

Abstract
A 24-year-old female presented with wasting and weakness of both hands and fasciculations over the chin since 12 years, followed by imbalance while walking and speech changes since 10 years. Her 12-year-old sister also had a similar clinical presentation. There were fasciculations over the chin, tongue, hands, back, thighs with wasting and weakness in tongue, and C7, C8, T1 segments in both upper limbs along with bipyramidal signs. There was limb and gait ataxia. Magnetic resonance imaging brain showed pancerebellar atrophy, and electromyography was suggestive of anterior horn cell involvement in bulbar, cervical, thoracic, and lumbar segments. Next-generation sequencing identified a novel likely pathogenic deletion mutation: chr6:152527389_152527399del, c.22711_22721del, and p.Ala7571ArgfsTer4 in exon 125 of synaptic nuclear envelope protein 1 (SYNE1) gene. This mutation leads to frameshift and premature termination of the protein 'Nesprin 1'. Amyotrophic lateral sclerosis-like presentation followed by cerebellar ataxia have been described with SYNE1 ataxia. This unique phenotype and novel deletion mutation of SYNE1 gene is the first case reported from India.
Keywords: Autosomal recessive cerebellar ataxia 1 ataxia, amyotrophic lateral sclerosis, motor neuron disease, spinocerebellar ataxia of recessive ataxia, synaptic nuclear envelope protein 1 ataxia
INTRODUCTION
Synaptic nuclear envelope protein 1 (SYNE1) ataxia is a slowly progressive autosomal recessive cerebellar ataxia first described in Quebec, Canada.[1] Later, similar cases were detected in France, Brazil, and Japan, with pure cerebellar ataxia.[2] We report a 24-year-old female who presented with cerebellar ataxia and amyotrophic lateral sclerosis (ALS)-like features with a novel SYNE1 gene deletion mutation. This first clinically and genetically confirmed case report from India indicates that this unique phenotype should alert clinicians to consider SYNE1 mutation in this part of the world.
CASE REPORT
A 24-year-old female born of non consanguineous marriage presented with a history of weakness and thinning of the right hand followed by the left hand since 11 years of age. Over the next two years, she noticed twitching over the chin and arms. By 14 years of age, she started having imbalance while walking, stiffness in legs, and dysarthria. Of seven siblings, her 12-year-old sister reported similar thinning, weakness in both hands with twitching over the chin and arms for two years followed by imbalance while walking for six months. No other generations were affected.
Examination showed normal higher mental functions. Speech was spastic dysarthric with nasal twang. There was gaze-evoked nystagmus. Pursuits and saccades were normal. There was tongue wasting with weakness and fasciculations. Gag reflex was normal. There was wasting of hand and forearm muscles and fasciculations over the chin, arms, back, and thighs. She had spasticity in both lower limbs with pure motor weakness in C7–T1 segments and normal power in both lower limbs.
There was generalized hyperreflexia with bilateral ankle clonus and extensor plantar reflex. Sensory examination was normal. She had limb ataxia with impaired finger-nose-finger test in the upper limbs. Gait was spastic ataxic with impaired tandem walking.
Serum electrolytes, thyroid, and parathyroid studies were normal. Serum alpha-fetoprotein, lipid profile, albumin levels, protein electrophoresis, vitamin E, and copper levels were normal. Fundus was normal. There was no Kayser–Fleischer ring on Slit-lamp examination. Magnetic resonance imaging (MRI) of the brain [Figure 1] showed severe pancerebellar atrophy.
Figure 1.
