

Abstract
Rationale:
Hydrogen sulfide (H2S) is a physiological mediator that regulates cardiovascular homeostasis. Three major enzymes contribute to the generation of endogenously produced H2S, namely cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS) and 3-mercaptopyruvate sulfurtransferase (3-MST). Although the biological roles of CSE and CBS have been extensively investigated in the cardiovascular system, very little is known about that of 3-MST. In the present study we determined the importance of 3-MST in the heart and blood vessels, using a genetic model with a global 3-MST deletion.
Results:
3-MST is the most abundant transcript in the mouse heart, compared to CSE and CBS. 3-MST was mainly localized in smooth muscle cells and cardiomyocytes, where it was present in both the mitochondria and the cytosol. Levels of serum and cardiac H2S species were not altered in adult young (2–3 months old) 3-MST−/− mice compared to WT animals. No significant changes in the expression of CSE and CBS were observed. Additionally, 3-MST−/− mice had normal left ventricular structure and function, blood pressure and vascular reactivity. Interestingly, genetic ablation of 3-MST protected mice against myocardial ischemia reperfusion injury, and abolished the protection offered by ischemic pre- and post-conditioning. 3-MST−/− mice showed lower expression levels of thiosulfate sulfurtransferase, lower levels of cellular antioxidants and elevated basal levels of cardiac reactive oxygen species. In parallel, 3-MST−/− mice showed no significant alterations in endothelial NO synthase or downstream targets. Finally, in a separate cohort of older 3-MST−/− mice (18 months old), a hypertensive phenotype associated with cardiac hypertrophy and NO insufficiency was observed.
Conclusions:
Overall, genetic ablation of 3-MST impacts on the mouse cardiovascular system in an age-dependent manner. Loss of 3-MST exerts a cardioprotective role in young adult mice, while with aging it predisposes them to hypertension and cardiac hypertrophy.
Keywords: 3-mercaptopyruvate transferase (3-MST), Myocardial infarction, Blood pressure, Reactive Oxygen Species (ROS), Nitric Oxide (NO), Aging
1. Introduction
Hydrogen sulfide (H2S) is an established signaling molecule that exerts multiple physiological effects in most tissues and organs throughout the body [1]. In the cardiovascular system H2S has been shown to promote angiogenesis, to trigger vasorelaxation and reduce blood pressure, to limit inflammation and atherosclerosis as well as to confer cardioprotection [2–4]. Reduced production of H2S has been observed in several animal models of cardiovascular disease, as well as in patient cohorts [5–7]. Endogenous production of H2S occurs both through non-enzymatic and enzymatic pathways. Cysteine can yield H2S in the presence of vitamin B6 and iron and alimentary polysulfides can be converted to H2S through the action of reducing agents, such as glutathione [5,8]. The main enzymatic sources of H2S include cystathionine-γ-synthase (CSE), cystathionine-β-synthase (CBS) and 3-mercaptopyruvate transferase (3-MST) [5,9]. More recently, cysteinyltRNA synthetase was reported to possess cysteine persulfide synthase activity generating hydrogen persulfides and H2S [10]. The bacterial gut flora is also known to contribute to the circulating and tissue H2S pools [11]. Although the contribution of non-enzymatic vs enzymatic sources of H2S remains poorly characterized, it is generally assumed that most of the H2S in cardiovascular tissues is enzymatically generated. CSE is believed to be the main enzyme present in the heart and the vasculature and most studies focused on its role in cardiovascular physiology and pathophysiology [12]. Indeed, mice with global deletion of CSE are hypertensive, more susceptible to atherosclerosis and express more severe phenotypes when subjected to stress compared to wild-type animals [2,13–15]. Fewer studies have investigated the role of CBS in the cardiovascular system, as the enzyme was thought to be more functionally relevant to the nervous system. However, the fact that CBS knockout mice die before adulthood (at 5–6 weeks), together with the lack of selective and potent CBS inhibitors make studies with CBS difficult to perform [16]. Nevertheless, CBS mutations that render the enzyme inactive or hypoactive lead to increased homocysteine levels, a known risk factor for cardiovascular disease [17].
3-MST (EC 2.8.1.2) is the third main enzyme involved in H2S bio-synthesis. It uses 3-mercaptopyruvate (3-MP) as substrate to produce H2S, persulfides and polysulfides [18]. 3-MST generates H2S and pyruvate in a two-step reaction i.e., it transfers sulfur from 3-MP to a cysteine 247 in its active site, yielding a 3-MST persulfide and a molecule of pyruvate [19]. The persulfide can either release H2S after reacting with thiols or reduced thioredoxin or be transferred to thiol protein or other small molecules [20]. In a study that was performed ten years ago, 3-MST was shown to be expressed in endothelial cells [21], indicating that this enzyme might have important biological roles in cardiovascular function. 3-MST has now been confirmed to be expressed in the heart, kidney and many blood vessels [22–24] but its function in the cardiovascular system remains enigmatic. Herein, we set out to evaluate the role of 3-MST in cardiac and vascular homeostasis using a global 3-MST−/− mouse.
2. Materials and methods
2.1. Materials
Most of the reagents were purchased by Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated.
2.2. Animals
Male C57Bl/6J WT and 3-MST−/− [25] of similar age (2–3 or 18 months old) were used in the current study. The mice were bred in the animal facilities of BRFAA (Greece), the Cardiovascular Center of Excellence, Louisiana State University Health Sciences Center, New Orleans, (USA) and the Department of Pharmacy, University of Naples “Federico II” (Italy). The genotype of the mice was confirmed for all individual mice by genotyping and western blot as previously described [25]. All experimental procedures reported here were approved by the veterinary authority of the Prefecture of Athens and in accordance to the National Registration (Presidential Decree 56/2013) in harmonization to the European Directive 63/2010 or by the Institute for Animal Care and Use Committee at Louisiana State University Health Sciences Center (USA) or according to the ARRIVE guidelines and authorized by Centro Servizi Veterinari Università degli Studi di Napoli ‘Federico II’. Mice were housed in a pathogen-free, temperature controlled (22 °C), 12-h light/dark cycle in accredited animal facilities and allowed free access to diets and water. To harvest tissues and blood the mice were first anesthetized under sevoflurane anesthesia (dose 3% v/v) and then sacrificed by cervical dislocation. The whole heart was excised, briefly dried from any surrounding water and weighed. Moreover, tibias from all mice were removed, cleaned from surrounding muscles and measured in length with a ruler. All tissues were snap frozen in liquid nitrogen and stored in −80 °C until the day of analysis.
2.3. Blood pressure measurements
Blood pressure was measured with the non-invasive plethysmography tail-cuff method (Kent Scientific, Torrington, CT, USA) as previously described [26]. Baseline blood pressure was measured in WT and 3-MST−/− mice for 3 days before actually beginning the formal measurements. This is the established training period that allows the mice to acclimatize with the technique and eliminate any stress response. Once, confirmed that all mice showed no signs of stress response, measurements for 2 consecutive days were performed and averaged for the calculation of mean, systolic and diastolic blood pressure.
