IR (10 Gy) treatment. Oval doublets represent two separate DNA motifs within 20 bp (black: p53‐binding candidate motifs; white: AML1‐binding motifs).
Figure 4.
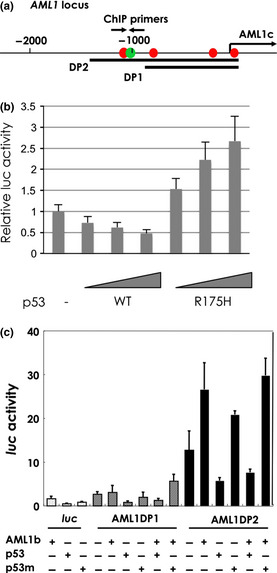
p53 inactivates AML1 promoter. (a) The indicated promoter regions of 900 bp (DP1) and 1400 bp (DP2) were cloned into luc reporter plasmids. The arrow labeled AML1c indicates the transcription start site. The opposing arrows indicate the positions of the primers used for ChIP analysis. (b, c) AML1 promoter is repressed by p53. SaOs2 cells were transfected with expression vector for wild‐type p53, p53R175H and/or AML1b together with a human AML1 distal promoter–luciferase (luc) reporter containing DP1 or DP2. Data shown are relative values of firefly luciferase activity normalized to Renilla luciferase activity. The data shown are the means ± standard error of the mean (SEM).
Depletion of AML1 blocks T‐cell proliferation
To determine whether the expression level of AML1 affects proliferation of T cells, we mated mutant animals carrying loxP‐flanked (Aml1 fl) alleles20 with ERT2‐Cre knock‐in mice21 to generate Aml1 fl/fl ERT2‐Cre mice. In these mice, the Aml1 fl allele could be effectively deleted in vitro in hematopoietic cells by the addition of 4‐hydroxytamoxifen (4‐OHT), which activates ERT‐Cre recombinase (Fig. 5a). After 5 days of 4‐OHT treatment, the Aml1 gene was deleted from almost all cells (Fig. 5b), and the Aml1 transcript was also almost completely absent, as determined by RT‐PCR (Fig. 5c). At this time, the expression level of Runx3, which might compensate for loss of AML1 function, was not altered (Fig. S4). We then examined T‐cell proliferation in these Aml1 fl/fl ERT2‐Cre mice. Splenic CD4 cells were isolated from the Aml1 fl/fl ERT2‐Cre mice and cultured in the presence of IL‐2 after stimulation with anti‐CD3e. Ten days after the addition of 4‐OHT, we compared the number of live cells from Aml1 fl/fl ERT2‐Cre and Aml1 w/w ERT2‐Cre mice. The proliferation of T‐cells from which Aml1 was deleted was clearly lower than that of T‐cells from Aml1 w/w ERT2‐Cre mice (Fig. 5d). This result is consistent with a previous report showing that conditional deletion of Aml1 results in decreased numbers of T cells.20 These observations suggest an important role for AML1 in T‐cell proliferation.
Figure 5.
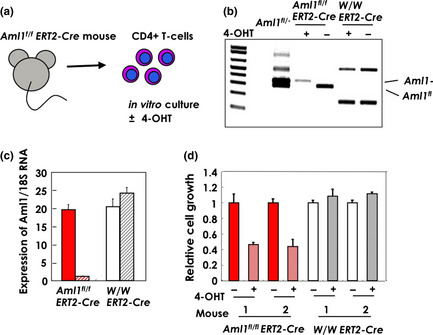
Depletion of AML1 inhibits the growth of CD4 positive T‐cells. (a) Splenic T‐cells of Aml1 fl/flERT2‐Cre mice were isolated using an antibody against CD4 and cultured for 5 days with or without 4‐hydroxytamoxifen (4‐OHT) in the presence of IL‐2. (b) Genotype of Aml1 was analyzed after 5‐day culture with or without 4‐OHT in the presence of IL‐2. Primers used for genotyping were as follows: f2: ACAAAACCTAGGTGTACCAGGAGAACAAGT, r1: GTCTACTCCTTGCCTCAGAAAACAAAAAC, fl20: CCCTGAAGACAGGAGAAGTTTCCA. (c) Expression of Aml1 was measured by quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) and correlated to 18S rRNA levels after 5‐day culture with or without 4‐OHT in the presence of IL‐2. (d) T‐cells with or without the Aml1 allele were cultured in the presence of IL‐2 for 10 days. The growth rate (live cells) was measured by the production of adenosine triphosphate (ATP). ATP production of cells from Aml1fl/flERT2‐Cre mice (red bars, diagonal stripe) treated with 4‐OHT was less than that of untreated cells (red bars, filled). The drug was not toxic to wild‐type cells (white and gray diagonal stripes: treated and non‐treated, respectively).
