

This model fits predicted pIs to empirical values for a diverse set of viruses. The results suggest that many previously reported discrepancies between theoretical and empirical virus pIs can be explained by coulombic neutralization of PBRs of the inner capsid. Given the diversity of virus capsid structures, this nonarbitrary, heuristic approach to predicting virus pI offers an effective alternative to a simplistic, one-size-fits-all charge model of the virion. The accurate, structure-based prediction of PBRs of the virus capsid employed here may also be of general interest to structural virologists.
KEYWORDS: capsid, DNA binding, electrostatic, modeling, point of zero charge, polynucleotide, RNA binding, DNA-binding proteins, RNA-binding proteins, electrostatic model, pI, prediction
ABSTRACT
Knowledge of the isoelectric points (pIs) of viruses is beneficial for predicting virus behavior in environmental transport and physical/chemical treatment applications. However, the empirically measured pIs of many viruses have thus far defied simple explanation, let alone prediction, based on the ionizable amino acid composition of the virus capsid. Here, we suggest an approach for predicting the pI of nonenveloped viruses by excluding capsid regions that stabilize the virus polynucleotide via electrostatic interactions. This method was applied first to viruses with known polynucleotide-binding regions (PBRs) and/or three-dimensional (3D) structures. Then, PBRs were predicted in a group of 32 unique viral capsid proteome sequences via conserved structures and sequence motifs. Removing predicted PBRs resulted in a significantly better fit to empirical pI values. After modification, mean differences between theoretical and empirical pI values were reduced from 2.1 ± 2.4 to 0.1 ± 1.7 pH units.
IMPORTANCE This model fits predicted pIs to empirical values for a diverse set of viruses. The results suggest that many previously reported discrepancies between theoretical and empirical virus pIs can be explained by coulombic neutralization of PBRs of the inner capsid. Given the diversity of virus capsid structures, this nonarbitrary, heuristic approach to predicting virus pI offers an effective alternative to a simplistic, one-size-fits-all charge model of the virion. The accurate, structure-based prediction of PBRs of the virus capsid employed here may also be of general interest to structural virologists.
INTRODUCTION
Electrostatic interactions between virus particles and their environment are integral to virus fate and transport in physical/chemical processes and in the natural environment. Virus surface charge varies between net negative and positive charge with increasing pH. The isoelectric point (pI) of a virus is defined as the pH at which the net virion charge is neutral. Knowing the pI of a virus enables prediction of whether a virus will be positively or negatively charged in the environment. Predicting the sign of virus surface charge can be important not only for enhancing understanding of virus deposition on surfaces such as soil particles, or virus destabilization via coagulants in water treatment, but also for understanding of virion-virion aggregation (1, 2). Viruses tend to aggregate near the pI due to negated electrostatic repulsion, and aggregation can significantly impair the efficacy of disinfection processes (3, 4). In addition, knowledge of virus pI can inform virus concentration and detection in environmental samples, e.g., via isoelectric focusing (5, 6).
Although the available data for virus pIs are sparse and include some outliers, the general consistency of empirically determined virus pIs between researchers and spanning decades is encouraging. In their indispensable review of virus pIs, Michen and Graule (7) note that the range of reported pIs for bacteriophages MS2 and ΦX174 can be limited to 0.8 and 0.1 pH units, respectively, by limiting for strain and purity. Empirical pIs are also similar between strains of a single virus species, as shown in Fig. 1. The similarity of pI between closely related viruses suggests that pI may be predictable based on conserved virion structure.
FIG 1.
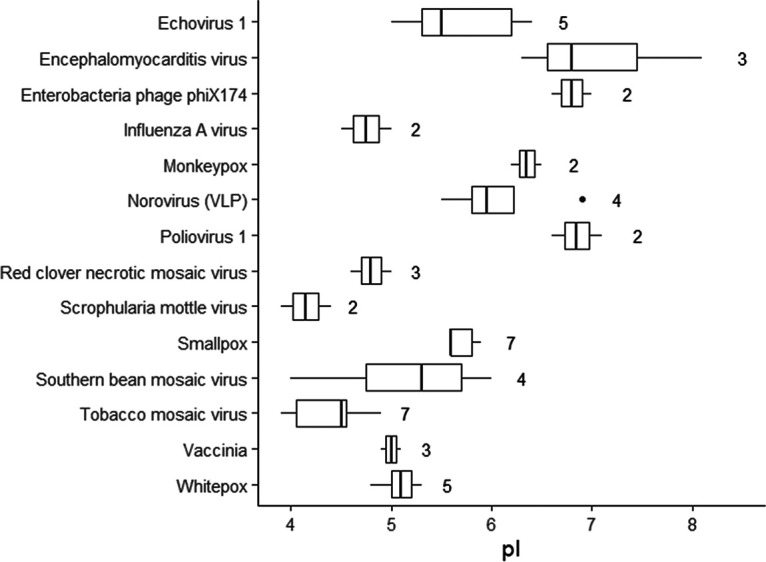
Relative conservation of isoelectric point (pI) in closely related virus strains. The plot shows empirical pI values for different strains of the same species, with the number of strains shown to the right of each box (data summarized from Michen and Graule [7]). To minimize differences due to experimental conditions, pI values for each virus along the y axis were obtained from single studies comparing different strains.
Attempts to model virion pI generally involve quantifying and modifying the charges of ionizable amino acids within capsid proteins (8–12). Altering the composition of ionizable amino acids within capsid proteins can have a predictable and measurable effect on virion pI (13). Modern recombinant techniques allow some degree of “charge tuning” of viral particles by adding or replacing ionizable amino acids within capsid proteins (14).
However, the sequence of ionizable amino acids alone appears insufficient to accurately predict pI. Based on analyses of hundreds of virus proteomes, theoretical virus pIs calculated from ionizable amino acid residues are tightly clustered near neutral pH, with an overall range between approximately pH 5.5 and 8 (15, 16). However, empirical virus pIs below pH 5 are frequently reported, and they have been measured as low as pH 2 (7). Given that the distribution of theoretical virus pIs (pH 5.5 to 8) is on the same order as the variation in empirical pIs between strains of a single virus species (<2 pH units; see Fig. 1), refined prediction of virus pI based on ionizable amino acids may not be possible at the species level. Rather, the primary goal for a model of virion pI should be to reliably predict which viruses will have pIs outside the expected circumneutral range.
Previous researchers have explained the differences between theoretical and empirical pIs by either supposing a strong negative influence from the viral polynucleotide (genome) at the virion core (12, 17–21), or by supposing that only the exterior capsid surface contributes to virion charge (9, 11). Given the low pKa (∼1) of the polynucleotide phosphodiester group (Table 1) and the porous nature of virus capsids, the assumption that the virion core influences overall charge is credible. However, DNA and RNA folding and compaction during encapsidation requires a cloud of counterions to overcome electrostatic self-repulsion (22–26), at least some of which are likely to be retained in the assembled virion core (27). In addition, experiments comparing the charges of whole virions to those of empty capsids lacking a genome (virus-like particles) have failed to account for major discrepancies in theoretical and empirical pIs (9, 19, 28, 29). The second proposed model, in which only exterior capsid residues contribute to virion charge, has been used to calculate predicted pIs, but only for structurally similar bacteriophages in the Leviviridae family (9, 11). Neither of these previous methods (presuming a negative charge from the virion core or selecting only exterior residues) has yet been demonstrated on a large, diverse set of viruses.
TABLE 1.
Acid dissociation constants (pKa) for protein and nucleic acid constituents
| Residue | pKa or pKa range | Charge |
|---|---|---|
| Proteinsa | ||
| Terminal carboxyl | 2.87 | Negative |
| Aspartic acid | 3.87 | Negative |
| Glutamic acid | 4.41 | Negative |
| Cysteine | 7.56 | Negative |
| Tyrosine | 10.85 | Negative |
| Histidine | 5.64 | Positive |
| Lysine | 9.05 | Positive |
| Terminal amine | 9.09 | Positive |
| Arginine | 11.84 | Positive |
| Nucleic acidsb | ||
| Primary phosphoryl | 1 | Negative |
| Deprotonated base (G, T, U) | 9.4–10 | Negative |
| Amino group (A, C, G) | 2.3–4.6 | Positive |
Values from the Isoelectric Point Calculator (http://isoelectric.org/) (71).
Values from Blackburn (72).
Furthermore, using either approach to develop a predictive model of pI would require fitting one or more variables to the empirical pI data, since there is no other empirical source of data to describe the influence of core charge or decision criterion for what constitutes an “exterior” residue in large and convoluted capsids. Given the limited empirical pI data and the bias toward viruses commonly used in research (e.g., Leviviridae and plant viruses), these approaches are likely to overfit a prediction to the available data. Preferably, a model for pI prediction would rely on a separate, independently verifiable criterion for what elements of virion structure contribute to the overall charge.
The goal of this study was to propose a simple model to improve pI prediction for nonenveloped viruses. Our hypothesis was that positive charges from basic protein residues in polynucleotide-binding regions (PBRs) of the capsid interior are neutralized by noncovalent bonding with the viral polynucleotide (genome) and therefore should not be considered in the capsid pI calculation. We approached this challenge by first modifying capsid protein sequences based on regions known or suspected to stabilize the viral genome and then calculated the predicted pI using a simple sum of charges method. This heuristic approach applies a rule for including and excluding amino acids from the pI calculation rather than imposing a simplified physical model on the virion structure. In addition, the heuristic is nonarbitrary, in that amino acids are excluded based on function rather than an attempt to fit the predicted pIs to empirical values. For this study, both three-dimensional (3D) structural models of virus capsids and capsid proteome sequences were evaluated with and without modifications. The implication of this approach is that while one simple structural model may not be applicable to all viruses, a descriptive model of virion pI can arise from a simple, nonarbitrary heuristic.
RESULTS
The evidence in support of PBR exclusion as a means of pI prediction comes from both known capsid structures and predicted PBRs. “Known structures” include both 3D capsid models and experimentally determined PBRs. Given the relatively few viruses for which both known structures and empirical pI values were available, PBRs were predicted for a larger set of viruses based on conserved structures apparent in the experimentally determined PBRs. The most accurate PBR predictions were then used to predict pIs for the extended virus set.
Known capsid structures.
Some researchers have suggested that only residues on the exterior capsid surfaces contribute to overall capsid charge (9, 11). However, for the set of viruses with available 3D structures and empirical pIs, residues on the exterior surface were a poor predictor of overall virion charge. As shown in Fig. 2, exterior surface residues identified via CapsidMaps (30) tended to be composed of only acidic or basic residues, with no correlation to empirical pI. Interestingly, removing only the residues on the interior capsid surface did result in a better fit between theoretical and empirical pIs compared to unmodified capsids (Fig. 2). Many virus capsids feature a concentration of basic residues toward the interior—notably the single-stranded RNA (ssRNA) phages of Leviviridae commonly used in research. Thus, the exterior residues of these viruses have a lower pI than the capsid as a whole. However, whether a given virus has this unbalanced distribution of ionizable amino acids has defied any straightforward prediction (10). Given the diverse virus sizes and morphologies represented in Fig. 2, one universal charge distribution model is unlikely to explain why only the innermost surface-exposed residues do not contribute to overall virion charge.