2.4. Cardiac ischemia/re-perfusion injury
Myocardial infarction was induced by ligation of the left coronary artery as previously described [27]. WT and 3-MST−/− male mice 8–12 weeks of age were anesthetized by intraperitoneal injection with a combination of ketamine, xylazine, and atropine (0.01 ml/g, final concentrations of ketamine, xylazine, and atropine 10 mg/ml, 2 mg/ml, 0.06 mg/kg, respectively). A tracheotomy was performed for artificial respiration at 120–150 breaths/min and PEEP 2.0 (0.2 ml tidal volume) (Flexivent rodent ventilator; Scireq, Montreal, QC). Electrocardiogram recordings were performed by a lead I ECG recording with PowerLab 4.0 (ADInstruments, Sydney, Australia). Recordings were analyzed by LabChart 7.0 software. A thoracotomy was then performed between the fourth and fifth ribs, and the pericardium was carefully retracted to visualize the left anterior descending coronary artery (LAD), which was ligated using a 8–0 Prolene (Ethicon, Somerville, NJ) monofilament polypropylene suture placed 1 mm below the tip of the left ventricle. The heart was stabilized for 15 min before ligation to induce ischemia. After the ischemic period (30 min), the ligature was released and allowed reperfusion of the myocardium for 2 h. Moreover, some mice were exposed to ischemic pre-conditioning (one cycle of 5 min ischemia followed by 5 min of reperfusion was applied before the sustained ischemia) or post-conditioning (3 cycles of 10 s of ischemia followed by 10 s of reperfusion was applied immediately after sustained ischemia). Throughout the experiments, body temperature was maintained at 37 ± 0.5 °C by using a heating pad. After reperfusion, hearts were rapidly excised from the mice and directly cannulated and washed with 2.5 ml of saline-heparin 1% for blood removal. Five milliliters of 1% TTC phosphate buffer 37 °C were infused via the cannula into the coronary circulation, followed by incubation of the myocardium for 5 min in the same buffer; 2.5 ml of 1% Evans blue, diluted in distilled water, was then infused into the heart. Hearts were kept in −20 °C for 24 h, sliced in 1-mm sections parallel to the atrioventricular groove, and then fixed in 4% formaldehyde overnight. Slices were then placed between glass plates 1 mm apart and photographed with a Cannon Powershot A620 Digital Camera through Zeiss 459,300 microscope and measured with the Scion Image program. Infarct and risk area volumes were expressed in cubic centimeters, and the percentage of infarct-to-risk area ratio (percentage of ischemia/reperfusion) was calculated. In an independent series of experiments performed at the Center of Excellence (LSU HSC, New Orleans), mice were subjected to 30 min of ischemia, followed by 24 hrs of reperfusion, as previously described [28].
2.5. Echocardiography
WT and 3-MST−/− mice were anaesthetized (ketamine, 100 mg/kg i.p) and underwent echocardiographic assessment of left ventricular (LV) function using an ultrasound system (Vivid 7; GE Healthcare, USA) with a 13-MHz linear transducer, as previously described [29]. Parameters such as heart rate (HR), interventricular septal-end diastole and end-systole dimensions (IVSd and IVSs), end-diastole and end-systole diameter (LVEDD, LVESD), LV posterior wall thickness at diastole and systole (PWd, PWs), fractional shortening [FS % = (EDD −ESD)/EDD * 100] and LV radius to LV posterior wall thickness ratio (r/h) were calculated.
2.6. Imaging on cardiac 3-MST expression
2.6.1. Cardiomyocyte isolation
Cardiomyocytes from WT mice were isolated as described [30]. Freshly isolated cells were plated in laminin coated plates for 15 min and treated with mitotracker (Thermo scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Subsequently were fixed with 4% PFA for 15 min at RT.
2.6.2. Immunostaining
Slides were incubated in a blocking buffer consisting of Triton X-100 (0.3% v/v) and BSA (0.5% w/v) in PBS for 2 h at room temperature (RT). Samples were washed with PBS, prior to the addition of primary antibodies and overnight incubation at 4 °C. Subsequently, samples were incubated with anti-CD31 (rat, BD Pharmingen, Franklin Lakes, NJ USA), anti-3MST (rabbit, ATLAS, Bromma, Sweden) and anti-CDH2 (mouse, Santacruz Biotechnology, Dallas, TX) in TritonX-100 (0.2% v/v in PBS). Thereafter, anti-rabbit, anti-rat and anti-mouse secondary antibodies (1:200 in PBS) supplemented with DAPI (10 ng/ml), were added. For the isolated cardiomyocytes Phalloidin (Alexa Fluo, Thermo Fisher Scientific, Waltham, MA, USA) and 3-MST were used. After washing, samples were mounted with Dako fluorescent mounting medium (Dako, Glostrup, Denmark). Images were taken using a confocal microscope (LSM-780; Zeiss, Jena, Germany) and ZEN software (Zeiss).
2.7. mRNA expression
2.7.1. Droplet Digital PCR (ddPCR)
ddPCR was performed as previously described [31]. In brief, absolute quantification of target cDNA was performed with the QX200 Droplet Digital PCR system (Bio–Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s instructions. cDNA was added to the ddPCR reaction setup together with 1xQX200 ddPCR EvaGreen supermix (Bio-Rad, 186–4034), 250 nM reverse and forward primers and nuclease-free water. The forward (F) and reverse (R) primers used were CSE: F GCACCAACAGGTACTTCAGGA and R AACGAAGCCGACCACT TGT; CBS: F TGCGGAACTACATGTCCAAG and R TTGCAGACTTCGTC TGATG; 3-MST: F CTTTTCCGAGCTCTGGTGTC and R TAGGCAGCATG TGGTCGTAG and for 18S rRNA: F CTTTGGTCGCTCGCTCCTC and R CTGACCGGGTTGGTTTTGAT. The ddPCR reaction mixture (20 μl) and 70 μl Droplet Generation Oil for EvaGreen (Bio–Rad, 1864005) were used for droplet generation in a QX200 Droplet Generator (Bio–Rad). Droplets were carefully transferred to a 96-Well plate and sealed with the PX1 PCR Plate Sealer (Bio-Rad). The PCR reaction was carried out in a C1000 Touch Thermal Cycler (Bio-Rad) (95 °C for 5 min, 40 cycles of 95 °C (30 s), 55 °C (20 s) and 72 °C (30 s) and the signals were measured with the QX200 Droplet Reader (Bio-Rad) using the QuantaSoft Reading software in the absolute quantification mode (Bio-Rad, Version 1.74). Analysis was performed with QuantaSoft AnalysisPro software (Bio-Rad) and all data shown were calculated above the threshold. All experiments included a no template control.
2.8. 2. Real time PCR
TST mRNA expression was measured by real time PCR. mRNA was reversed transcribed to cDNA according to the instructions of the TaKaRa cDNA synthesis kit (RR037A, TaKaRa, Kusatsou, Japan). 15 ng of cDNA were used per PCR reaction in a 2-step real time PCR (T annealing = 60 °C) where SYBR Green was used as the detection probe. The mRNA expression levels of TST, was measured by using β-actin as housekeeping gene and were quantified by the 2−ΔCt method. The primers used for TST are as follows: FCCTGCTGTAGGTTCACCTTTTAGG, RGGAGGCACCAAG-AGCAATTCTAA; and for β-actin: FTGGAAGGTGGA CAGTGAGGC and RCCCAGGCATTGCTGACAGG.
2.9. Myocardial and serum levels of H2S, sulfane sulfur and polysulfides
H2S and sulfane sulphur in heart and serum was measured by gas chromatography (GC) as previously described [32]). The Agilent 7890 GC gas chromatography system and G660XA Series chemiluminescence detector were used. Free H2S in fresh blood and tissue was liberated by incubating in 1 M sodium citrate buffer (pH 6.0) at 37 °C for 10 min. Sulfane sulfur was released by incubating 100 μl of sample in an equal volume of 15 mM DTT for 50 min, followed by the addition of 400 μl of 1 M sodium citrate at 37 °C. The resultant headspace gases were analyzed using the GC system. Cardiac polysulfides were measured by using the SSP4 (Dojingo Molecular Technologies, Shanghai, China) probe followed by LC-MS/MS detection as described before [33]. In addition, H2S levels in serum and heart were measured by the methylene blue assay as previously described [34], with some slight modifications. In brief, 25 μl of serum was mixed with 75 μl Potassium Phosphate Buffer (PPB, 100 mM, pH 7.4), followed by the addition of 100 μl zinc acetate 1% w/v solution and 20 μl 50% TCA. The mixture was then centrifuged at 12,000g, 5 min, 4 °C and the supernatant was then mixed with 49 μl DMPD [N,N-dimethyl-p-phenylenediamine-sulfate] solution (20 mM in 7.2 N HCl), followed by the addition of 49 μl FeCl3 (30 mM, in 1.2 N HCl). After incubation at RT, in dark for 15 min, 100 μl/well were transferred in a transparent clear bottom 96-well plate and the absorbance was measured at 680 nm in a TECAN infinite 200 PRO reader (Tecan, Switzerland). The concentration of H2S was estimated by a parallel run of a Na2S standard curve (range 1.56–100 μM). The protocol for the cardiac tissue was similar to the one followed for serum with the exception of the following modifications: a) about 30 mg of cardiac tissue lyophilized with mortar and pestle were dissolved in 150 μl PPB buffer, followed by subsequent centrifugation at 12,000g, 5 min, 4 °C. The supernatant was then mixed with substrates of H2S producing enzymes (8 μM pyridoxal-5′-phosphate [PLP], 0,4 mM L-cysteine, 0,08 mM Homocysteine and 8 μM 3-mercaptopyruvate [3-MP]) and incubated at 37 °C for 30 min. b) the reaction was terminated by adding 15 μl zinc acetate 1% w/v solution and 15 μl 50% TCA, c) at the end an equal volume of tissue homogenate was mixed with an equal volume of DMPD (20 mM in 7.2 N HCl) and an equal volume of FeCl3 (30 mM, in 1.2 N HCl).