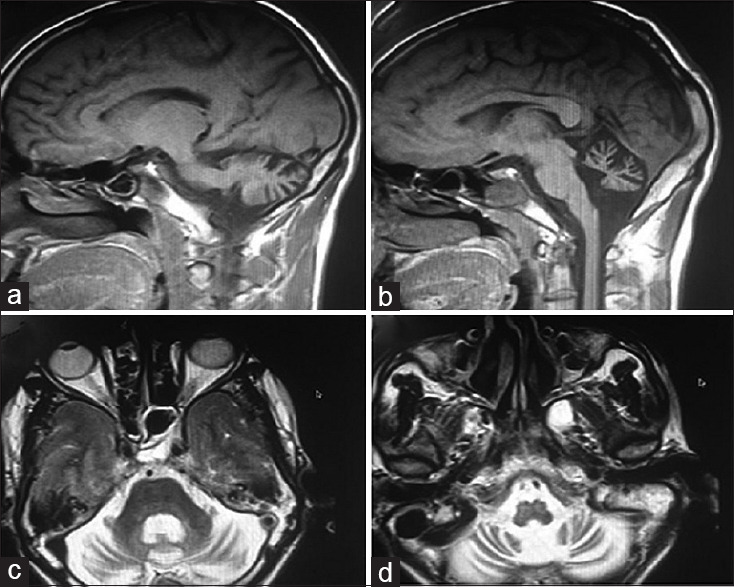
(a and b) T1-weighted sagittal magnetic resonance imaging brain shows severe cerebellar atrophy and brainstem is normal. (c and d) T2-weighted axial magnetic resonance imaging I brain shows pancerebellar atrophy affecting vermis and cerebellar hemispheres equally with preserved brainstem
Electrophysiology showed normal sensory nerve action potentials. Distal compound motor action potentials in the upper limbs showed axonal motor involvement and normal study in the lower limbs. Electromyography (EMG) showed active denervation (fasciculations, fibrillations, and positive sharp waves) in bulbar and active plus chronic denervation (high amplitude and long duration motor unit action potentials) in cervical, thoracic, and lumbar segments. EMG and clinical assessment were suggestive of clinically definite ALS.[3] Similar examination and EMG findings were seen in younger sister.
The possibility of inherited autosomal recessive cerebellar ataxia with amyotrophy and bipyramidal involvement or spastic ataxia spectrum disease[4] was considered. SCA3 (spinocerebellar ataxia of autosomal dominant) pattern was also considered – SCA3 showed phenotypic similarity with our case such as amyotrophy, pyramidal involvement, and cerebellar ataxia with MRI showing cerebellar atrophy. However, family history and age of onset in our patient and her younger sibling (age of onset first decade) were favoring autosomal recessive inheritance.
Next-generation sequencing (NGS) was performed using Clinical Exome Panel (TruSight One, Illumina) covering coding exons and flanking intronic sequences of >4800 genes associated with known inherited diseases.
Genomic DNA was used for the library preparation for NGS and sequenced on the NextSeq platform (Illumina). Variations were identified using the STRAND® NGS and Strand Omics™ software.[5] Variant annotation with suggestive American College of Medical Genetics and Genomics (ACMG) labels was done by automated pipelines in StrandOmics.
A homozygous deletion mutation, chr6:152527389_152527399del, c.22711_22721del, and p. Ala7571ArgfsTer4, was detected in exon 125 of the SYNE1 gene. The identified variant has not been described in literature or in any public database, such as 1000 Genome database, dbSNP, EVS, ExAC, and gnomAD, therefore, considered as “novel” variant. The variant causes 11- base deletion at alanine (7571) to alanine (7574) and leads to a frameshift at alanine (7571) and consequent truncation after four amino acids. The resulting protein product has length 7573 as opposed to the original length of 8749. The variant is predicted to cause a frameshift mutation and consequent premature termination of the protein, which will likely result in loss of function. Sanger's sequencing confirmed a homozygous deletion of 11 nucleotides at position c.22711_22721 in SYNE1 gene (RefSeq id:NM_NM_033071). This sequence was confirmed by sequencing with both forward and reverse primers. This variant was considered as novel and “likely pathogenic” based on ACMG standards interpretation and reporting of sequence variation.