FIG 2.
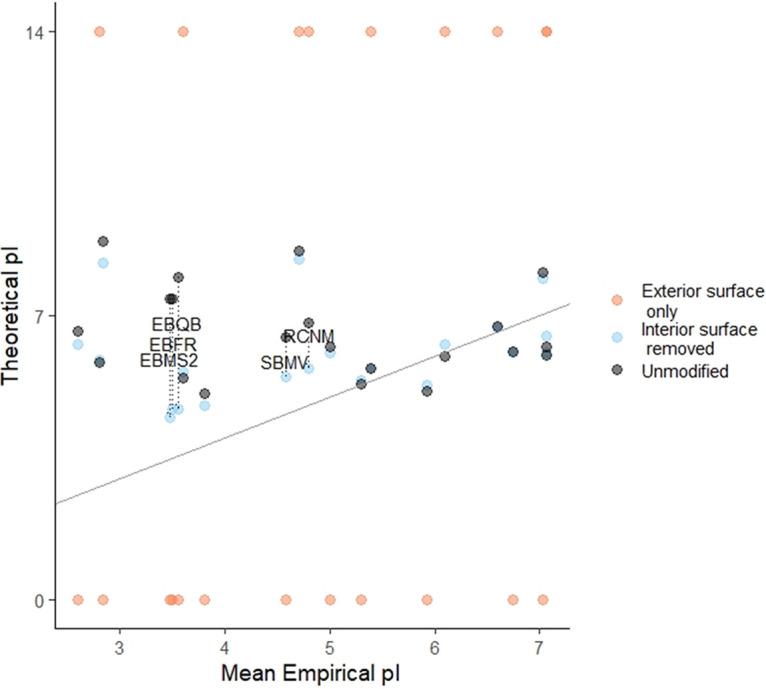
Impact of including only exterior residues in predicted pI calculation using 3D capsid structures. Exterior residues are defined by inclusion of only the exterior capsid surface or exclusion of the interior capsid surface, as identified via CapsidMaps (30). Exterior capsid surfaces were composed of entirely acidic or basic residues; these pIs are shown as approaching neutral charge at a pH of 0 or 14, respectively. The mean empirical pI value for each unique virus is shown in comparison to predicted pI of the virus calculated from exterior capsid residues, as defined in the legends. The diagonal line represents equivalent theoretical and empirical pIs. Viruses showing the greatest improvement in predicted pI after removing interior surfaces are labeled; a guide to virus abbreviations is provided in Table 2.
A review of the viruses most positively impacted by removal of interior residues provides some insight. The following five viruses with the greatest improvement in pI estimation are labeled in Fig. 2: Leviviridae bacteriophages fr (EBFR), MS2 (EBMS2), and Qβ (EBQB), and the ssRNA plant viruses red clover necrotic mosaic virus (RCNM), and southern bean mosaic virus (SBMV). Leviviridae spp. feature a highly conserved assembly mechanism by which RNA binds to planar beta sheets on the capsid protein interior, forming subunits from which the capsid self-assembles (31–34). Thus, these phages have large, basic surfaces on the capsid interior devoted to RNA binding. The other two viruses, SBMV and RCNM, are ssRNA plant viruses with highly basic, disordered N termini. These disordered regions also function to stabilize the viral polynucleotide (35–37). Therefore, the viruses showing the greatest impact from interior residue exclusion all feature highly basic interior residues that stabilize the viral genome. Both of these basic, interior capsid features are noncovalently bound to the viral RNA (31, 33, 35, 36), and therefore their positive contribution to virion charge is likely negated by the negatively charged polynucleotide.
Known polynucleotide-binding regions.
To determine the impact of PBRs on virus pI, known PBRs were identified for 15 viruses from annotations in the UniProt database (38) and the literature (see Table S3 in the supplemental material). Theoretical virus pIs were compared before and after modification by excluding PBRs from the charge calculation. As shown in Fig. 3, excluding PBRs improved pI estimation for the majority of viruses. The predicted pIs before modification deviated from empirical values by 2.2 ± 2.4 pH units; after modification, predicted pIs deviated from empirical pIs by 1.4 ± 1.5 pH units. This reduction in mean deviation after PBR exclusion was significant to a high degree of confidence (paired t value = 5.81 [14 degrees of freedom (df)], P = 5 × 10−5). In comparison, excluding interior surface residues also decreased the deviation between theoretical and empirical pIs to 1.0 ± 1.7 pH units, though the improvement was slightly less significant (paired t = 2.82 [20 df], P = 0.01). However, the identification of capsid surfaces may not be practical for larger, layered capsids due to complex structure, computational burden, or lack of entire 3D structures.
FIG 3.

Effect of modifications on theoretical capsid pI. Box and whisker plots represent the range of empirical pIs found in the literature, while circles represent predicted pIs. The predicted pIs reflect the theoretical capsid charges: without modification, after excluding known viral polynucleotide-binding regions (“PBRs removed”) and after removing capsid interior surfaces using 3D structures [“Interior surface removed (3-D)”]. Both modifications were not possible for some viruses due to unknown PBR locations, unavailable 3D structures, and/or large size. A key to the 23 virus abbreviations (x axis) is provided in Table 2.
Modification via PBR exclusion and interior surface exclusion appeared to be complementary. Many viruses shown in Fig. 3 lacked either known PBRs or capsid structures, so only one method was possible with the available data. In only a few cases, either PBR or interior surface exclusion produced a far better prediction than the other method. For example, pI estimation for bacteriophage GA (EBGA) was far closer to empirical values via PBR exclusion. The interior of the bacteriophage GA capsid protein contains both acidic and basic residues (34), so nonspecific removal of the entire interior surface has little net effect on capsid pI. On the other hand, southern bean mosaic virus (SBMV) was more accurately predicted after interior surface exclusion, possibly indicating that the full extent of the PBR was not known for this virus.
In this study, PBRs with the greatest impact on predicted pI could be divided into the following three primary categories: predominately basic beta sheets and associated turns, disordered polypeptide termini (primarily N termini), and histone-like proteins. Basic beta sheets were typical PBRs for the Leviviridae family of ssRNA bacteriophages routinely used as model viruses in research. Some Leviviridae phages (fr, MS2, and SP) also had basic alpha helices serving as PBRs in their maturation and minor capsid proteins. In the two representatives of Allolevivirus (bacteriophages Qβ and SP), basic residues occurred primarily on the turns between beta sheets on the major capsid protein, rather than on the beta sheets themselves. Four other ssRNA viruses in this study—cowpea chlorotic mosaic virus (CCMV), cucumber mosaic virus (CMV), red clover necrotic mosaic virus (RCNM), and southern bean mosaic virus (SBMV)—featured PBRs within disordered, highly basic N termini (35, 36). Some of these N termini also feature alpha helices that bind to RNA stem-loops (39). Human adenovirus 5 (HAdV5) was the only virus in this study that featured histone-like proteins, and the removal of these proteins improved the accuracy of the pI prediction by 3.6 pH units, as shown in Fig. 3. Reovirus type 3 (REO3) also has a protein thought to act as a spool for RNA within the capsid (40); however, exclusion of this protein did not impact the predicted pI.
In addition to common PBR structures, PBR sequences in this study had high arginine and lysine fractions (0.42 ± 0.29). These arginine- and lysine-rich regions are often indicative of RNA and DNA binding (41–44). The beta sheets of Leviviridae capsid proteins had lower arginine/lysine fractions (0.11 ± 0.01) than other PBRs, as basic amino acids tend to be distributed over a noncontiguous surface rather than within one short sequence. A full list of viral proteins and their PBRs is provided in the supplemental material (Table S3). The set of viruses evaluated in Fig. 3 was limited by the availability of empirical pIs, as well as curated proteome sequences and 3D structures. Here, as in other virus pI research, Leviviridae spp. in particular are overrepresented, preempting the conclusion that the PBR exclusion approach is universally applicable. Nonetheless, excluding PBRs explained multiple discrepancies in predicted pIs with a single, nonarbitrary heuristic and was thus a promising direction for a predictive model of virus pI. However, PBRs would have to be predicted within virus proteome sequences in order to apply this method to a larger set of viruses.
Predicted polynucleotide-binding regions.
Unfortunately, excluding PBRs by the above methods required either a high-resolution 3D capsid model or the full extent of the capsid PBR(s). A method of predicting PBRs based on capsid protein sequences would be far preferable, as capsid proteomes are available for a wide range of viruses. Furthermore, PBRs are often discovered by point mutations/deletions of select amino acids, so the full extent of the PBR may not be known. As a first attempt at a predictive model of virion pI based on PBR exclusion, PBRs were predicted in a diverse group of 32 viruses based on the conserved PBR features discussed in “Known polynucleotide-binding regions” (above). Specifically, PBR predictions attempted to capture basic beta sheets and associated beta turns, disordered polypeptide termini, and arginine-rich regions. (Although both arginine and lysine contributed to the predicted PBRs, the common term “arginine-rich” is used here to describe these basic regions, since arginine is the dominant residue.) In addition, two Web-based tools for detecting nucleic acid (NA)-binding residues, Pprint (45, 46) and DRNApred (47, 48), were evaluated for virus PBR prediction. Both tools modeled the likelihood of amino acids binding to RNA or DNA based on position within the primary sequence. All PBR predictions were evaluated against the known PBRs discussed above (“Known polynucleotide-binding regions”), as well as against a validation set of 40 other capsid proteins. Predicted pIs were then calculated by excluding the predicted PBRs; predicted pIs were compared to empirical values.
Prediction of polynucleotide-binding regions.
Overall, searching for conserved structures offered the most reliable PBR prediction. As a single predictor, arginine-rich regions had the greatest predictive power (Matthews correlation coefficient [MCC] = 0.32 ± 0.29). Prediction of PBRs via beta structures (sheets, MCC = 0.05 ± 0.23; turns, MCC = 0.07 ± 0.20) and disordered termini (MCC = 0.16 ± 0.31) had lower MCCs than the arginine-rich region prediction. However, these low metrics may reflect the relatively low prevalence of beta sheets and disordered termini within the training and validation sets rather than a poor match to experimental data. These structures were intended to complement one another rather than to predict all regions with a single structure. When evaluated only on proteins containing beta sheet PBRs, the beta sheet predictor performed far better (MCC = 0.45 ± 0.16). The disordered termini also performed better against a set of only proteins with PBRs located on disordered termini (MCC = 0.35 ± 0.32), although the standard deviation indicates that the fit was not universal or specific. A summary of MCCs for all predictions is provided in Table S1 in the supplemental material.