2.10. Western blotting
Cardiac tissue was lyophilized with mortar and pestle and then homogenized in lysis Buffer (50 mM Tris-HCl pH 7.4, 1% v/v NP-40, 0.5% w/v Na-deoxycholate, 0.1% w/v SDS, 150 mM NaCl, 2 mM EDTA) supplemented with 1% v/v protease and phosphatase inhibitor cocktail (Calbiochem-Novagen; Houston, TX). Protein amount was quantified using the Bradford assay and concentration was normalized before western blot analysis. Samples were separated on 10% w/v SDS–PAGE and transferred to a nitrocellulose membrane (Macherey-Nagel; Düren, Germany). The membranes were blocked and probed with the following antibodies at the dilution as indicated: anti-CBS (1:1000, ProteinTech, Rosemont, IL, USA), anti-CSE (1:1000, Abcam, Cambridge, UK), anti-3-MST (1:500, Atlas Antibodies, Bromma, Sweden), anti-eNOS (1:1000, Cell Signaling Technologies, 9572S, Danvers, MA, USA), anti-PKG-I (1:1000, Cell Signaling Technologies, 3248S, Danvers, MA, USA), anti-sGCα1 (1:1000, Cayman chemicals, 160895, Ann Arbor, MI, USA), anti-sGCβ1 (1:1000, Cayman chemicals, 160897, Ann Arbor, MI, USA), anti-GAPDH (1:3000, Cell Signaling Technologies, 2118S, Danvers, MA, USA). Immunoblots were next processed with anti-Rabbit secondary antibody (1:4000 Millipore, Burlington, MA, USA) and visualized using the Western HRP substrate (Millipore, Burlington, MA, USA). Quantification of western blots was performed using ImageJ software (NIH Image, National Institutes of Health, USA) and ratios of the respective protein band/GAPDH were used for statistical analysis.
2.11. Measurement of serum Troponin-I
Serum was obtained from mice after 30 min of Left Coronary Artery Ligation (ischemia) and 24 h of reperfusion (re-oxygenation) to measure the cardiac-specific isoform of troponin-I using a mouse-specific ELISA kit (Life Diagnostics Inc, West Chester, PA, USA).
2.12. Measurement of myocardial hydrogen peroxide
Cytoplasmic levels of NADPH-driven H2O2 production in the cytoplasm were measured by Ampliflu RedTM fluorescence as previously described with some slight modifications [35]. In particular, whole hearts from WT and 3-MST−/− were homogenized with mortar and pestle in PBS (0.5 g/ml), centrifuged (2000g, 20 min, 4 °C) and then the supernatant was collected and snap frozen for further analysis. In all cases, appropriate blank samples without tissue, substrate or the fluorescent probe were used for validating the reliability of the method. The final RFU signal was normalized according to the protein concentration, which was on average 0.53 mg/well by using the Bradford method. The fluorescence was measured in TECAN infinite 200 PRO reader under the following settings Ex = 535 nm, Em = 595 nm, Gain = 70.
2.13. Measurement of myocardial superoxide
Myocardial superoxide levels were used by using the MitoSOX probe (Abcam, Cambridge, UK) in hearts, homogenized in mitochondria isolation medium-MIM (250 mM Sucrose, 10 mM HEPES, 1 mM EGTA, 1 g/L BSA pH: 7.4) in the presence of protease and phosphatase inhibitors (1:1000 dilution). The homogenate was subjected to a number of serial centrifugations as follows: 2000 g, 10 min, 4 °C (supernatant collected), 10,000 g, 10 min, 4 °C (pellet collected and resuspended in 50 μl MIM), 7000 g, 5 min, 4 °C (pellet collected and resuspended in 300 μl PBS). At the end 80 μl homogenate/well were added in combination with 20 μl diluted (5 μl of the dissolved in DMSO MitoSOX probe further diluted into 2 ml PBS). The fluorescence was measured in a TECAN infinite 200 PRO reader under the following settings: Ex = 540 nm, Em = 590 nm, Gain = 90.
2.14. Measurement of myocardial nitric oxide
Levels of myocardial nitric oxide (NO) was measured by DAF-FM DA fluorescence as previously reported for the liver [26] with slight modifications for the cardiac tissue used in the present study. In particular, 143x w/v parts of the cardiac tissue were homogenized in 250 mM sucrose/10 mM TRIS-buffer pH 7.4 supplemented with protease and phosphatase inhibitors (1:1000 dilution). The supernatant was collected, snap frozen and stored in −80 °C until the day of analysis. On the day of the experiment, the fluorescence was measured in a TECAN infinite 200 PRO reader under the following settings: Ex = 480 nm, Em = 512 nm, Gain = 90.
2.15. Measurement of myocardial RONS (DHE staining)
DHE is a cell permeable probe that reacts with superoxide and NO derivatives to form ethidium bromide, which intercalates in DNA and exhibits a bright red color detectable by fluorescence microscopy. Hearts from wild type and 3-MST−/− mice were incubated with dihydroethidium (DHE, 10 μmol/L) for 20 min in the dark, before being snap frozen in liquid nitrogen. LC-MS/MS of DHE derivatives was performed on a 1290 Infinity UHPLC system (Agilent, Waldbronn, Germany) coupled to a 5500 QTrap triple quadrupole mass spectrometer with a TurboV electro spray ionization source (Sciex Deutschland GmbH, Darmstadt, Germany). Detection of DHE oxidation products (superoxide and ethidium) was achieved by multiple reaction monitoring (MRM) in positive ion mode. The method allowed the detection of DHE degradation product ethidium and the superoxide-specific product. System control and analytical data analysis were processed by Analyst software 1.6.2 and MultiQuant 3.0, respectively.
2.16. Measurement of cellular antioxidants (NADPH and GSH)
The measurement was performed as previously described [36]. Powdered tissues (~20 mg) were lysed with the RIPA buffer. NADPH/NADP+ and GSH/GSSG ratios were measured using commercially available kits (ab176724 and ab138881, Abcam, Cambridge, UK) according to the manufacturer’s instructions. Tissue weight was used for normalization.