Discussion
RUNX family members function as oncogenes when they are ectopically overexpressed.26, 27, 28, 29 In this study, we elucidated a novel relationship between AML1 and p53 in vivo using mice that carried mutations in one or both genes, and we present genetic evidence for a mechanism underlying the overexpression of AML1.
The lower frequency of onset of T‐cell lymphoma in association with the Aml1 +/− p53 −/− genotype may indicate that the presence of two copies of the Aml1 gene are needed for T‐cell transformation and/or proliferation, and that one allele of Aml1 confers no growth advantage to malignant cells. Because we did not examine the expression level of Aml1 at the early stage of T‐cell transformation, we cannot fully resolve the importance of AML1 in T‐cell malignancy. Moreover, a genetic defect in p53 affects the regulation of a large number of genes. Therefore, Aml1 hemizygosity might not be the factor that directly prevents malignant transformation in a p53‐null background.
Aml1 might function as an oncogene in a lymphoma that develops due to a defect in p53. Consistent with this idea, expression levels of Aml1 were higher in p53 −/− lymphocytes than those observed in wild type cells (Fig. 2c,d). However, expression levels of Aml1 were not constantly high in lymphomas (Fig. 1b). These results suggest that high levels of Aml1 expression might be important for development, but not for maintenance, of lymphomas. The mechanism of this enhanced expression of Aml1 remains unclear, although the data support the hypothesis that a high level of Aml1 is critical for lymphoma development. In addition, the depletion or deletion of Aml1 induced the expression of Fas (Fig. S5), suggesting that AML1 plays some roles in cell survival, as reported.30 In the early stages of lymphoma development, anti‐apoptotic regulation by AML1 might also be important.
The primary role of p53 in the cell‐cycle checkpoint in hematopoietic cells is probably to repress stem cell division, thereby maintaining the quiescent status of these cells in the steady state.16 Another recent report also indicated that the reduced p53 activity of p53 +/− mice is associated with higher numbers of proliferating hematopoietic stem and progenitor cells in older mice.15 Furthermore, a p53‐null genotype promotes expansion of long‐term proliferating stem cells in BM and activates HSCs.15, 16 However, the target of p53 that induces HSC proliferation is unknown. Here we showed that AML1 is at least one of the targets of p53 in hematopoietic stem or progenitor cells. AML1 plays a positive role in cell‐cycle progression in mouse myeloid cells.31, 32 Thus, the data in this study indicates that p53 may repress AML1 in HSCs in the steady state, but induce AML1 once HSCs are exposed to stress.
The mechanism by which p53 represses AML1 gene expression may differ from the known mechanisms by which p53 regulates other genes. Our results support a novel mechanism of p53 gene regulation, in which p53 binding to the target‐gene locus represses transcription, but release of p53 activates transcription. The ability of p53 to respond to stress is usually associated with modification of p53 by phosphorylation, acetylation, or sumoylation.33, 34 However, it is difficult to use this model to explain our results concerning the stress response of p53 and AML1 transcription, because p53 normally represses the AML1 gene but activates it when the cell is exposed to various stresses. We also observed constitutive activation of AML1 gene in cells with a p53‐null genotype. It is likely that, once activated (and probably modified), p53 dissociates from the repressor complex on the Aml1 gene locus.
In future studies, it will be important to analyze the AML1 level in human T‐ALL specimens caused by p53 deficiency or mutation. AML1 has important functions not only in hematopoiesis, but also in neurogenesis and skeletal muscle.23, 35, 36 Therefore, a broad‐based study of the relationship between the two important transcription factors p53 and AML1 in various malignant diseases could provide information that would aid in understanding how these tumors are generated. If the same results are obtained in human hematopoietic stem cells as in mouse cells, then it may be possible to design drugs that inhibit AML1 activity for use in treating T‐cell malignancy.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Phenotypes of lymphomas.