Most combinations of structures failed to improve on the prediction via arginine-rich regions alone. PBR prediction based on arginine-rich regions alone covered 18% of residues predicted based on beta sheets, 39% of beta turns, and 26% of disordered termini. Since only the combination of arginine-rich regions and beta turns provided a better fit to training and validation data, beta sheet and disordered terminus predictions likely contributed more false positives than did the arginine-rich regions alone. Selection of arginine-rich regions also successfully identified known alpha helix PBRs in maturation proteins of Leviviridae phages fr (EBFR) and MS2 (EBMS2), as well as 61% of residues in the histone-like protein of human adenovirus 5 (HadV5). All PBR predictions based on beta sheets, beta turns, disordered termini, and arginine-rich regions are provided at the end of the supplemental material document.
However, beta turns in particular were complementary to the arginine-rich search, likely because bases in these regions may be adjacent in the tertiary structure but distant in the primary sequence. Fittingly, a combination of arginine-rich regions and beta turns was optimal for predicting the known PBRs in this study (MCC = 0.34 ± 0.28). Arginine-rich regions alone had a slightly higher mean MCC (0.27 ± 0.32) for the validation set than the combination of arginine-rich and beta turn regions (MCC = 0.26 ± 0.29). However, the lower variance of the latter indicated fewer poor predictions. Therefore, the combination of arginine-rich and beta turn regions was used as the preferred PBR prediction method for this study.
The naive, structure-based PBR prediction method used here performed well compared to other predictors of NA-binding residues. For reference, MCCs for other available NA-binding prediction tools range from approximately 0.14 to 0.23 for RNA-binding and 0.14 to 0.35 for DNA-binding (49). The two sequence-specific predictors compared here, Pprint (MCC = 0.22 ± 0.27) and DRNApred (MCC = 0.17 ± 0.23), performed within this range for virus PBRs as well. Pprint performed better than DRNApred in this study, possibly because Pprint was optimized to the PBR training set, while DRNApred used a default decision criterion. However, neither tool’s prediction exceeded the median MCC of the structure-based predictions considered here. Neither tool was designed with virus polynucleotides or capsid proteins in mind; thus, poor performance is more a reflection of the application than the tools themselves. However, the structure-based PBR prediction used here (MCC = 0.34 ± 0.28) also performed comparably to maximum reported performance of Pprint (MCC = 0.32) (50) and DRNApred (MCC = 0.31 [DNA] and 0.36 [RNA]) (48) on their intended data sets (as determined by the respective authors).
Impact of predicted polynucleotide-binding regions on predicted isoelectric point.
The impact of excluding predicted PBRs in pI calculations for individual viruses can be seen in Fig. 4. Since the combination of arginine-rich regions and beta turns provided the best prediction of PBRs, the PBRs predicted under these parameters were excluded for the “structure-based prediction” of pI. The overall improvement in accuracy of the modified predictions compared to the unmodified predictions is shown in the histogram in Fig. 5. The differences between theoretical and empirical pI values decreased in both magnitude and variance after modification, from 2.1 ± 2.4 to 0.1 ± 1.7 pH units. This difference was significant to a high degree of confidence (paired t = 7.24 [31 df], P = 4 × 10−8). In addition, the histogram shifted from a bimodal to a normal distribution, indicating again that the PBR exclusion method accounts for deviations in one group (viruses with PBRs) without dramatically shifting predictions for the other (viruses with covalently bound or free polynucleotides).
FIG 4.

Effect of excluding predicted polynucleotide-binding regions (PBRs) on theoretical capsid pIs (colored circles) for 32 viruses. Box and whisker plots represent the range of empirical pIs found in the literature, while circles represent predicted pIs calculated without modification, as well as after excluding predicted PBRs. PBRs were predicted either via the structure-based prediction developed in this study identifying arginine-rich regions and beta turns, or by one of two available NA-binding prediction tools, Pprint (45, 46) or DRNApred (47, 48). A key to the virus abbreviations (x axis) is provided in Table 2.
FIG 5.
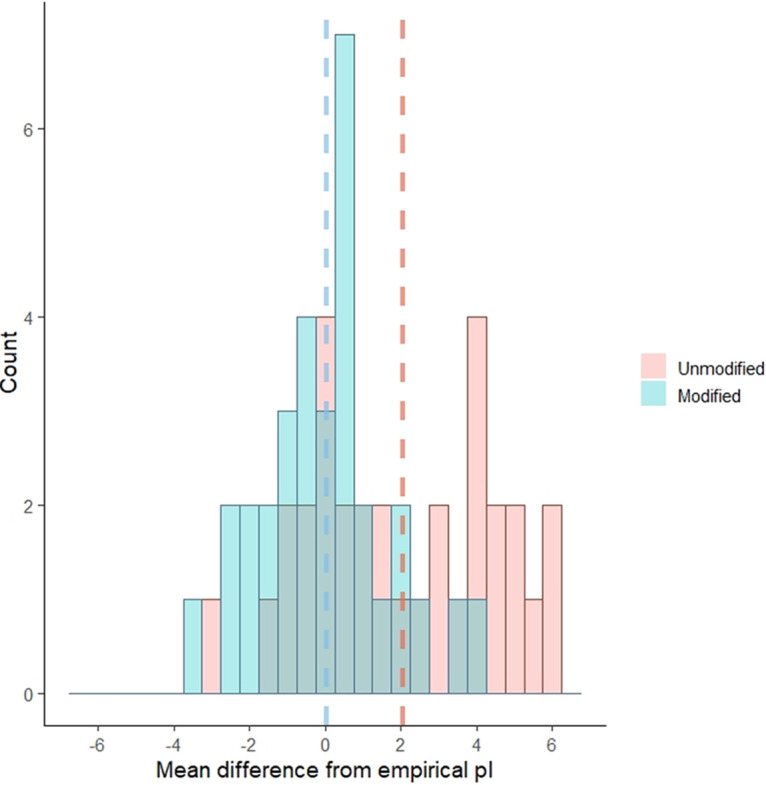
Histogram showing the shift in difference between predicted and mean empirical pI with and without modification by removal of predicted polynucleotide-binding regions (PBRs; including both arginine-rich regions and beta turns). Dashed lines represent the mean of means for each category (modified or unmodified), by color. The differences between theoretical and empirical pI values decreased significantly after modification, from 2.1 ± 2.4 to 0.1 ± 1.7 pH units.
Perhaps more impressively, accurate pI estimation was strongly correlated with the mean MCC of PBR prediction, as demonstrated in Fig. 6. MCCs for all PBR predictions used in this study negatively correlated with the mean absolute difference between theoretical and empirical pI (Spearman’s correlation ρ = −0.84; P < 2 × 10−16). Although training of the PBR prediction tools occurred independently of any impact on virus pI, better PBR prediction resulted in better pI prediction.
FIG 6.
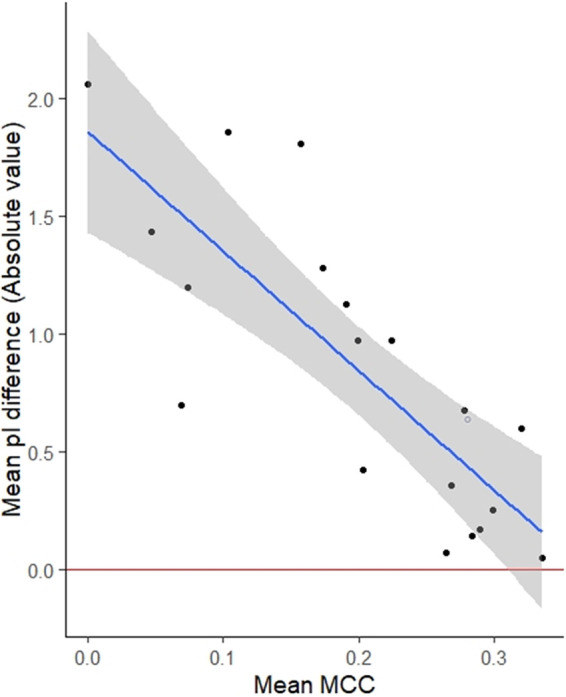
Correlation between polynucleotide-binding region prediction (represented as mean Matthews correlation coefficient [MCC] on the x axis) and pI prediction (mean pI difference on the y axis). Mean MCCs were calculated for all polynucleotide-binding prediction methods evaluated for this study. The blue line indicates the least-squares linear regression, with a shaded 95% confidence interval.
The impact of other conserved structure predictions on pI largely aligned with known PBRs, as shown in Fig. 7. This confirmation of structure-based pI prediction among closely related viruses further supports the case for excluding PBRs from pI calculation. Allolevivirus phages Qβ (EBQB) and SP (EBSP), as well as the tymovirus turnip yellow mosaic virus (TYMV), all had prominent known PBRs located on beta turns. Accordingly, the beta turn prediction showed some of the greatest improvements in pI prediction for EBQB, EBSP, and TYMV. Predictions for other tymoviruses improved as well, including those for belladonna mottle virus (BDMV), Erysimum latent virus (ELV), and Scrophularia mottle virus (ScrMV). Excluding beta sheets (independent of beta turns) also improved pI prediction for Leviviridae (EBFR, EBGA, EBMS2, EBQB, and EBSP) and tymoviruses (ELV and ScrMV), as expected from known PBRs. However, neither beta-sheet nor beta-turn prediction performed as well overall as arginine-rich region prediction, even for Leviviridae spp.
FIG 7.

Effect of excluding particular types of predicted polynucleotide-binding regions (PBRs) on theoretical capsid pIs. Box and whisker plots represent the range of empirical pIs found in the literature, while circles represent predicted pIs calculated without modification, as well as after excluding predicted PBRs. Several structures were used to predict PBRs: disordered N and C termini (disordered termini), arginine-rich regions (ARG rich), beta sheets, and beta turns (i.e., turns between adjacent beta sheets). A key to the 32 virus abbreviations (x axis) is provided in Table 2.
Several viruses had known PBRs on disordered N or C termini, namely cowpea chlorotic mosaic virus (CCMV), cucumber mosaic virus (CMV), cottontail rabbit polyomavirus (CRPV), red clover necrotic mosaic virus (RCNM), and southern bean mosaic virus (SBMV). These viruses were among the very few to show improved pI after removal of disordered termini, as shown in Fig. 7. The specificity of pI improvement on removal of disordered termini both verifies the connection between PBRs and pI, and it also indicates that the occurrence of disordered termini is not particular to PBRs and is therefore a poor predictor.
DISCUSSION
This study proposed a heuristic approach to pI prediction, in which only capsid residues bound to the viral polynucleotide were selectively excluded from the capsid charge calculation. This approach was based on the hypothesis that the charges of ionizable amino acids in these regions are neutralized by coulombic interactions with the virus polynucleotide. The PBR exclusion approach proposes a conceptual and conditional model for virion charge distribution, rather than a simplistic universal model. Furthermore, this approach excludes capsid regions only on the basis of polynucleotide binding, rather than for a desired impact on pI, and therefore avoids arbitrary removal of capsid regions to fit empirical pI values. A nonarbitrary approach is especially important given the relatively few viruses for which empirical pIs and capsid structures are known. With such a small data set, arbitrary variables are likely to overfit the model to the training set. In addition, the variety of capsid structures among even nonenveloped, icosahedral viruses may defy a universal model.