2.17. Measurement of vascular reactivity
Vascular reactivity and responses to phenylephrine, acetylcholine and the NO donor DEA-NONOate were measured in aortic rings as previously described [37]. Animals were sacrificed with CO2 and thoracic aortas were rapidly harvested, dissected, and cleaned of adherent connective and fat tissue. Rings of about 1 mm length were cut and placed in organ baths (3 ml) filled with oxygenated (95% O2 −5% CO2) Krebs solution maintained at 37 °C. The rings were connected to an isometric transducer (type 7006, Ugo Basile, Comerio, Italy) and changes in tension were recorded continuously with a computerized system (Data Capsule 17400, Ugo Basile, Comerio, Italy). The rings were stretched until a resting tension of 1.5 g was reached and allowed to equilibrate for at least 45 min, during which time tension was adjusted, as necessary, to 1.5 g and bathing solution was periodically changed. In each experiment, rings were first challenged with PE (1 μM) until the responses were reproducible. The contractile response of the aortic rings to PE was first measured (10−9–10−5.5M). In separate series of experiments, the rings were contracted with PE (1 μM) and, once a plateau was reached, a cumulative concentration-response curve of the following drugs was performed: Acetylcholine (10−8–10−4.5M), DEA-NONOate (10−8–10−4.5M)
2.18. Statistical analysis
Data are presented as means ± S.E.M. Differences were analyzed using two-tailed unpaired Student’s t-test for comparisons between two-groups. For comparisons between three or more groups ANOVA analysis was performed followed by an appropriate post-hoc test. All statistical calculations were made using Graphpad Prism statistical software (8.0b, La Jolla, CA, USA). Sample sizes are reported in all figure captions. P-value was considered significant when it was less than 0.05.
3. Results
3.1. Expression of 3-MST in the heart
Initially, we sought to determine the RNA copies of 3-MST in the heart compared to that of CSE and CBS. More steady state 3-MST than CSE or CBS mRNA transcript levels were detected in the mouse heart. Genetic ablation of the 3-MST gene depleted 3-MST mRNA without inducing any compensatory increase in CSE or CBS (Fig. 1A). However, the deletion of 3-MST did result in a significant decrease in the expression of thiosulfate sulfurtransferase TST, an enzyme capable of generating polysulfides from thiosulfate in the mitochondria [38] (Fig. 1B). To examine the cellular and subcellular localization of 3-MST in the heart, we performed immunofluorescence staining of heart tissue and isolated cardiomyocytes. 3-MST was abundantly expressed in the mouse heart in various cell types including cardiomyocytes and endothelial cells (Fig. 1C). In isolated cardiomyocytes, 3-MST localized to both the cytosol and the mitochondria (Fig. 1D).
Fig. 1.
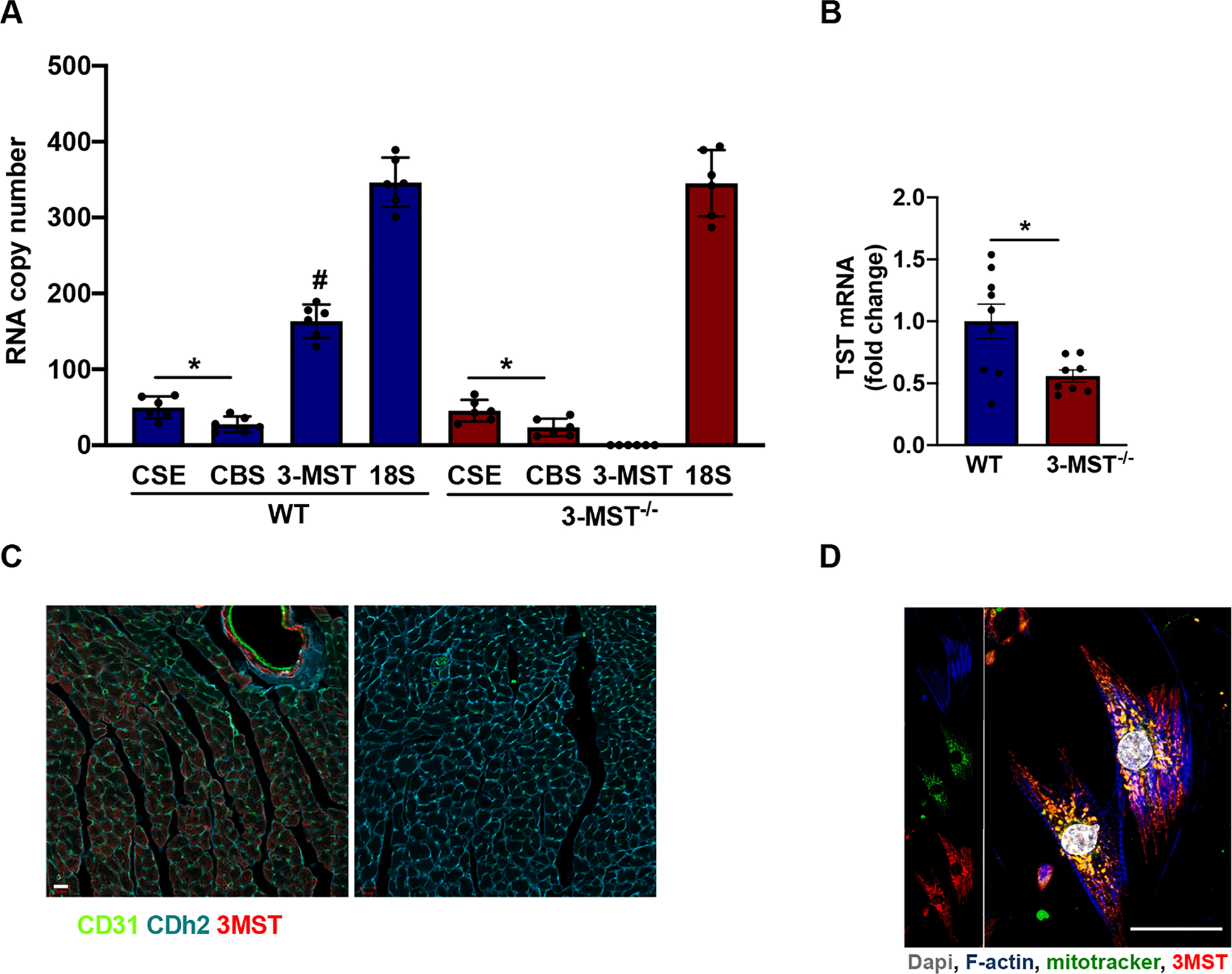
Expression of 3-MST in the heart. mRNA copy number of H2S producing enzymes (CSE, CBS, 3-MST) compared to the copy numbers of 18S rRNA in the heart of WT and 3-MST−/− mice (A). Steady state mRNA levels of TST in the heart of WT and 3-MST−/− mice (B). Immunostaining of CD31 (green, endothelial cell marker), Cdh2 (blue, cardiomyocyte marker) and 3-MST (red) in WT heart cross-sections. Comparable results were obtained in 5 additional animals, bar = 20 μm (C). Subcellular localization of 3-MST in cardiomyocytes. Representative immunostaining of primary isolated murine cardiomyocytes from WT hearts. F-actin (blue), Dapi (grey), mitotracker (green) and 3-MST (red). The co-localization of 3-MST and mitotracker signals was approximately 60% of the total, bar = 25 μm. The panel on the right is a negative control (no primary Ab) for the 3-MST staining (D). Values are shown as mean ± SEM, n = 6/group (A), n = 8–9/group (B) *p < 0.05 vs indicated group, #p < 0.05 vs CSE and CBS mRNA copies in WT.
3.2. No detectable changes in sulfur pools in 3-MST−/− mice
To assess the levels of H2S and related species in 3-MST−/− mice, we utilized a number of methods that measure distinct pools in the serum and heart tissue. Namely, we employed the widely used methylene blue assay, gas chromatography to determine H2S and sulfane sulfur respectively and SSP4-based fluorescence to detect hydrogen polysulfides. Interestingly, myocardial free or bound H2S (sulfane sulfur, hydrogen persulfides and polysulfides) was not reduced in the cardiac tissue of the 3-MST−/− mice compared to WT (Fig. 2A–D). Similarly, no difference between the two genotypes was observed in serum of free H2S and sulfane sulfur (Fig. 2E–G). In line with what we observed at the mRNA level, CSE and CBS protein expression in the heart was similar in WT and 3-MST−/− mice (Fig. 3 E–F, I).
Fig. 2.