Fig. S2. Effects of p53 shRNA on AML1 expression.
Fig. S3. Active chromatin modifications in the distal promoter regions of the mouse and human AML1 genes.
Fig. S4. Effect of AML1 depletion on the Runx3 gene expression.
Fig. S5. Effect of AML1 depletion on Fas gene expression.
Table S1. Type of diseases and latency seen in p53‐null mice.
Acknowledgments
We thank Dr T. Okuda for providing Aml1 knockout mice, Dr H. Koseki for Trp53 knockout mice, and Dr M. Enari for the sh‐p53 plasmid. We acknowledge C. Hatanaka and M. Shino for technical assistance. We also thank Dr T. Katsumoto and Dr K. Yamagata for technical instruction. This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Health, Labor, and Welfare and from the Ministry of Education, Culture, Sports, Science, and Technology, and by the National Cancer Center Research and Development Fund.
(Cancer Sci 2013; 104: 1033–1038)
References
- 1. Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci USA 1991; 88: 10431–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Golub TR, Barker GF, Bohlander SK et al Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA 1995; 92: 4917–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Osato M, Asou N, Abdalla E et al Biallelic and heterozygous point mutations in the runt domain of the AML1/PEBP2alphaB gene associated with myeloblastic leukemias. Blood 1999; 93: 1817–24. [PubMed] [Google Scholar]
- 4. Imai Y, Kurokawa M, Izutsu K et al Mutations of the AML1 gene in myelodysplastic syndrome and their functional implications in leukemogenesis. Blood 2000; 96: 3154–60. [PubMed] [Google Scholar]
- 5. Song WJ, Sullivan MG, Legare RD et al Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet 1999; 23: 166–75. [DOI] [PubMed] [Google Scholar]
- 6. Niini T, Kanerva J, Vettenranta K, Saarinen‐Pihkala UM, Knuutila S. AML1 gene amplification: a novel finding in childhood acute lymphoblastic leukemia. Haematologica 2000; 85: 166–75. [PubMed] [Google Scholar]
- 7. Dal Cin P, Atkins L, Ford C et al Amplification of AML1 in childhood acute lymphoblastic leukemias. Genes Chromosom Cancer 2001; 30: 407–9. [DOI] [PubMed] [Google Scholar]
- 8. Mikhail FM, Serry KA, Hatem N et al AML1 gene overexpression in childhood acute lymphoblastic leukemia. Leukemia 2002; 16: 658–68. [DOI] [PubMed] [Google Scholar]
- 9. Harewood L, Robinson1 H, Harris R et al Amplification of AML1 on a duplicated chromosome 21 in acute lymphoblastic leukemia: a study of 20 cases. Leukemia 2003; 17: 547–53. [DOI] [PubMed] [Google Scholar]
- 10. Soulier J, Trakhtenbrot L, Najfeld V et al Amplification of band q22 of chromosome 21, including AML1, in older children with acute lymphoblastic leukemia: an emerging molecular cytogenetic subgroup. Leukemia 2003; 17: 1679–82. [DOI] [PubMed] [Google Scholar]
- 11. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8: 275–83. [DOI] [PubMed] [Google Scholar]
- 12. Shounan Y, Dolnikov A, MacKenzie KL, Miller M, Chan YY, Symonds G. Retroviral transduction of hematopoietic progenitor cells with mutant p53 promotes survival and proliferation, modifies differentiation potential and inhibits apoptosis. Leukemia 1996; 10: 1619–28. [PubMed] [Google Scholar]
- 13. Shaulsky G, Goldfinger N, Peled A, Rotter V. Involvement of wild‐type p53 in pre‐B‐cell differentiation in vitro . Proc Natl Acad Sci USA 1991; 88: 8982–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kastan MB, Radin AI, Kuerbitz SJ et al Levels of p53 protein increase with maturation in human hematopoietic cells. Cancer Res 1991; 51: 4279–86. [PubMed] [Google Scholar]
- 15. Dumble M, Moore L, Chambers SM et al The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood 2007; 109: 1736–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y, Elf SE, Miyata Y et al p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009; 4: 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prokocimer M, Rotter V. Structure and function of p53 in normal cells and their aberrations in cancer cells: projection on the hematologic cell lineages. Blood 1994; 84: 2391–411. [PubMed] [Google Scholar]