For example, bacteriophage MS2 has often been the exemplar of the effect of the viral genome on pI. The predicted pI of MS2 based on charged capsid moieties has been estimated to be between pH 7 (8) and 9 (51) (in this study, pH 7.4), while the estimated pI of MS2’s single-stranded RNA genome is approximately 3 (51). In contrast, the measured pI of MS2 is approximately 3.5 (7), closer to the estimated RNA pI than the estimated capsid pI. Structural models both including and excluding inner core charges have been proposed to explain this discrepancy. In MS2, the outermost shell of ssRNA lies directly beneath the capsid, and much of the capsid interior is devoted to binding the ssRNA genome (approximately 57% of the MS2 capsid protein) (32, 52). This highly basic region can be negated, thus decreasing the calculated pI to near the empirical value, by supposing either a strong negative influence from the core or that only exterior residues are relevant. In this way, bacteriophage MS2 is also an exemplar of the danger of overfitting a model.
Modifications based on known capsid structure.
The first evidence in support of the PBR exclusion hypothesis came from 3D capsid structures. While exterior surface residues were not correlated with overall virion charge, removing interior capsid surfaces improved overall pI prediction for a diverse set of viruses (Fig. 2). The exclusion of the interior capsid residues was most beneficial for viruses with interior PBRs. These viruses (Leviviridae phages and viruses with disordered termini) also show some of the greatest discrepancies between empirical and predicted pIs (Fig. 3). Because these typically basic, arginine-rich PBRs stabilize the negatively charged polynucleotide via coulombic forces (35, 36, 39, 41, 43), PBRs should therefore contribute no net charge to the virion. The degree to which major capsid proteins are involved in polynucleotide binding differs greatly between viruses and is generally greatest for viruses with single-stranded genomes (32, 33, 36, 52). This tendency is corroborated by the greater effect of both PBR exclusion and interior capsid surface exclusion on pI, but the trend has exceptions. As shown in Fig. 3 and 4, one double-stranded virus showing a major change (>2 pH units) in predicted pI after PBR exclusion is human adenovirus 5 (HadV5). The PBRs on HadV5 are located on core proteins, however, rather than on the capsid shell. Another exception is the double-stranded DNA (dsDNA) virus cottontail rabbit papillomavirus (CRPV), which has PBRs on capsid proteins themselves. This variable response, even when accounting for genome, also indicates that a universal approach of removing the capsid interior may not yield the best overall fit to empirical pI values. Nonetheless, the results shown in Fig. 3 demonstrate that removing the capsid interior may have negligible effect on viruses with evenly distributed ionizable amino acids.
Following these insights from 3D capsid structures, known PBRs were identified for exclusion from the capsid charge calculation, based on reports from the literature or conserved PBR structures. Known PBRs were found for 15 viruses. Predicted pI values were calculated for both the unmodified proteomes of these viruses, as well as the proteomes after PBR exclusion. Exclusion of known PBRs yielded additional improvements in predicted pIs, especially for viruses with unavailable 3D structures (Fig. 3). PBRs with the greatest impact on capsid pI fell into the following three broad structural categories: interior beta sheets, turns between beta sheets, and disordered termini. In addition, PBRs tended to have a high fraction of arginine and lysine (0.42 ± 0.29 arginine and/or lysine), simply termed “arginine-rich” here and elsewhere in the literature. These similarities indicated that PBRs could potentially be predicted via conserved primary and/or secondary structures.
Selective exclusion of predicted polynucleotide-binding regions.
Based on the improvement in predicted pIs after excluding known PBRs, we attempted to predict PBRs based on these conserved structures. These predicted PBRs would then be excluded from predicted pI calculations. The structure-based PBR prediction method performed better than existing RNA- and DNA-binding prediction tools. Selection of arginine-rich regions was the most comprehensive predictor (MCC = 0.32 ± 0.29), although this prediction improved by also selecting basic beta turns (MCC = 0.34 ± 0.28). (The MCC values for all predictions are provided in Table S1 in the supplemental material.) Unlike disordered termini and many basic beta sheets, beta turns may be adjacent in tertiary structure and constitute a region of basic charges, while still being distant in the protein sequence. Therefore, many beta-turn PBRs did not satisfy the conditions of arginine-rich prediction.
Two of the most conclusive findings from the predictive models were that (i) pI prediction improved most for groups of viruses known to have certain conserved PBR structures, and (ii) better PBR prediction led to better pI prediction. When comparing pI predictions based on individual PBR structure predictions (Fig. 7), pIs improved for both viruses known to have those PBRs and for closely related viruses. For example, leviviruses and tymoviruses, which feature PBRs along beta sheets and beta turns (53–55), showed the greatest pI improvements from beta sheet and turn predictions. Viruses with PBRs along disordered termini also showed the greatest pI improvement after exclusion of disordered termini. The strong correlation between PBR and pI prediction was true across the variety of prediction methods evaluated in this study, even though PBR prediction was conducted independently of the eventual impact on pI. Therefore, these PBR predictions further validate the results from known PBRs (Fig. 3) to support the hypothesis that PBRs do not contribute to overall virion charge. Thus, unlike previous models for pI prediction (9, 11, 12, 17), the PBR exclusion method is a nonarbitrary method of predicting capsid pI, in that no part of the model was adjusted for the effect on pI.
While the PBR exclusion method explained many of the biggest discrepancies between empirical and predicted pIs, several exceptions indicate the need for further research and refinement. As shown in Fig. 1, different strains of the same virus may deviate in pI by as much as 2 pH units. Of the 32 viruses considered here, seven predicted pI values remained more than 2 pH units from their empirical pIs after modification (Fig. 5), those for adeno-associated virus 4 (AAV4), bacteriophage PM2 (PM2), simian rotavirus A (SRVA), bacteriophage SP (EBSP), poliovirus 1 (POL1), southern bean mosaic virus (SBMV), and Scrophularia mosaic virus (ScrMV). The pI predictions for EBSP, SBMV, and ScrMV all improved with modification, though further refinements may be needed to bring predictions closer to empirical pI values. Unfortunately, three of the seven viruses (AAV4, PM2, and SRVA) are each represented by a single empirical pI reference, so the expected range of pIs for different strains and different experimental methods is unknown. Of these, both SRVA and PM2 are large viruses (diameter, >75 nm) with multilayered capsid structures, including an internal phospholipid bilayer in PM2. SRVA capsid proteins VP6 and VP7 also feature several calcium-binding regions (38). Polyvalent cations can be integral to the structure of some capsids and may influence virion charge (56). However, whether integral cations contribute to overall charge differently than nonintegral counterions in solution has yet to be determined.
Besides POL1, other members of the genus Enterovirus also had somewhat poorer pI predictions after removing predicted PBRs (Fig. 4): coxsackievirus A21 (CXA21), coxsackievirus B5 (CXB5), echovirus 1 (ECV1), human rhinovirus 2 (HRV2), and norovirus 1 (NOR1), as well as the closely related Mengo encephalomyocarditis virus (MEV). The PBR prediction is likely to generate some false positives as well as false negatives. In a review of the literature, PBRs were only found for POL1, which has three interior, NA-binding arginines (57). After removing these residues, the predicted pI of POL1 was slightly less accurate (Fig. 3). In contrast, excluding the capsid interior slightly decreased the divergence between theoretical and empirical pIs for this group (Fig. 3). Thus, Enterovirus may share a similar capsid structure that was not explained by this attempt at PBR prediction. Hepatitis A virus (HHAV) is in the same family as enteroviruses (Picornaviridae), yet, pI prediction for HHAV improved after excluding PBRs (Fig. 4). However, unlike other picornaviruses, HHAV occurs in cell culture and infected tissues in both enveloped and nonenveloped forms (26), and the sole source for HHAV pI is a brief with minimal information on methods (58). Therefore, further confirmation of empirical pIs for enveloped and nonenveloped HHAV is a research priority.
Picornaviruses (including enteroviruses) have covalently bound genomes (26), and may rely less on electrostatic binding than other ssRNA viruses. During assembly, picornaviruses form procapsids. Although there is disagreement over the precise mechanism of encapsidation, the procapsid contains the ssRNA polynucleotide, and the mature virion is condensed around the core via cleavage and restructuring of capsid proteins (26). However, the interaction that stabilizes the virion is likely between capsid proteins and proteins involved in genome replication, i.e., a protein-protein interaction rather than a protein-RNA interaction (59). This alternate mechanism of stabilizing the ssRNA capsid may explain the poor performance of the PBR exclusion model for enteroviruses in particular. However, this exception supports the fundamental hypothesis that polynucleotide binding is responsible for the greatest pI discrepancies; for viruses known to lack PBRs, the PBR exclusion method is neither necessary nor appropriate. The more that is known about the diversity of virion morphogenesis, the more detailed our model of virion charge structure can become.
Michen and Graule (7) noted that different methods of measuring pI may be responsible for some of the variation in empirical pIs reported in the literature. The available empirical pIs for these enteroviruses were all determined via isoelectric focusing, However, extrapolation of pI from electrophoretic mobility is generally considered a more accurate method for determining pI of monodispersed viral particles (56). Virus aggregation in isoelectric focusing may also lead to inaccurate estimation of pI, since aggregates can be subject to gravitational/buoyant forces in addition to electrophoresis (60). However, observation of electrophoretic mobility via dynamic light scattering requires high titers (>109 PFU/ml) that are not reasonable for many viruses. For the viruses referenced in this study, empirical pIs determined via electrophoretic mobility and isoelectric focusing methods tended to agree for the few viruses tested using both methods, as shown in Fig. S1 in the supplemental material. However, isoelectric focusing was used for all of the high pI values (above pH 7) included in this study, while electrophoretic mobility measurements skew toward lower pIs. Since many of the high virus pIs have not been confirmed via multiple methods (including those of Enterovirus spp., as well as AAV4, PM2, and SRVA), further validation of empirical pIs is needed.
Future research needs.
The PBR exclusion approach outlined in this study demonstrated strong potential to predict the pI of nonenveloped viruses based on structural features. The insight of PBR exclusion provides researchers three avenues for predicting the pI of a virus, in order of preference: (i) if the virus has a well-defined capsid with known PBRs, the known PBRs may be excluded, (ii) PBRs may be predicted via the definitions of conserved structures (arginine-rich regions and beta turns) provided in this study, or (iii) if the virus has a well-defined 3D structure, excluding the interior residues may approximate PBR exclusion. Where information is limited, researchers should cross-confirm using multiple methods.
We further stress that this method is likely not valid for enveloped viruses such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The effect of the envelope on charge and ion gradients is not considered here, and the composition of the lipid envelope itself is not encoded in the viral genome. In addition, the pIs of large viruses with multilayered capsid structures were poorly predicted in this study. Although the PBR exclusion principle should apply regardless of virion shape, one of the poorly modeled large viruses was a tailed bacteriophage (PM2). Since icosahedral virions typical of waterborne human viruses are most represented in the empirical data, a thorough assessment was not possible across all virion morphologies. Tailed bacteriophages and asymmetrically shaped virions may also have unevenly distributed charge concentrations not predicted by the overall pI. These localized charge concentrations could have important implications for interactions with the surrounding environment.