No changes in sulfur pools in 3-MST−/− mice. Myocardial H2S measured by methylene blue (A), H2Sn measured by SSP4 (B), sulfane sulfur (C) and free H2S measured by gas chromatography (D) in WT and 3-MST−/− mice. Serum H2S measured by methylene blue (E), sulfane sulfur (F) and free H2S measured by gas chromatography (G) in WT and 3-MST−/− mice. Values are shown as mean ± SEM, n = 6–9/group (A, B, E), n = 15/group (C, D, F, G).
Fig. 3.

Expression levels of NO/cGMP pathway and CSE, CBS, 3-MST proteins in the heart. Quantification of protein levels of eNOS (A), PKG-I (B), sGCα1 (C), sGCβ1 (D), CSE (E), CBS (F) and 3-MST (H). Representative blots for each indicated protein (I). Values are shown as mean ± SEM, n = 8/group (A-H). *p < 0.05 vs indicated group.
3.3. 3-MST−/− mice have normal LV structure and function
Left ventricular structure and function in young 3-MST−/− mice was similar to that of WT animals as indicated by echocardiography (Table 1). It is interesting to note that although the ejection fraction (EF) and fraction shortening (FS) were not statistically different between WT and 3-MST−/− there was a trend towards reduced values in the 3-MST−/− mice (p-value = 0.09). This trend could potentially reflect a diminishing cardiac function that would progress into heart failure under stress conditions or with advances in age.
Table 1.
Cardiac function parameters analyzed by echocardiography in 10–12 weeks old WT and 3-MST−/− mice.
| WT (n = 12) | 3-MST−/− (n = 11) | p-value | |
|---|---|---|---|
| HR | 618.27 ± 25.74 | 631.09 ± 40.87 | 0.37 |
| ISVd (mm) | 0.79 ± 0.04 | 0.79 ± 0.04 | 0.97 |
| ISVs (mm) | 1.30 ± 0.05 | 1.27 ± 0.06 | 0.32 |
| EDD (mm) | 3.13 ± 0.13 | 3.12 ± 0.17 | 0.90 |
| ESD (mm) | 1.75 ± 0.10 | 1.79 ± 0.14 | 0.40 |
| PWd (mm) | 0.79 ± 0.04 | 0.79 ± 0.04 | 0.97 |
| PWs (mm) | 1.30 ± 0.05 | 1.27 ± 0.06 | 0.32 |
| FS (%) | 44.10 ± 1.79 | 42.61 ± 2.32 | 0.10 |
| EF (%) | 82.48 ± 1.64 | 81.02 ± 2.31 | 0.09 |
| h/r | 1.99 ± 0.16 | 1.98 ± 0.18 | 0.93 |
The full name of the measured parameters is given in the abbreviation list. Values are shown as mean ± SEM
3.4. The NO/cGMP pathway is not significantly altered in 3-MST−/− mice
Given the interplay between H2S and NO, we determined the expression of key components of the NO/cGMP pathway in the hearts of 3-MST−/− mice. Although there was a trend for increased endothelial NO synthase (eNOS) levels in 3-MST−/− mice (Fig. 3A), this did not reach statistical significance (p = 0.1). Also, no differences in the expression of the α1 and β1 subunits of soluble guanylyl cyclase (sGC) were detected (Fig. 3C–D). Interestingly, levels of cGMP-dependent protein kinase I (PKG-I) were elevated in 3-MST−/− mice (Fig. 3B). In addition, we measured the levels of cardiac NO by using the fluorescent probe DAF-FM DA; as shown in Fig. 9E, no difference in DAF-FM DA fluorescence was evident between young 3-MST−/− vs WT mice.
Fig. 9.

Hypertension and cardiac hypertrophy in aged 3-MST−/− mice. Systolic (A), Diastolic (B), Mean (C) Arterial Pressure and heart weight (normalized to the tibia length) (D) in aged (18 months old) WT and 3-MST−/− mice. Myocardial NO production measured by DAF-FM DA fluoroscence and presented as RFU/mg protein (E). Values are shown as mean ± SEM, n = 5–6/group. *p < 0.05 vs indicated group.
3.5. 3-MST−/− mice have normal blood pressure, heart weight and vascular reactivity
To further explore the cardiovascular phenotype of the 3-MST−/− mice under basal conditions, we measured the blood pressure and vascular reactivity in these mice. 3-MST−/− mice had systolic, diastolic and mean blood pressure that was similar to that of WT mice (Fig. 4A–C). In agreement with the echocardiography data, no change in cardiac weight was observed in 3-MST−/− mice (Fig. 4D). Moreover, both the phenylephrine (PE)-induced contraction (Fig. 5A) and acetylcholine (ACh)-induced relaxation (Fig. 5B) of isolated aortic rings, as well as responses to the NO donor DEA-NONOate (Fig. 5C) were comparable in vessels from WT and 3-MST−/− mice.
Fig. 4.
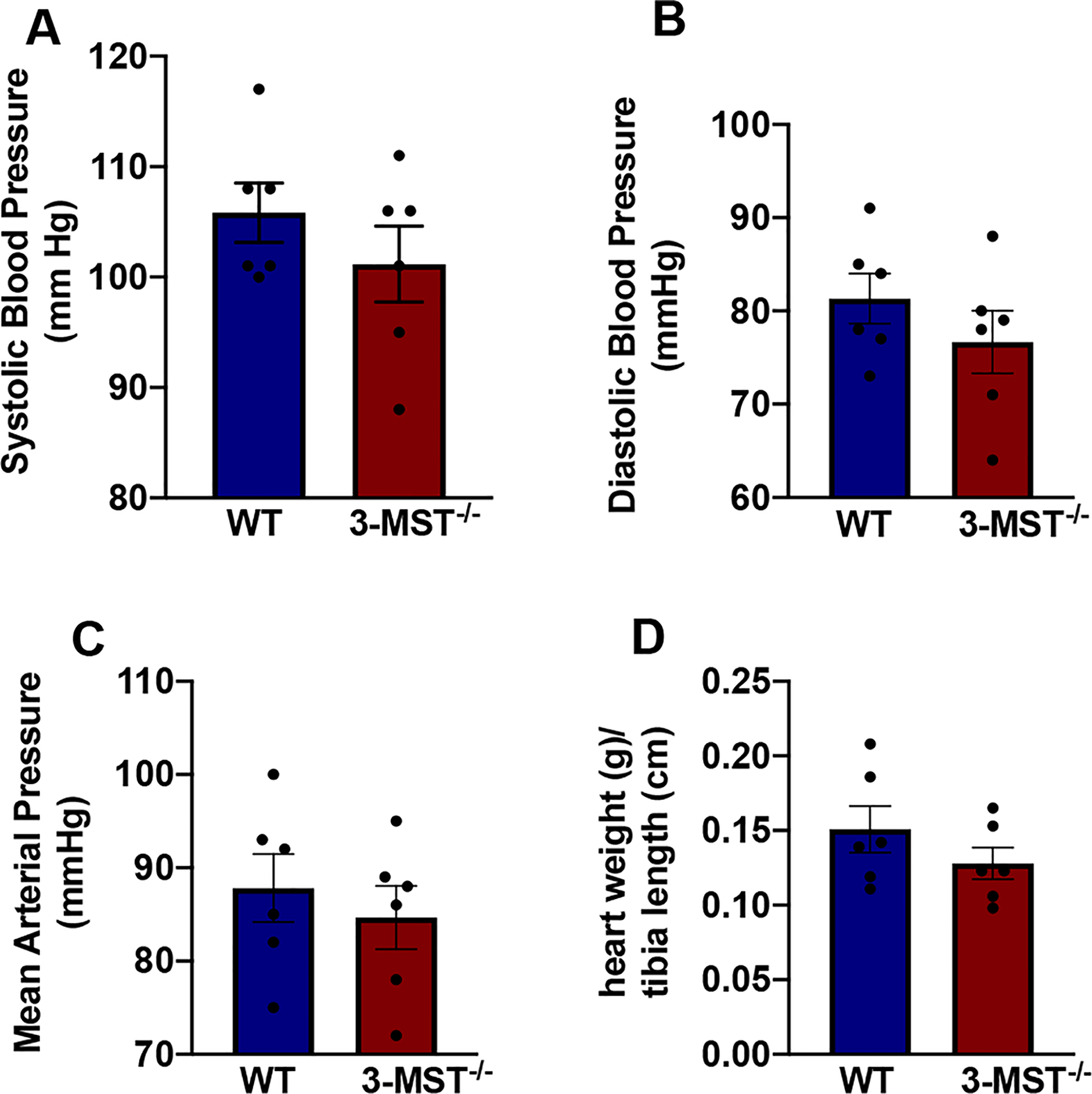
3-MST−/− mice have normal blood pressure. Systolic (A), Diastolic (B), Mean (C) Arterial Pressure and heart weight (normalized to the tibia length) (D) in WT and 3-MST−/− mice. Values are shown as mean ± SEM, n = 6/group.