- 18. Hatta Y, Yamada Y, Tomonaga M, Said JW, Miyosi I, Koeffler HP. Allelotype analysis of adult T‐Cell leukemia. Blood 1998; 92: 2113–7. [PubMed] [Google Scholar]
- 19. Jacks T, Remington L, Williams BO et al Tumor spectrum analysis in p53‐ mutant mice. Curr Biol 1994; 4: 1–7. [DOI] [PubMed] [Google Scholar]
- 20. Ichikawa M, Asai T, Saito T et al AML‐1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med 2004; 10: 299–304. [DOI] [PubMed] [Google Scholar]
- 21. Seibler J, Zevnik B, Küter‐Luks B. Rapid generation of inducible mouse mutants. Nucleic Acids Res 2003; 31: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alberts AS, Geneste O, Treisman R. Activation of SRF‐Regulated chromosomal templates by Rho‐family GTPases requires a signal that also induces H4 hyperacetylation. Cell 1998; 92: 475–87. [DOI] [PubMed] [Google Scholar]
- 23. Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996; 84: 321–30. [DOI] [PubMed] [Google Scholar]
- 24. Miyoshi H, Ohira M, Shimizu K et al Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res 1995; 23: 2762–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Telfer JC, Rothenberg EV. Expression and function of a stem cell promoter for the murine CBFalpha2 gene: distinct roles and regulation in natural killer and T cell development. Dev Biol 2001; 229: 363–82. [DOI] [PubMed] [Google Scholar]
- 26. Wotton SF, Blyth K, Kilbey A et al RUNX1 transformation of primary embryonic fibroblasts is revealed in the absence of p53. Oncogene 2004; 23: 5476–86. [DOI] [PubMed] [Google Scholar]
- 27. Kurokawa M, Tanaka T, Tanaka K et al Overexpression of the AML1 proto oncoprotein in NIH3T3 cells leads to neoplastic transformation depending on the DNA‐binding and transactivation potencies. Oncogene 1996; 12: 883–92. [PubMed] [Google Scholar]
- 28. Wotton SF, Stewart M, Blyth K et al Proviral insertion indicates a dominant oncogenic role for Runx1/AML‐1 in T‐cell lymphoma. Cancer Res 2002; 62: 7181–5. [PubMed] [Google Scholar]
- 29. Blyth K, Terry A, Mackay N et al Runx2: a novel oncogenic effector revealed by in vivo complementation and retroviral tagging. Oncogene 2001; 20: 295–302. [DOI] [PubMed] [Google Scholar]
- 30. Fujii M, Hayashi K, Niki M et al Overexpression of AML1 renders a T hybridoma resistant to T cell receptor‐mediated apoptosis. Oncogene 1998; 17: 1813–20. [DOI] [PubMed] [Google Scholar]
- 31. Bernardin‐Fried F, Kummalue T, Leijen S, Collector MI, Ravid K, Friedoman AD. AML1/RUNX1 increases during G1 to S cell‐cycle progression independent of cytokine‐dependent phosphorylation and induces cyclin D3 gene expression. J Biol Chem 2004; 279: 15678–87. [DOI] [PubMed] [Google Scholar]
- 32. Growney JD, Shigematsu H, Li Z et al Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005; 106: 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bode AM, Dong Z. Post‐translational modification of p53 in tumorigenesis. Nat Rev Cancer 2004; 4: 793–805. [DOI] [PubMed] [Google Scholar]
- 34. Dai C, Gu W. p53 post‐translational modification: deregulated in tumorigenesis. Trends Mol Med 2010; 16: 528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Theriault FM, Roy P, Stifani S. AML1/Runx1 is important for the development of hindbrain cholinergic branchiovisceral motor neurons and selected cranial sensory neurons. Proc Natl Acad Sci USA 2004; 101: 10343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X, Blagden C, Fan J et al Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev 2005; 19: 1715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phenotypes of lymphomas.
Fig. S2. Effects of p53 shRNA on AML1 expression.
Fig. S3. Active chromatin modifications in the distal promoter regions of the mouse and human AML1 genes.
Fig. S4. Effect of AML1 depletion on the Runx3 gene expression.
Fig. S5. Effect of AML1 depletion on Fas gene expression.
Table S1. Type of diseases and latency seen in p53‐null mice.