Using the PBR exclusion approach with either known or predicted PBRs successfully explained many of the largest discrepancies between theoretical and empirical pIs. Given the variation in empirical pIs reported for different virus strains (Fig. 1), accounting for such major discrepancies may be the limit of a predictive model at this time. Although the structure-based prediction used in this study outperformed existing NA-binding prediction tools, the main hurdle to a predictive pI model is that the available empirical pI and PBR data are poorly corroborated and overrepresent certain virus groups (e.g., Leviviridae). Further research is needed to identify the full extent of PBRs in virus capsid proteins, as well as to determine or corroborate empirical pIs for a broader range of viruses. In addition, the identification of beta turns used in this method relied on NetSurfP-2.0 to predict secondary structure (61). However, the conservation of structure in viral PBRs (e.g., interior beta sheets, disordered termini, and associated alpha helices) suggests that a sequence-based prediction tool specifically trained to viral capsid PBR motifs could be successful in identifying PBRs directly from proteome sequences.
MATERIALS AND METHODS
Prediction of isoelectric point.
The sum of ionizable amino acid charges (Q) for a given capsid protein, i, was calculated using the Henderson-Hasselbalch formula:
| (1) |
where pKaAA is the pKa value for a given amino acid, zpos is the number of positively charged amino acids in the capsid, and zneg is the number of negatively charged amino acids in the capsid. Amino acid pKa values are given in Table 1. The sum of charges for the entire capsid was considered by multiplying the sum of charges (including C and N termini) for each of m capsid proteins by the number of copies, n, of that protein within the capsid:
| (2) |
A summary of capsid proteins and copy numbers is presented in Table S3 in the supplemental material. Amino acids identified for exclusion (whether based on 3D capsid structure or PBRs) were not included in the sum of charges, including terminal amine and carboxyl charges where appropriate. Values of the predicted pI were calculated using the sum of charges for each capsid from pH 0 to 14 in order to find the pH value at which the charge approximated 0.
Empirical values for virus pIs were gathered from the literature, including Michen and Graule’s previous review (7), as well as all primary articles for additional information on experimental methods and conditions. A summary of empirical pIs is provided in Table S2 in the supplemental material. Multiple pIs have been reported by several sources using isoelectric focusing for mengovirus, human coxsackievirus A and B, human echovirus 1, and poliovirus 1 and 2 (7). While initially researchers suggested the different pIs represented different viral forms (62), Vrijsen et al. (60) demonstrated that the fraction of poliovirus 1 appearing near a pH of 4 to 5 in the pH gradient was separable by low centrifugation and did not appear when the virus sample was added near the primary pI (pH, ∼7) in an established pH gradient. Therefore, the secondary pI for poliovirus 1 was likely due to aggregated virus, while the primary pI represented monodispersed virus (60). Since the secondary band did not form when polioviruses were added at the pI (where some virus aggregation would also likely occur), the ampholytes used in isoelectric focusing as charge carriers may have destabilized viruses by charge neutralization and/or hydrophobic interactions. Ampholines used for isoelectric focusing have been shown to aggregate with acidic polysaccharides around pH 5 (63). Human enteroviruses B and C and mengovirus have also shown two distinct bands in isoelectric focusing with secondary pIs between pHs 4.4 and 4.8 (7). However, Chlumecka et al. (64) later found that the lower pI of mengovirus could be eliminated by adding ethylene glycol to promote dispersion. Here, all secondary pIs near pH 4 to 5 are likewise assumed to be an artifact of isoelectric focusing due to complexation with the ampholine buffer.
Our method of excluding polynucleotide-binding amino acids from the predicted pI calculation was evaluated by comparing the difference between predicted pIs and mean empirical values for each virus. The mean and standard deviation of these differences was compared before and after modifying the protein sequences by removing PBRs. The R stats package was used to perform a paired, two-tailed t test between the modified and unmodified samples.
Sources of capsid structures.
Amino acid sequences for all proteins composing the virus capsids were accessed via the UniProt database, as well as information about protein copy number, location within the capsid, and PBRs within capsid proteins, except as noted in the supplemental material (Table S3) (38). Sequences were analyzed in FASTA format using scripts written in-house using the R language (65). Only regions known to bind to the viral genome were considered, not regions binding host ATP/nucleotides. For viruses in the family Leviviridae, the entire interior-facing beta sheet was considered polynucleotide binding, as this region and RNA-associated assembly are highly conserved (31, 34). The beta sheet region was identified via literature values for bacteriophages MS2 (EBMS2) and Qβ (EBQB) (31, 52). The beta sheets for bacteriophages fr and GA were identified by cross-confirming results of visualization via PyMOL (66), secondary structure prediction via NetSurfP-2.0 (61, 67), and sequence similarity via the UniProt database. For human adenovirus 5 (HAdV5), two of the proteins in the UniProt database were entirely located within the virion core and closely associated with the host genome, histone-like nucleoprotein (GenPept accession number P68951 [https://www.uniprot.org/blast/?about=P68951%5b25-198%5d&key=Chain&id=PRO_0000036580]) and core protein X (accession number Q2KS10 [https://www.uniprot.org/blast/?about=Q2KS10%5b33-51%5d&key=Peptide&id=PRO_0000421138]). The entire sequences of these two short proteins (173 and 19 amino acids, respectively) was considered polynucleotide binding.
Viruses with available 3D capsid structures were identified via the VIPERdb icosahedral virus capsid database (68). The impact of 3D structure on capsid pI was evaluated by defining exterior residues as (i) including only the exterior capsid surface, or (ii) excluding only the interior capsid surface. Residues on the interior and exterior capsid surfaces were identified using the CapsidMaps tool via the VIPERdb website (30). In this study, 27 viruses were initially identified for having available 3D structures as well as empirical isoelectric point values in the literature. Of these 27 viruses, the surface residues of the 7 largest viruses (BP29, EBT4, HAdV5, PM2, PRD1, REO3, and SRVA) could not be accessed via the CapsidMaps tool due to large size and/or insufficient detail. A summary of viruses evaluated in this study is provided in Table 2.
TABLE 2.
Classification and abbreviations for viruses used in this study
| Abbreviation | Species (strain) | NCBI taxon IDa | PDB IDb (reference) | Genus | Family | Nucleic acidd | Host kingdom |
|---|---|---|---|---|---|---|---|
| AAV4 | Adeno-associated virus 4 | 57579 | 2G8G (73) | Dependoparvovirus | Parvoviridae | ssDNA | Animalia |
| BDMV | Belladonna mottle virus | 12149 | Tymovirus | Tymoviridae | ssRNA | Plantae | |
| BP29 | Bacillus phage Φ29 | 10756 | Salasvirus | Podoviridae | dsDNA | Bacteria | |
| CCMV | Cowpea chlorotic mottle virus | 12303 | 1CWP (74) | Bromovirus | Bromoviridae | ssRNA | Plantae |
| CMV | Cucumber mosaic virus (FNY) | 12307 | 1F15 (75) | Cucumovirus | Bromoviridae | ssRNA | Plantae |
| CPaV2 | Canine parvovirus 2 | 10790 | Protoparvovirus | Parvoviridae | ssDNA | Animalia | |
| CPaV2c | Feline panleukopenia virus | 10787 | 1C8G (76) | Protoparvovirus | Parvoviridae | ssDNA | Animalia |
| CRPV | Cottontail rabbit papillomavirus (Kansas) | 31553 | Kappapapillomavirus | Papillomaviridae | dsDNA | Animalia | |
| CRPVc | Human papillomavirus 16 | 333760 | 5KEQ (77) | Alphapapillomavirus | Papillomaviridae | dsDNA | Animalia |
| CXA21 | Human coxsackievirus A21 | 12070 | 1Z7S (78) | Enterovirus | Picornaviridae | ssRNA | Animalia |
| CXB5 | Human coxsackievirus B5 (Peterborough) | 103907 | Enterovirus | Picornaviridae | ssRNA | Animalia | |
| CXB5c | Human coxsackievirus B3 | 103904 | 1COV (79) | Enterovirus | Picornaviridae | ssRNA | Animalia |
| EBFR | Enterobacteria phage fr | 12017 | 1FRS (80) | Levivirus | Leviviridae | ssRNA | Bacteria |
| EBGA | Enterobacteria phage GA | 12018 | 1GAV (34) | Levivirus | Leviviridae | ssRNA | Bacteria |
| EBMS2 | Enterobacteria phage MS2 | 329852 | 2MS2 (81) | Levivirus | Leviviridae | ssRNA | Bacteria |
| EBQB | Enterobacteria phage Qβ | 39803 | 5VLY (82) | Allolevivirus | Leviviridae | ssRNA | Bacteria |
| EBSP | Enterobacteria phage SP | 12027 | Allolevivirus | Leviviridae | ssRNA | Bacteria | |
| EBT4 | Enterobacteria phage T4 | 10665 | 5VF3 (83) | Tequatrovirus | Myoviridae | dsDNA | Bacteria |
| ECV1 | Echovirus 1 (Farouk) | 103908 | 1EV1 (84) | Enterovirus | Picornaviridae | ssRNA | Animalia |
| ELV | Erysimum latent virus | 12152 | Tymovirus | Tymoviridae | ssRNA | Plantae | |
| HAdV5 | Human adenovirus 5 | 28285 | 4V4U (85) | Mastadenovirus | Adenoviridae | dsDNA | Animalia |
| HHAV | Hepatitis A virus (HM175) | 12098 | 4QPI (86) | Hepatovirus | Picornaviridae | ssRNA | Animalia |
| HRV2 | Human rhinovirus 2 | 12130 | 1FPN (87) | Enterovirus | Picornaviridae | ssRNA | Animalia |
| MEV | Mengo encephalomyocarditis virus | 12107 | 2MEV (88) | Cardiovirus | Picornaviridae | ssRNA | Animalia |
| NOR1 | Norwalk virus (Funabashi) | 524364 | 1IHM (89) | Norovirus | Caliciviridae | ssRNA | Animalia |
| PHIX | Enterobacteria phage ΦX174 (Sanger) | 10847 | 2BPA (90) | Sinsheimervirus | Microviridae | ssDNA | Bacteria |
| PM2 | Pseudoalteromonas phage PM2 | 10661 | 2W0C (91) | Corticovirus | Corticoviridae | dsDNA | Bacteria |
| POL1 | Poliovirus (Mahoney) | 12081 | 1HXS (92) | Enterovirus | Picornaviridae | ssRNA | Animalia |
| PRD1 | Enterobacteria phage PRD1 | 10658 | 1W8X (93) | Alphatectivirus | Tectiviridae | dsDNA | Bacteria |
| RCNM | Red clover necrotic mosaic virus | 12267 | 6MRM (94) | Dianthovirus | Tombusviridae | ssRNA | Plantae |
| REO3 | Reovirus 3 (Dearing) | 10886 | 2CSE (95) | Orthoreovirus | Reoviridae | dsRNA | Animalia |
| SBMV | Southern bean mosaic virus | 652938 | 4SbV (96) | Sobemovirus | Solemoviridae | ssRNA | Plantae |
| ScrMV | Scrophularia mottle virus | 312273 | Tymovirus | Tymoviridae | ssRNA | Plantae | |
| SRVA | Simian rotavirus A (SA11) | 450149 | 4V7Q (97) | Rotavirus | Reoviridae | dsRNA | Animalia |
| TBMV | Tobacco mosaic virus (Vulgare) | 12243 | Tobamovirus | Virgaviridae | ssRNA | Plantae | |
| TYMV | Turnip yellow mosaic virus | 12154 | 1AUY (98) | Tymovirus | Tymoviridae | ssRNA | Plantae |
National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov) taxonomic identifier (ID) (99).