Fig. 5.

No changes in vascular reactivity in 3-MST−/− mice. Vascular reactivity of aortic rings from WT and 3-MST−/− mice (A-C). Contractile responses to PE (A), dilatory responses to Ach (B) and the NO donor DEA-NONOate (C) in PE-precontracted vessels. Values are shown as mean ± SEM, n = 5–8/group.
3.6. Genetic ablation or pharmacological inhibition of 3-MST protects against myocardial IRI
To determine the consequences of 3-MST deletion under conditions of stress, we used a myocardial ischemia reperfusion injury model that involved a 30 min occlusion of the left anterior descending coronary artery followed by 2 h of re-perfusion. 3-MST−/− mice exhibited a significantly reduced infarct area compared to WT mice exposed to the same ischemic stimulus (Fig. 6A). The areas at risk (%) were similar between the two genotypes (Fig. 6B). In line with the data from the genetic model, administration of I 3MT-3, a small molecule inhibitor of 3-MST [39], protected the heart from ischemia reperfusion injury (Fig. 6C). The areas at risk (%) were similar between the vehicle and the I3MT-3 groups (Fig. 6D). Since the favorable outcome of 3-MST deletion was unexpected the experiments were repeated in an independent laboratory, using a different mouse colony. In this set of experiments, the left anterior descending coronary artery was ligated for 30 min but reperfusion was allowed to continue for 24 h. Under these conditions, infarct size (Fig. 7 A) and plasma levels of troponin-I levels were lower in 3-MST−/− mice than in wild-type mice 24 h post reperfusion (Fig. 7C). There were no differences in the area at risk between the two genotypes (Fig. 7 B).
Fig. 6.
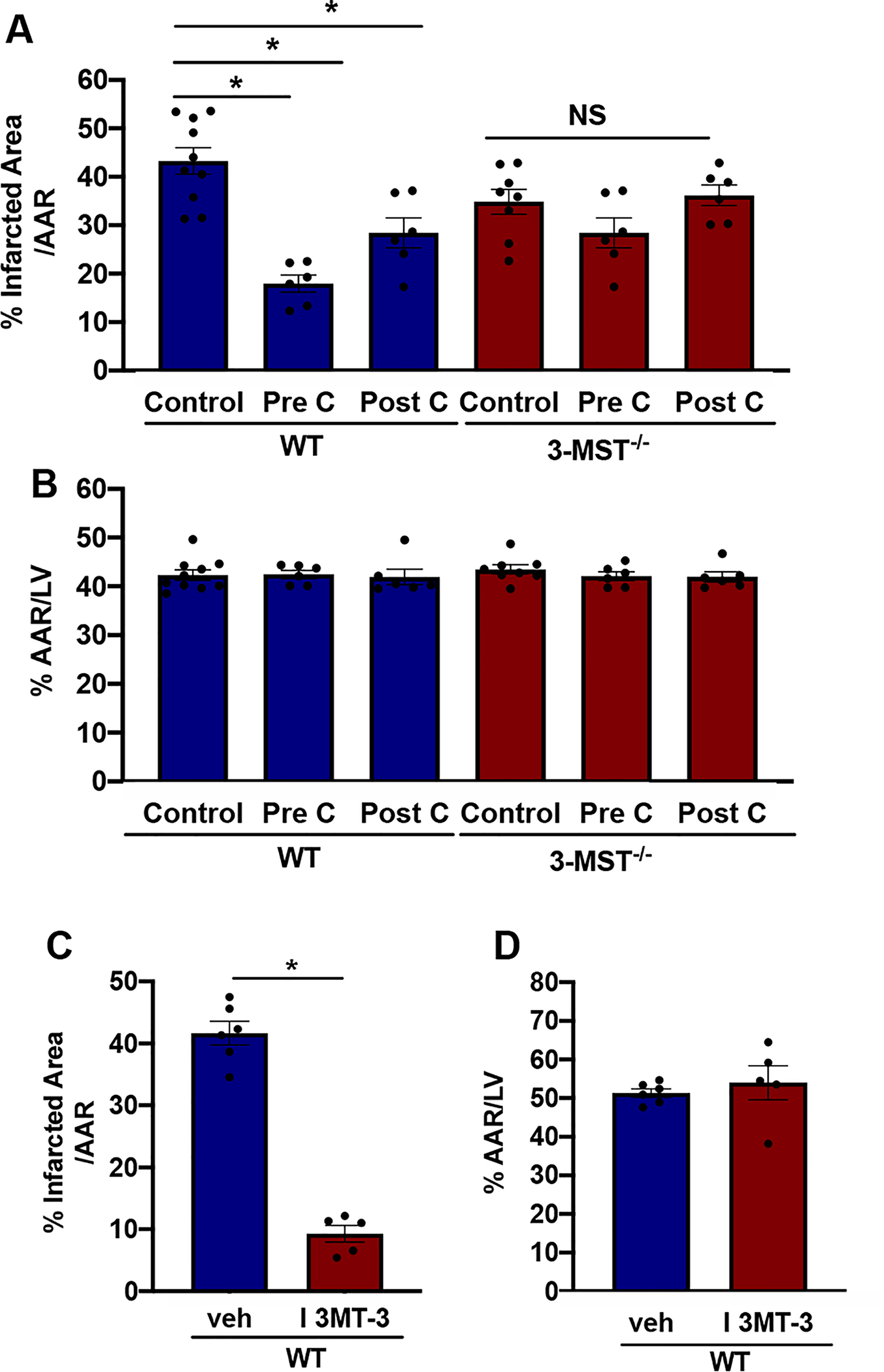
Genetic or pharmacological inhibition of 3-MST reduces ischemia/reperfusion injury. % Infarcted Area (A) and % Area At Risk (AAR) (B) measured as described in the Methods in WT and 3-MST−/− mice subjected to 30 min ischemia followed by 2 h reperfusion under the following conditions: Control, PreC (ischemic pre-conditioning), PostC (ischemic post-conditioning). % Infarcted Area (C) and % Area At Risk (D) in WT mice treated with either vehicle (veh) or the pharmacological inhibitor of 3-MST, I 3MT-3. Values are shown as mean ± SEM, n = 8–10/group (Control), n = 5–6/group (WT (veh), WT (I 3MT-3), PreC and PostC). *p < 0.05 vs indicated group.
Fig. 7.

Reduced infarct size and troponin levels in 3-MST−/− mice. % Infarcted Area (A) and % Area At Risk (AAR) (B) in WT and 3-MST−/− subjected to 30 min ischemia followed by 24 h reperfusion (A). Serum levels of Troponin-I in WT and 3-MST−/− subjected to 30 min ischemia followed by 24 h reperfusion (C). Values are shown as mean ± SEM, n = 9/group (A-B), n = 9–13/group (C). *p < 0.05 vs indicated group.