DISCUSSION
SYNE1 is one of the largest genes comprising 146 exons encoding the 8797 amino-acid SYNE1.[1] This protein is also known as Nesprin 1 (nuclear envelope spectrin 1) which is a part of the spectrin family of structural proteins responsible for linking the plasma membrane to the actin cytoskeleton.[1]
It was discovered in 2007 as the gene causing spinocerebellar ataxia of recessive type 8 or autosomal recessive cerebellar ataxia 1 which was thought to be pure cerebellar ataxia. Recently, Japanese families with SYNE1 mutations were associated with a motor neuron phenotype[6] in addition to cerebellar ataxia. It is also being considered as a spastic ataxia disease.[4] According to a multicenter study[7] for SYNE1 mutation, the age of disease onset ranges from 6 to 42 years (median: 14 years). The disease starts with noncerebellar features in 50% of patients.[7] This includes ALS-like motor neuron features. Almost all patients showed “cerebellar plus phenotype', motor neuron disease being the most frequent.
Nesprin 1 is a structural protein with widespread functional role and hence has varied manifestations such as cognitive decline, slow saccades, ophthalmoparesis or brainstem manifestations, and musculoskeletal abnormalities such as kyphoscoliosis, pes cavus, and arthrogryposis multiplex congenita syndrome.[7]
Thus SYNE1 mutation has a unique phenotype of ataxia with ALS-like features.
This report supports that motor neuron involvement is a frequently associated manifestation and may be the initial presentation of this disease. To the best of our knowledge, this is a novel and likely pathogenic deletion mutation in SYNE1 gene. This is the first clinically and genetically confirmed case of SYNE1 ataxia from India. NGS methodology allows for massively parallel reading of sequenced millions to billions short DNAs. When multiple genes are associated with disease condition, such as ataxia, then NGS test is comparatively inexpensive as compared to Sanger sequencing. It is accurate, fast, and cost-effective. NGS technologies have helped researchers discovering new genetic mutations by whole genome or whole exome sequencing methods. It can improve the diagnostic rate of clinically heterogeneous genetic neurodegenerative disorders leading to better patient care.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the father has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The father understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We would like to thank the patient and her family for their patience and cooperation.
Dean TNMC, Nair Hospital for assisting in getting the genetic tests done.
REFERENCES
- 1.Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmüller C, Baets J, et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: A large multi-centre study. Brain. 2016;139:1378–93. doi: 10.1093/brain/aww079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noreau A, Bourassa CV, Szuto A, Levert A, Dobrzeniecka S, Gauthier J, et al. SYNE1 mutations in autosomal recessive cerebellar ataxia. JAMA Neurol. 2013;70:1296–31. doi: 10.1001/jamaneurol.2013.3268. [DOI] [PubMed] [Google Scholar]
- 3.de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119:497–503. doi: 10.1016/j.clinph.2007.09.143. [DOI] [PubMed] [Google Scholar]
- 4.Synofzik M, Schüle R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov Disord. 2017;32:332–45. doi: 10.1002/mds.26944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mannan AU, Singh J, Lakshmikeshava R, Thota N, Singh S, Sowmya TS, et al. Detection of high frequency of mutations in a breast and/or ovarian cancer cohort: Implications of embracing a multi-gene panel in molecular diagnosis in India. J Hum Genet. 2016;61:515–22. doi: 10.1038/jhg.2016.4. [DOI] [PubMed] [Google Scholar]
- 6.Yoshinaga T, Nakamura K, Ishikawa M, Yamaguchi T, Takano K, Wakui K, et al. A novel frameshift mutation of SYNE1 in a Japanese family with autosomal recessive cerebellar ataxia type 8. Hum Genome Var. 2017;4:17052. doi: 10.1038/hgv.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mademan I, Harmuth F, Giordano I, Timmann D, Magri S, Deconinck T, et al. Multisystemic SYNE1 ataxia: Confirming the high frequency and extending the mutational and phenotypic spectrum. Brain. 2016;139:e46. doi: 10.1093/brain/aww115. [DOI] [PMC free article] [PubMed] [Google Scholar]