Protein Data Bank (https://www.rcsb.org/) ID used for 3D structural comparisons (100).
Alternate species/strain used for 3D structure only.
ssDNA, single-stranded DNA; ssRNA, single-stranded RNA; dsDNA, double-stranded DNA; dsRNA, double-stranded RNA.
3D structures for closely related viruses were used when complete structures for strains used in empirical pI measurements were not available: feline parvovirus (PDB identifier [ID] 1C8G) was used for canine parvovirus (CPAV2; 98.7% capsid protein sequence identity), human coxsackievirus B3 (PDB ID 1COV) was used for human coxsackievirus B5 (CXB5; 90.4% genome polyprotein sequence identity), and human papillomavirus (PDB ID 5KEQ) was used for cottontail papillomavirus (CRPV; 43.4% capsid protein sequence identity). Despite the relatively poor sequence similarity between the human and cottontail papillomavirus, both had similar pIs for both unmodified (7.25 ± 0.05) and (known) PBR-excluded (6.15 ± 0.15) proteome sequences. Papillomaviruses share a DNA-binding C terminus on the L1 major capsid protein and may also have a less essential PBR on the N terminus of the L2 minor capsid protein (69, 70).
Predictive methods.
Potential PBRs were identified based on proteome sequences alone in an attempt to predict virion pI by the PBR exclusion method for a group of 32 viruses. PBRs were first predicted by identifying conserved PBR structures, including: predominately basic beta sheets and associated beta turns, disordered C and N termini, and arginine- and lysine-rich regions. Prediction of secondary structures (beta sheets, beta turns, and disordered termini) from proteome sequences was performed using a deep-learning protein structure prediction tool, NetSurfP-2.0 (61, 67). In addition to identifying PBRs via conserved structures, two Web-based tools for position-specific prediction of NA binding by residues were evaluated, Pprint for RNA binding (45, 46, 50), and DRNApred (47, 48) for DNA- and RNA-binding regions (48). However, neither tool was developed specifically for viral polynucleotide binding prediction.
All PBR predictions were evaluated based on the mean Matthews correlation coefficient (MCC) for all proteins in the training or validation set (49) according to the following equation:
| (3) |
where true positives (TP) are correctly predicted residues within PBRs, false positives (FP) are incorrectly predicted residues outside known PBRs, true negatives (TN) are residues outside known PBRs not predicted by the function, and false negatives (FN) are residues within known PBRs not predicted by the function. The MCC returns a value between −1 and 1, and it has the benefit of evaluating predictions of both positive and negative values. Capsid proteins containing known PBRs for the viruses used in this study were used as a training set for identifying PBRs. Predictions were then evaluated against a validation set of 40 different viral capsid proteins selected from the UniProt database which contained DNA- or RNA-binding regions (38). A summary of capsid proteins in the training and validation sets is presented in Tables S3 and S4 in the supplemental material. For each prediction method, the mean MCC for all viruses was compared to the mean absolute difference between theoretical and empirical pI for all viruses. The correlation between MCC and absolute pI difference was calculated via Spearman’s rank correlation using the R stats package (65).
Methods for predicting arginine-rich regions, beta sheets and turns, disordered termini, and RNA binding via Pprint were optimized based on known PBR sequences. Variables taking an integer value (e.g., amino acid counts or distances along a sequence) were optimized via brute force calculation, while noninteger variables were optimized using a 1-dimensional optimization function, “optimize,” in the R stats package (65). Via optimization, arginine-rich regions were defined as regions consisting of at least 24% arginine and/or lysine, with a minimum of 5 arginines/lysines and a maximum separation of 9 amino acids between consecutive arginines/lysines. Although arginine was the predominate amino acid in these regions (thus the name “arginine-rich”), lysine also has a strongly basic side chain and was frequently present in the same regions.
Beta sheets were defined as regions of contiguous predicted beta sheet structure at least 12 amino acids in length and composed of net neutral or basic ionizable amino acids (i.e., at least as many basic ionizable amino acids [ARG, LYS, HIS] as strong acidic ionizable amino acids [ASP, GLU, CYS]). Prediction of beta-turn PBRs was optimized by searching for contiguous regions that include a predicted beta turn and contain at least 2 basic ionizable amino acids (ARG, LYS, and/or HIS) separated by at most 3 amino acids. Disordered termini were defined via optimization as regions that include the C or N terminus and contain only residues with a disorder probability of 0.66 or greater, based on randomness predictions by NetSurfP-2.0 (61, 67).
The optimal RNA-binding likelihood for Pprint (46), based on the support vector machine (SVM) score (Pprint output), was determined to be 0.57. Unlike Pprint, DRNApred classified residues using default parameters, so no optimization was required (47). However, the DRNApred prediction was better when a positive hit for either DNA or RNA binding was considered positive (mean MCC, 0.17 ± 0.23) than when only hits matching the virus genome type were considered positive (mean MCC, 0.10 ± 0.24). The poor classification of viral capsid PBRs based on nucleic acid type may result from DRNApred not being intended for use specifically with viral genomes, which may be single- or double-stranded DNA or RNA.
Data availability.
All 3D structures, proteome sequences, and empirical pI data used in this study are publicly available. National Center for Biotechnology Information (NCBI) and Protein Data Bank (PDB) identifiers and citations for the viruses referenced in this study are provided in Table 2. A detailed summary of all capsid protein sequences, as well as UniProt entries and citations, is provided in Table S3. Secondary structures for proteome sequences were predicted via the NetSurfP-2.0 webtool (61, 67). PBRs were predicted using both in-house code to identify conserved structures based on NetSurfP output and the freely available webtools Pprint (46) and DRNApred (47). In-house R scripts used to identify conserved PBR structures are provided in the supplemental material (section S5). Empirical pI data and citations are provided in Table S2.
Supplementary Material
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Heffron J, Mayer BK. 2016. Virus mitigation by coagulation: recent discoveries and future directions. Environ Sci: Water Res Technol 2:443–459. doi: 10.1039/C6EW00060F. [DOI] [Google Scholar]
- 2.Heffron J, McDermid B, Maher E, McNamara P, Mayer BK. 2019. Mechanisms of virus mitigation and suitability of bacteriophages as surrogates in drinking water treatment by iron electrocoagulation. Water Res 163:114877. doi: 10.1016/j.watres.2019.114877. [DOI] [PubMed] [Google Scholar]
- 3.Mattle MJ, Crouzy B, Brennecke M, Wigginton KR, Perona P, Kohn T. 2011. Impact of virus aggregation on inactivation by peracetic acid and implications for other disinfectants. Environ Sci Technol 45:7710–7717. doi: 10.1021/es201633s. [DOI] [PubMed] [Google Scholar]
- 4.Gerba CP, Betancourt WQ. 2017. Viral aggregation: impact on virus behavior in the environment. Environ Sci Technol 51:7318–7325. doi: 10.1021/acs.est.6b05835. [DOI] [PubMed] [Google Scholar]
- 5.Horká M, Kubíček O, Růžička F, Holá V, Malinovská I, Šlais K. 2007. Capillary isoelectric focusing of native and inactivated microorganisms. J Chromatogr A 1155:164–171. doi: 10.1016/j.chroma.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 6.Brorson K, Shen H, Lute S, Pérez JS, Frey DD. 2008. Characterization and purification of bacteriophages using chromatofocusing. J Chromatogr A 1207:110–121. doi: 10.1016/j.chroma.2008.08.037. [DOI] [PubMed] [Google Scholar]
- 7.Michen B, Graule T. 2010. Isoelectric points of viruses. J Appl Microbiol 109:388–397. [DOI] [PubMed] [Google Scholar]
- 8.Mayer BK, Yang Y, Gerrity DW, Abbaszadegan M. 2015. The impact of capsid proteins on virus removal and inactivation during water treatment processes. Microbiol Insights 8:15–28. doi: 10.4137/MBI.S31441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armanious A, Aeppli M, Jacak R, Refardt D, Sigstam T, Kohn T, Sander M. 2016. Viruses at solid-water interfaces: a systematic assessment of interactions driving adsorption. Environ Sci Technol 50:732–743. doi: 10.1021/acs.est.5b04644. [DOI] [PubMed] [Google Scholar]
- 10.Božič AL, Šiber A, Podgornik R. 2012. How simple can a model of an empty viral capsid be? Charge distributions in viral capsids. J Biol Phys 38:657–671. doi: 10.1007/s10867-012-9278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penrod SL, Olson TM, Grant SB. 1996. Deposition kinetics of two viruses in packed beds of quartz granular media. Langmuir 12:5576–5587. doi: 10.1021/la950884d. [DOI] [Google Scholar]
- 12.Schaldach CM, Bourcier WL, Shaw HF, Viani BE, Wilson WD. 2006. The influence of ionic strength on the interaction of viruses with charged surfaces under environmental conditions. J Colloid Interface Sci 294:1–10. doi: 10.1016/j.jcis.2005.06.082. [DOI] [PubMed] [Google Scholar]
- 13.Weichert WS, Parker JSL, Wahid ATM, Chang SF, Meier E, Parrish CR. 1998. Assaying for structural variation in the parvovirus capsid and its role in infection. Virology 250:106–117. doi: 10.1006/viro.1998.9352. [DOI] [PubMed] [Google Scholar]
- 14.Brunk NE, Uchida M, Lee B, Fukuto M, Yang L, Douglas T, Jadhao V. 2019. Linker-mediated assembly of virus-like particles into ordered arrays via electrostatic control. ACS Appl Bio Mater 2:2192–2201. doi: 10.1021/acsabm.9b00166. [DOI] [PubMed] [Google Scholar]