3.7. Loss of conditioning, higher cardiac ROS levels and lower cellular antioxidants in 3-MST−/−
One mechanism implicated in ischemic preconditioning is the generation of low levels of reactive oxygen species that activate signaling pathways to promote cardiac survival following a longer ischemic insult, as well as during the early reperfusion phase [40]. To evaluate the effect of 3-MST on cellular redox state we measured the levels of cardiac RONS and antioxidants using dihydroethidium (DHE), Amplex Red fluorescence (detection of H2O2), MitoSOX fluorescence (detection of mitochondrial superoxide), as well as NADPH/NADP+ and GSH/GSSG ratios. 3-MST−/− mice had significantly higher levels of cardiac RONS (Fig. 8A) and H2O2 (Fig. 8B) as well as a clear trend towards elevated cardiac superoxide anion generation (Fig. 8C). At the same time, hearts from 3-MST−/− mice contained significantly lower levels of the cellular antioxidants NADPH (Fig. 8D) and the reduced form of glutathione (Fig. 8 E).
Fig. 8.

Increased ROS production in the hearts of 3-MST−/−. Cardiac RONS levels as estimated by DHE staining (A). Quantification of the differences in DHE fluorescence between WT and 3-MST−/− (Panel A Left). Representative images of DHE staining in cardiac sections from WT and 3-MST−/− mice, bar = 200 μm (Panel A, Right Top). Heat map of the DHE products in samples as in B (Panel A, Right Bottom). Cardiac H2O2 levels measured by Amplex Red Fluorescence in the heart homogenates of WT and 3-MST−/− mice (B). Cardiac superoxide (O2−) levels as measured by MitoSOX Fluorescence in the mitochondrial fraction of hearts from WT and 3-MST−/− mice (C). NADPH/NADP+ (D) and GSSH/GSSG (E) ratios in the heart of WT and 3-MST−/−. Values are shown as mean ± SEM, n = 5–6/group. *p < 0.05 vs indicated group.
3.8. Aged 3-MST−/− mice exhibit elevated blood pressure, cardiac weight and lower cardiac NO levels.
To evaluate the effect of aging on cardiovascular function in a 3-MST null background, we measured blood pressure and cardiac weight in 18 months old mice. The aged 3-MST−/− mice were hypertensive, characterized by a significantly higher systolic, diastolic and mean arterial pressure (Fig. 9A–C) and cardiac hypertrophy, as indicated by the significantly increased heart weight/tibia length ratio (Fig. 9D). Aged 3-MST−/− mice also had lower cardiac NO levels compared to WT littermates (Fig. 9E).
4. Discussion
The major findings of our study are that 1) 3-MST is highly expressed in the heart and it is present both in the cytosol and the mitochondria, 2) lack of 3-MST does not lead to any change in the free H2S, sulfane sulfur or polysulfides levels in the heart, 3) lack of 3-MST does not alter vascular reactivity, blood pressure and LV structure and function in young adult animals, 4) genetic ablation of 3-MST leads to an increase in cellular ROS and a reduction in anti-oxidant molecules in the heart, 5) genetic ablation of pharmacological inhibition of 3-MST increases cardioprotection after myocardial IRI in young mice and 6) loss of 3-MST results in hypertension and cardiac hypertrophy in aged mice.
There have been a number of reports demonstrating expression of 3-MST in aortic and mesenteric artery homogenates, as well as in the heart [22,24]. To determine the relative expression of 3-MST compared to the other two enzymes that produce H2S, we measured steady state mRNA transcripts. 3-MST was more abundantly expressed compared to CSE and CBS in the mouse heart. Moreover, 3-MST was detected in cardiomyocytes and endothelial cells of large vessels but not in cardiac capillaries. This observation confirms findings from cell cultures of the above mentioned cell types and is in agreement with the fact that the 3-MST promoter has features of a typical housekeeping gene [41]. 3-MST has been found in both the cytosolic fraction and the mitochondria of astrocytes and hepatocytes [5]. In the present study, we confirmed that 3-MST is also equally distributed among the cytosol and the mitochondria of cardiomyocytes.
3-MST is capable of producing cysteine- and glutathione-persulfides (Cys-SSH and GSSH), hydrogen persulfides and polysulfides, along with H2S [18,42]. To evaluate the impact 3-MST deletion on different H2S pools in the heart, a combination of analytical assays that detect distinct sulfur species was used. H2S levels measured by the classic methylene blue assay [24], H2Sn polysulfide species measured by the fluorescence probe SSP4 [33] and free H2S and sulfane sulfur measured by gas chromatography [23], all failed to detect a difference between WT and 3-MST−/− mice. Similarly, no changes in H2S pools were observed in the serum of 3-MST−/− animals. This is in contrast with reports showing that 3-MST−/− mice have lower brain levels of H2S and H2S3 than WT mice [42]. As we observed no upregulation of CSE or CBS, the overproduction of sulfur species by these enzymes is unlikely to compensate for the absence of 3-MST. It should be noted that Nagahara et al. [38] reported an upregulation of TST in the liver of 3-MST KO. However, we noted a reduction in TST expression in the heart of 3-MST KO mice, indicating differential regulation of 3-MST expression among tissues. What could then explain the unaltered levels of H2S and polysulfides in 3-MST−/−? One possibility is that enzymes responsible for H2S degradation are downregulated in 3-MST−/−, or that 3-MST activity is low under basal conditions producing minimal amounts of H2S in the heart. Alternatively, 3-MST might affect a pool of H2S that is inaccessible to the analytical approaches used. Resolving this apparent paradox will require additional experimentation. It should be kept in mind that using the same techniques, we and others have shown marked reductions in H2S pools in CSE KO mice [5,30–31].
Extensive cross-talk has been shown to occur between the H2S and NO pathways [43–44]. H2S-induced vasorelaxation, angiogenesis and cardioprotection are all dependent on NO [32,45]. H2S has also been reported to regulate eNOS expression and increase NO bioavailability [43]. Moreover, 3-MST was found to enhance cGMP/PKG signaling in endothelial cells [46]. We, thus, evaluated the effect of lack of 3-MST on the NO/cGMP pathway in the heart. With the exception of PKG-I upregulation, we observed no significant biochemical (expression of eNOS/sGC), or functional (changes in vasorelaxation to ACh and a NO donor, or blood pressure) consequences of 3-MST deletion.
Since, baseline cardiovascular characteristics were similar between WT and 3-MST−/− mice, we asked whether responses of the two strains might differ under stressful, pathophysiological conditions. Given that a large body of evidence indicates that H2S exhibits cardioprotective actions [2], we subjected mice to ischemia reperfusion injury and assessed cardiac damage by using histochemical and biochemical readouts. In these experiments, we found that lack of 3-MST or inhibition of 3-MST activity is beneficial, as it leads to reduced infarct size. This finding is in contrast to the previously reported protective role of CSE in IRI [28,32]. Interestingly, the protection afforded by the mechanical maneuvers-pre and postconditioning- were abolished in the 3-MST−/− animals. Based on the fact that pre- and post-conditioning protect the heart by promoting a mild elevation of ROS [40] and that H2S/3-MST exert antioxidant effects [1], we evaluated the cellular redox state in mice lacking 3-MST.
H2S has been shown to exert a variety of direct and indirect anti-oxidant effects. For example, it readily reacts with superoxide anions at rates that are higher than classic antioxidants, such as glutathione and cysteine [47]. The physiological significance of the direct scavenging effects has been questioned due to the low cellular concentration of H2S compared to that of GSH or cysteine. In addition to its direct effects, H2S activates Nrf2, a master regulator of antioxidant gene expression by sulfhydrating Keap1 to release the inhibitory effect of Keap1 on Nrf2 [48]. Indeed, 3-MST−/− mice, like CSE−/− [32], show enhanced levels of ROS and reduced amounts of antioxidant molecules. Given that no change in H2S levels was detected in hearts from 3-MST−/− mice, it seems that any changes in redox homeostasis observed are independent of H2S. However, 3-MST has been shown to participate in redox cycling and to contribute to detoxification from cellular oxidants [20]. Also, 3-MST has been proposed to exert antioxidant effects via the production of persulfidated species such as cysteine- and glutathione-persulfide [18]. An alternative mechanism for the anti-oxidant effects of 3-MST would involve changes in the sulfhydration and therefore the activity of key proteins involved in redox balance, such as Mn-superoxide dismutase [49].