- 15.Kozlowski LP. 2017. Proteome-pI: proteome isoelectric point database. Nucleic Acids Res 45:D1112–D1116. doi: 10.1093/nar/gkw978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiraga J, Mackiewicz P, Mackiewicz D, Kowalczuk M, Biecek P, Polak N, Smolarczyk K, Dudek MR, Cebrat S. 2007. The relationships between the isoelectric point and: length of proteins, taxonomy and ecology of organisms. BMC Genomics 8:163–116. doi: 10.1186/1471-2164-8-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langlet J, Gaboriaud F, Gantzer C, Duval JFL. 2008. Impact of chemical and structural anisotropy on the electrophoretic mobility of spherical soft multilayer particles: the case of bacteriophage MS2. Biophys J 94:3293–3312. doi: 10.1529/biophysj.107.115477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langlet J, Gaboriaud F, Duval JFL, Gantzer C. 2008. Aggregation and surface properties of F-specific RNA phages: implication for membrane filtration processes. Water Res 42:2769–2777. doi: 10.1016/j.watres.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Dika C, Duval JFL, Ly-Chatain HM, Merlin C, Gantzer C. 2011. Impact of internal RNA on aggregation and electrokinetics of viruses: comparison between MS2 phage and corresponding virus-like particles. Appl Environ Microbiol 77:4939–4948. doi: 10.1128/AEM.00407-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dika C, Duval JFL, Francius G, Perrin A, Gantzer C. 2015. Isoelectric point is an inadequate descriptor of MS2, Phi X 174 and PRD1 phages adhesion on abiotic surfaces. J Colloid Interface Sci 446:327–334. doi: 10.1016/j.jcis.2014.08.055. [DOI] [PubMed] [Google Scholar]
- 21.Duval JFL, Merlin J, Narayana PAL. 2011. Electrostatic interactions between diffuse soft multi-layered (bio)particles: beyond Debye-Hückel approximation and Deryagin formulation. Phys Chem Chem Phys 13:1037–1053. doi: 10.1039/c004243a. [DOI] [PubMed] [Google Scholar]
- 22.Gao T, Zhang W, Wang Y, Yang G. 2019. DNA compaction and charge neutralization regulated by divalent ions in very low pH solution. Polymers (Basel) 11:337. doi: 10.3390/polym11020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murthy VL, Rose GD. 2000. Is counterion delocalization responsible for collapse in RNA folding? Biochemistry 39:14365–14370. doi: 10.1021/bi001820r. [DOI] [PubMed] [Google Scholar]
- 24.Todd BA, Rau DC. 2008. Interplay of ion binding and attraction in DNA condensed by multivalent cations. Nucleic Acids Res 36:501–510. doi: 10.1093/nar/gkm1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuller DN, Rickgauer JP, Jardine PJ, Grimes S, Anderson DL, Smith DE. 2007. Ionic effects on viral DNA packaging and portal motor function in bacteriophage φ29. Proc Natl Acad Sci U S A 104:11245–11250. doi: 10.1073/pnas.0701323104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang P, Liu Y, Ma H-C, Paul AV, Wimmer E. 2014. Picornavirus morphogenesis. Microbiol Mol Biol Rev 78:418–437. doi: 10.1128/MMBR.00012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angelescu DG, Linse P. 2008. Modelling of icosahedral viruses. Curr Opin Colloid Interface Sci 13:389–394. doi: 10.1016/j.cocis.2007.10.004. [DOI] [Google Scholar]
- 28.Salo RJ, Mayor HD. 1978. Isoelectric focusing of parvoviruses. Intervirology 10:87–93. doi: 10.1159/000148972. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen TH, Easter N, Gutiérrez L, Huyett L, Defnet E, Mylon SE, Ferri JK, Viet NA. 2011. The RNA core weakly influences the interactions of the bacteriophage MS2 at key environmental interfaces. Soft Matter 7:10449–10456. doi: 10.1039/c1sm06092a. [DOI] [Google Scholar]
- 30.Carrillo-Tripp M, Montiel-García DJ, Brooks CL, Reddy VS. 2015. CapsidMaps: protein-protein interaction pattern discovery platform for the structural analysis of virus capsids using Google Maps. J Struct Biol 190:47–55. doi: 10.1016/j.jsb.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rumnieks J, Tars K. 2014. Crystal structure of the bacteriophage Qβ coat protein in complex with the rna operator of the replicase gene. J Mol Biol 426:1039–1049. doi: 10.1016/j.jmb.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 32.Toropova K, Basnak G, Twarock R, Stockley PG, Ranson NA. 2008. The three-dimensional structure of genomic RNA in bacteriophage MS2: implications for assembly. J Mol Biol 375:824–836. doi: 10.1016/j.jmb.2007.08.067. [DOI] [PubMed] [Google Scholar]
- 33.Basnak G, Morton VL, Rolfsson Ó, Stonehouse NJ, Ashcroft AE, Stockley PG. 2010. Viral genomic single-stranded RNA directs the pathway toward a T = 3 capsid. J Mol Biol 395:924–936. doi: 10.1016/j.jmb.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tars K, Bundule M, Fridborg K, Liljas L. 1997. The crystal structure of bacteriophage GA and a comparison of bacteriophages belonging to the major groups of Escherichia coli leviviruses. J Mol Biol 271:759–773. doi: 10.1006/jmbi.1997.1214. [DOI] [PubMed] [Google Scholar]
- 35.Lee SK, Hacker DL. 2001. In vitro analysis of an RNA binding site within the N-terminal 30 amino acids of the southern cowpea mosaic virus coat protein. Virology 286:317–327. doi: 10.1006/viro.2001.0979. [DOI] [PubMed] [Google Scholar]
- 36.Belyi VA, Muthukumar M. 2006. Electrostatic origin of the genome packing in viruses. Proc Natl Acad Sci U S A 103:17174–17178. doi: 10.1073/pnas.0608311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park S-H, Sit TL, Kim K-H, Lommel SA. 2013. The red clover necrotic mosaic virus capsid protein N-terminal amino acids possess specific RNA binding activity and are required for stable virion assembly. Virus Res 176:107–118. doi: 10.1016/j.virusres.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 38.The UniProt Consortium. 2019. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ford RJ, Barker AM, Bakker SE, Coutts RH, Ranson NA, Phillips SV, Pearson AR, Stockley PG. 2013. Sequence-specific, RNA-protein interactions overcome electrostatic barriers preventing assembly of satellite tobacco necrosis virus coat protein. J Mol Biol 425:1050–1064. doi: 10.1016/j.jmb.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reinisch KM, Nibert ML, Harrison SC. 2000. Structure of the reovirus core at 3.6 Å resolution. Nature 404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- 41.García-García C, Draper DE. 2003. Electrostatic interactions in a peptide-RNA complex. J Mol Biol 331:75–88. doi: 10.1016/S0022-2836(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 42.Bayer TS, Booth LN, Knudsen SM, Ellington AD. 2005. Arginine-rich motifs present multiple interfaces for specific binding by RNA. RNA 11:1848–1857. doi: 10.1261/rna.2167605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grieger JC, Snowdy S, Samulski RJ. 2006. Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J Virol 80:5199–5210. doi: 10.1128/JVI.02723-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao K, Wu Y, Chen Q, Zhang Z, Chen X, Zhang Y. 2016. The arginine/lysine-richelement within the DNA-binding domain is essential for nuclear localization and function of the intracellular pathogen resistance 1. PLoS One 11:e0162832. doi: 10.1371/journal.pone.0162832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar M, Gromiha MM, Raghava GPS. 2008. Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins Struct Funct Bioinforma 71:189–194. doi: 10.1002/prot.21677. [DOI] [PubMed] [Google Scholar]
- 46.Kumar M, Gromiha MM, Raghava GPS. 2008. Pprint: for prediction of RNA-interacting amino acid residues. https://webs.iiitd.edu.in/raghava/pprint/.
- 47.Yan J, Kurgan L. 2017. DRNApred—DNA- and RNA-binding residues predictor. http://biomine.cs.vcu.edu/servers/DRNApred/. [DOI] [PMC free article] [PubMed]
- 48.Yan J, Kurgan L. 2017. DRNApred, fast sequence-based method that accurately predicts and discriminates DNA-and RNA-binding residues. Nucleic Acids Res 45:e84. doi: 10.1093/nar/gkx059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan J, Friedrich S, Kurgan L. 2016. A comprehensive comparative review of sequence-based predictors of DNA- and RNA-binding residues. Brief Bioinform 17:88–105. doi: 10.1093/bib/bbv023. [DOI] [PubMed] [Google Scholar]
- 50.Kumar M, Gromiha MM, Raghava GPS. 2011. SVM based prediction of RNA-binding proteins using binding residues and evolutionary information. J Mol Recognit 24:303–313. doi: 10.1002/jmr.1061. [DOI] [PubMed] [Google Scholar]
- 51.Duval JFL, Gaboriaud F. 2010. Progress in electrohydrodynamics of soft microbial particle interphases. Curr Opin Colloid Interface Sci 15:184–195. doi: 10.1016/j.cocis.2009.12.002. [DOI] [Google Scholar]
- 52.Rolfsson Ó, Middleton S, Manfield IW, White SJ, Fan B, Vaughan R, Ranson NA, Dykeman E, Twarock R, Ford J, Kao CC, Stockley PG. 2016. Direct evidence for packaging signal-mediated assembly of bacteriophage MS2. J Mol Biol 428:431–448. doi: 10.1016/j.jmb.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ehresmann B, Briand J‐P, Reinbolt J, Witz J. 1980. Identification of binding sites of turnip yellow mosaic virus protein and RNA by crosslinks induced in situ. Eur J Biochem 108:123–129. doi: 10.1111/j.1432-1033.1980.tb04702.x. [DOI] [PubMed] [Google Scholar]
- 54.Bink HHJ, Roepan SK, Pleij CWA. 2004. Two histidines of the coat protein of turnip yellow mosaic virus at the capsid interior are crucial for viability. Proteins Struct Funct Bioinforma 55:236–244. doi: 10.1002/prot.10600. [DOI] [PubMed] [Google Scholar]
- 55.Lim F, Spingola M, Peabody DS. 1996. The RNA-binding site of bacteriophage Qβ coat protein. J Biol Chem 271:31839–31845. doi: 10.1074/jbc.271.50.31839. [DOI] [PubMed] [Google Scholar]
- 56.Schäfer R, Hinnen R, Franklin RM. 1974. Structure and synthesis of a lipid‐containing bacteriophage: properties of the structural proteins and distribution of the phospholipid. Eur J Biochem 50:15–27. doi: 10.1111/j.1432-1033.1974.tb03868.x. [DOI] [PubMed] [Google Scholar]
- 57.Ansardi DC, Luo M, Morrow CD. 1994. Mutations in the poliovirus P1 capsid precursor at arginine residues VP4-ARG34, VP3-ARG223, and VP1-ARG129 affect virus assembly and encapsidation of genomic RNA. Virology 199:20–34. doi: 10.1006/viro.1994.1094. [DOI] [PubMed] [Google Scholar]
- 58.Nasser AM, Battagelli D, Sobsey MD. 1992. Isoelectric focusing of hepatitis A virus in sucrose gradients. Isr J Med Sci 28:73. [Google Scholar]
- 59.Liu Y, Wang C, Mueller S, Paul AV, Wimmer E, Jiang P. 2010. Direct interaction between two viral proteins, the nonstructural protein 2CATPase and the capsid protein VP3, is required for enterovirus morphogenesis. PLoS Pathog 6:e1001066. doi: 10.1371/journal.ppat.1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vrijsen R, Rombaut B, Boeye A. 1983. pH-dependent aggregation and electrofocusing of poliovirus. J Gen Virol 64:2339–2342. doi: 10.1099/0022-1317-64-10-2339. [DOI] [PubMed] [Google Scholar]
- 61.DTU Bioinformatics. 2019. NetSurfP-2.0. http://www.cbs.dtu.dk/services/NetSurfP.
- 62.Murray JP, Parks GA. 1980. Poliovirus adsorption on oxide surfaces, p 97–133. In Kavanaugh MC, Leckie JO (ed), Particulates in water. American Chemical Society, Washington, DC. [Google Scholar]
- 63.Righetti PG, Brown RP, Stone AL. 1978. Aggregation of ampholine on heparin and other acidic polysaccharides in isoelectric focusing. Biochim Biophys Acta 542:232–244. doi: 10.1016/0304-4165(78)90019-3. [DOI] [PubMed] [Google Scholar]
- 64.Chlumecka V, D’Obrenan P, Colter JS. 1977. Isoelectric focusing studies of Mengo virus variants, their protein structure units and constituent polypeptides. J Gen Virol 35:425–437. doi: 10.1099/0022-1317-35-3-425. [DOI] [Google Scholar]
- 65.R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 66.Schrödinger LLC. 2019. The PyMOL molecular graphics system. 2.3.2.
- 67.Klausen MS, Jespersen MC, Nielsen H, Jensen KK, Jurtz VI, Sønderby CK, Sommer MOA, Winther O, Nielsen M, Petersen B, Marcatili P. 2019. NetSurfP-2.0: improved prediction of protein structural features by integrated deep learning. Proteins Struct Funct Bioinforma 87:520–527. doi: 10.1002/prot.25674. [DOI] [PubMed] [Google Scholar]
- 68.Ho PT, Montiel-Garcia DJ, Wong JJ, Carrillo-Tripp M, Brooks CL, Johnson JE, Reddy VS. 2018. VIPERdb: a tool for virus research. Annu Rev Virol 5:477–488. doi: 10.1146/annurev-virology-092917-043405. [DOI] [PubMed] [Google Scholar]
- 69.Schäfer F, Florin L, Sapp M. 2002. DNA binding of L1 is required for human papillomavirus morphogenesis in vivo. Virology 295:172–181. doi: 10.1006/viro.2002.1361. [DOI] [PubMed] [Google Scholar]
- 70.Wang JW, Roden RBS. 2013. L2, the minor capsid protein of papillomavirus. Virology 445:175–186. doi: 10.1016/j.virol.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kozlowski LP. 2016. IPC—Isoelectric Point Calculator. Biol Direct 11:1–16. doi: 10.1186/s13062-016-0159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blackburn GM. 2006. Nucleic acids in chemistry and biology, 3rd ed Royal Society of Chemistry, Cambridge, UK. [Google Scholar]
- 73.Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA, Agbandje-McKenna M. 2006. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J Virol 80:11556–11570. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Speir JA, Munshi S, Wang G, Baker TS, Johnson JE. 1995. Structures of the native and swollen forms of cowpea chlorotic mottle virus determined by X-ray crystallography and cryo-electron microscopy. Structure 3:63–78. doi: 10.1016/s0969-2126(01)00135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith TJ, Chase E, Schmidt T, Perry KL. 2000. The structure of cucumber mosaic virus and comparison to cowpea chlorotic mottle virus. J Virol 74:7578–7586. doi: 10.1128/jvi.74.16.7578-7586.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simpson AA, Chandrasekar V, Hébert B, Sullivan GM, Rossmann MG, Parrish CR. 2000. Host range and variability of calcium binding by surface loops in the capsids of canine and feline parvoviruses 1. J Mol Biol 300:597–610. doi: 10.1006/jmbi.2000.3868. [DOI] [PubMed] [Google Scholar]
- 77.Guan J, Bywaters SM, Brendle SA, Ashley RE, Makhov AM, Conway JF, Christensen ND, Hafenstein S. 2017. Cryoelectron microscopy maps of human papillomavirus 16 reveal L2 densities and heparin binding site. Structure 25:253–263. doi: 10.1016/j.str.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 78.Xiao C, Bator-Kelly CM, Rieder E, Chipman PR, Craig A, Kuhn RJ, Wimmer E, Rossmann MG. 2005. The crystal structure of coxsackievirus A21 and its interaction with ICAM-1. Structure 13:1019–1033. doi: 10.1016/j.str.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 79.Muckelbauer JK, Kremer M, Minor I, Tong L, Zlotnick A, Johnson JE, Rossmann MG. 1995. Structure determination of coxsackievirus B3 to 3.5 Å resolution. Acta Crystallogr D Biol Crystallogr 51:871–887. doi: 10.1107/S0907444995002253. [DOI] [PubMed] [Google Scholar]
- 80.Liljas L, Fridborg K, Valegård K, Bundule M, Pumpens P. 1994. Crystal structure of bacteriophage fr capsids at 3.5 A resolution. J Mol Biol 244:279–290. doi: 10.1006/jmbi.1994.1729. [DOI] [PubMed] [Google Scholar]
- 81.Golmohammadi R, Valegard K, Fridborg K, Liljas L. 1993. The refined structure of bacteriophage MS2 at 2.8 A resolution. J Mol Biol 234:620–639. doi: 10.1006/jmbi.1993.1616. [DOI] [PubMed] [Google Scholar]
- 82.Cui Z, Gorzelnik KV, Chang JY, Langlais C, Jakana J, Young R, Zhang J. 2017. Structures of Qβ virions, virus-like particles, and the Qβ–MurA complex reveal internal coat proteins and the mechanism of host lysis. Proc Natl Acad Sci U S A 114:11697–11702. doi: 10.1073/pnas.1707102114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Z, Sun L, Zhang Z, Fokine A, Padilla-Sanchez V, Hanein D, Jiang W, Rossmann MG, Rao VB. 2017. Cryo-EM structure of the bacteriophage T4 isometric head at 3.3-Å resolution and its relevance to the assembly of icosahedral viruses. Proc Natl Acad Sci U S A 114:E8184–E8193. doi: 10.1073/pnas.1708483114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Filman DJ, Wien MW, Cunningham JA, Bergelson JM, Hogle JM. 1998. Structure determination of echovirus 1. Acta Crystallogr D Biol Crystallogr 54:1261–1272. doi: 10.1107/s0907444998002790. [DOI] [PubMed] [Google Scholar]
- 85.Fabry CMS, Rosa-Calatrava M, Conway JF, Zubieta C, Cusack S, Ruigrok RWH, Schoehn G. 2005. A quasi-atomic model of human adenovirus type 5 capsid. EMBO J 24:1645–1654. doi: 10.1038/sj.emboj.7600653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X, Ren J, Gao Q, Hu Z, Sun Y, Li X, Rowlands DJ, Yin W, Wang J, Stuart DI, Rao Z, Fry EE. 2015. Hepatitis A virus and the origins of picornaviruses. Nature 517:85–88. doi: 10.1038/nature13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Verdaguer N, Blaas D, Fita I. 2000. Structure of human rhinovirus serotype 2 (HRV2)11. J Mol Biol 300:1179–1194. doi: 10.1006/jmbi.2000.3943. [DOI] [PubMed] [Google Scholar]
- 88.Krishnaswamy S, Rossmann MG. 1990. Structural refinement and analysis of Mengo virus. J Mol Biol 211:803–844. doi: 10.1016/0022-2836(90)90077-Y. [DOI] [PubMed] [Google Scholar]
- 89.Prasad BVV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290. doi: 10.1126/science.286.5438.287. [DOI] [PubMed] [Google Scholar]
- 90.McKenna R, Xia D, Willingmann P, Ilag LL, Krishnaswamy S, Rossmann MG, Olson NH, Baker TS, Incardona NL. 1992. Atomic structure of single-stranded DNA bacteriophage ΦX174 and its functional implications. Nature 355:137–143. doi: 10.1038/355137a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abrescia NGA, Grimes JM, Kivelä HM, Assenberg R, Sutton GC, Butcher SJ, Bamford JKH, Bamford DH, Stuart DI. 2008. Insights into virus evolution and membrane biogenesis from the structure of the marine lipid-containing bacteriophage PM2. Mol Cell 31:749–761. doi: 10.1016/j.molcel.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 92.Miller ST, Hogle JM, Filman DJ. 2001. Ab initio phasing of high-symmetry macromolecular complexes: successful phasing of authentic poliovirus data to 3.0 Å resolution. J Mol Biol 307:499–512. doi: 10.1006/jmbi.2001.4485. [DOI] [PubMed] [Google Scholar]
- 93.Abrescia NGA, Cockburn JJB, Grimes JM, Sutton GC, Diprose JM, Butcher SJ, Fuller SD, San Martín C, Burnett RM, Stuart DI, Bamford DH, Bamford JKH. 2004. Insights into assembly from structural analysis of bacteriophage PRD1. Nature 432:68–74. doi: 10.1038/nature03056. [DOI] [PubMed] [Google Scholar]
- 94.Sherman MB, Guenther R, Reade R, Rochon D, Sit T, Smith TJ. 2019. Near-atomic-resolution cryo-electron microscopy structures of cucumber leaf spot virus and red clover necrotic mosaic virus: evolutionary divergence at the icosahedral three-fold axes. J Virol 94:e01439-19. doi: 10.1128/JVI.01439-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang X, Ji Y, Zhang L, Harrison SC, Marinescu DC, Nibert ML, Baker TS. 2005. Features of reovirus outer capsid protein μ1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Å resolution. Structure 13:1545–1557. doi: 10.1016/j.str.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Silva AM, Rossmann MG. 1985. The refinement of southern bean mosaic virus in reciprocal space. Acta Crystallogr B Struct Sci 41:147–157. doi: 10.1107/S0108768185001781. [DOI] [Google Scholar]
- 97.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. 2011. Atomic model of an infectious rotavirus particle. EMBO J 30:408–416. doi: 10.1038/emboj.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Canady MA, Larson SB, Day J, McPherson A. 1996. Crystal structure of turnip yellow mosaic virus. Nat Struct Biol 3:771–781. doi: 10.1038/nsb0996-771. [DOI] [PubMed] [Google Scholar]
- 99.Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Mizrachi I, Ostell J, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Yaschenko E, Ye J. 2009. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 37:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res 28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All 3D structures, proteome sequences, and empirical pI data used in this study are publicly available. National Center for Biotechnology Information (NCBI) and Protein Data Bank (PDB) identifiers and citations for the viruses referenced in this study are provided in Table 2. A detailed summary of all capsid protein sequences, as well as UniProt entries and citations, is provided in Table S3. Secondary structures for proteome sequences were predicted via the NetSurfP-2.0 webtool (61, 67). PBRs were predicted using both in-house code to identify conserved structures based on NetSurfP output and the freely available webtools Pprint (46) and DRNApred (47). In-house R scripts used to identify conserved PBR structures are provided in the supplemental material (section S5). Empirical pI data and citations are provided in Table S2.