Age is a risk factor for cardiovascular disease [50]. To determine if changes occur in the phenotype of 3-MST−/− mice with aging, we measured arterial blood pressure in 18 months old WT and 3-MST−/−. Unlike, what was seen in young animals, both diastolic and systolic blood pressure were increased in 3-MST−/− mice. 3-MST−/− mice also exhibited increased heart weight compared to WT animals and had reduced NO levels in the heart. The observed cardiac hypertrophy could result from hypertension, could also be attributed to biochemical changes in the heart of 3-MST−/− mice, or a combination of the above. Similar to 3-MST−/− mice, animals with targeted disruption of the CSE locus also develop hypertension, albeit at a younger age [13].
In summary, this is the first study that addresses the role of 3-MST in the cardiovascular system. Genetic ablation of 3-MST shifts cellular redox balance toward a pro-oxidant state. However, the resulting oxidative stress does not translate into functional changes in baseline cardiovascular parameters in young mice. Interestingly, the low-level oxidative stress in 3-MST−/− mice could possibly act as an ischemic preconditioning mechanism and be associated with the protection seen after ischemia reperfusion injury in young 3-MST−/− mice. In addition, aged 3-MST−/− mice exhibit hypertension, perhaps owing to exhaustion of anti-oxidant defense mechanisms in the older animals. Future studies in disease models are required to understand the role of 3-MST in cardiovascular pathologies.
5. Funding sources
The research work was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (Project number: HFRI-FM17-886 to A.P.). This work was also supported by the Deutsche Forschungsgemeinschaft (CRC1366/1 B1 Project # 39,404,578 to I.F. and S.-I.B. and the Cardio-Pulmonary Institute, EXC 2026, Project ID: 390649896).
Abbreviations:
- 3-MP
3-Mercaptopyruvate
- 3-MST
3-Mercaptopyruvate Sulfur Transferase
- 3-MST−/−
3-Mercaptopyruvate Sulfur Transferase Knock Out
- Ach
Acetylcholine
- BSA
Bovine Serum Albumin
- CBS
Cystathionine beta synthase
- CD 31
Cluster of Differentiation 31
- CDH2
Cadherin-2
- cGMP
cyclic guanosine monophosphate
- CO
Carbon Monoxide
- CSE
Cystathionine gamma lyase
- DAF-FM DA
4-Amino-5-methylamino- 2′,7′-difluorofluorescein diacetate
- DEA-NONOate
DEA/NO diethylammonium salt
- DHE
Dihydroethidium
- DMPD
N,N-dimethyl-p-phenylenediamine-sulfate
- DMSO
Dimethyl sulfoxide
- DTT
DL-Dithiothreitol
- EDD
End Diastolic Diameter
- EF (%)
Ejection Fraction (%)
- EGTA
Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid
- eNOS
endothelial Nitric Oxide Synthase
- ESD
End Systolic Diameter
- FS (%)
Fraction Shortening (%)
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GC
Gas Chromatography
- GSH
Reduced glutathione
- h/r
thickness/radius ratio of the left ventricle
- H2S
Hydrogen Sulfide
- H2Sn
Hydrogen polysulfides
- H2O2
Hydrogen Peroxide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
High Performance Liquid Chromatography
- HR
Heart Rate
- IVSd and IVSs
Interventricular Septum Thickness at diastole
- LAD
Left Coronary Artery Ligation
- LC-MS/MS
Liquid chromatography-mass spectrometry (double detector MS)
- LV
Left Ventricle
- MIM
Mitochondria Isolation Medium
- MRM
Multiple Reaction Monitoring
- NADPH
Nicotinamide Adenine Dinucleotide Phosphate
- Na2S
sodium sulfide
- NO
Nitric Oxide
- NP-40
Nonidet P-40
- O2−
Superoxide
- PBS
Phosphate-buffered Saline
- PE
Phenylephrine
- PEEP
Positive end-expiratory pressure
- PKG-I
Protein Kinase G-I
- PLP
Pyridoxal phosphate
- PWd
Posterior Wall thickness at diastole
- PWs
Posterior Wall thickness at systole
- RIPA
Radioimmunoprecipitation assay buffer
- ROS
Reactive Oxygen Species
- SDS
Sodium dodecyl sulfate
- sGC
soluble Guanyl Cyclase
- SSP4
Sulfane Sulfur Probe 4
- TCA
Trichloroacetic acid
- TRIS
Trisamine
- TTC
Triphenyl tetrazolium chloride
- WT
Wild Typ
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Murphy B, Bhattacharya R, Mukherjee P, Hydrogen sulfide signaling in mitochondria and disease, FASEB J. (2019) fj201901304R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Li Z, Polhemus DJ, Lefer DJ, Evolution of hydrogen sulfide therapeutics to treat cardiovascular disease, Circ. Res 123 (2018) 590–600. [DOI] [PubMed] [Google Scholar]
- [3].Kanagy NL, Szabo C, Papapetropoulos A, Vascular biology of hydrogen sulfide, Am. J. Physiol. Cell Physiol 312 (2017) C537–C549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bibli SI, Hu J, Sigala F, Wittig I, Heidler J, Zukunft S, Tsilimigras DI, Randriamboavonjy V, Wittig J, Kojonazarov B, Schürmann C, Siragusa M, Siuda D, Luck B, Abdel Malik R, Filis KA, Zografos G, Chen C, Wang DW, Pfeilschifter J, Brandes RP, Szabo C, Papapetropoulos A, Fleming I, Cystathionine γ lyase sulfhydrates the rna binding protein human antigen r to preserve endothelial cell function and delay atherogenesis, Circulation 139 (2019) 101–114. [DOI] [PubMed] [Google Scholar]
- [5].Szabo C, Papapetropoulos A, International union of basic and clinical pharmacology. Cii: Pharmacological modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors, Pharmacol. Rev 69 (2017) 497–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang R, Szabo C, Ichinose F, Ahmed A, Whiteman M, Papapetropoulos A, The role of H2S bioavailability in endothelial dysfunction, Trends Pharmacol. Sci 36 (2015) 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rajpal S, Katikaneni P, Deshotels M, Pardue S, Glawe J, Shen X, Akkus N, Modi K, Bhandari R, Dominic P, Reddy P, Kolluru GK, Kevil CG, Total sulfane sulfur bioavailability reflects ethnic and gender disparities in cardiovascular disease, Redox Biol 5 (2018) 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang J, Minkler P, Grove D, Wang R, Willard B, Dweik R, Hine C, Non-enzymatic hydrogen sulfide production from cysteine in blood is catalyzed by iron and vitamin B, Commun. Biol 2 (2019) 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kabil O, Banerjee R, Enzymology of H2S biogenesis, decay and signaling, Antioxid. Redox Signal 20 (2014) 770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Akaike T, Ida T, Wei FY, Nishida M, Kumagai Y, Alam MM, Ihara H, Sawa T, Matsunaga T, Kasamatsu S, Nishimura A, Morita M, Tomizawa K, Nishimura A, Watanabe S, Inaba K, Shima H, Tanuma N, Jung M, Fujii S, Watanabe Y, Ohmuraya M, Nagy P, Feelisch M, Fukuto JM, Motohashi H, Cysteinyl-trna synthetase governs cysteine polysulfidation and mitochondrial bioenergetics, Nat. Commun 8 (2017) 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shen X, Carlström M, Borniquel S, Jädert C, Kevil CG, Lundberg JO, Microbial regulation of host hydrogen sulfide bioavailability and metabolism, Free Radic. Biol. Med 60 (2013) 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang R, Physiological implications of hydrogen sulfide: a whiff exploration that blossomed, Physiol. Rev 92 (2012) 791–896. [DOI] [PubMed] [Google Scholar]
- [13].Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R, H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gammalyase, Science 322 (2008) 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rose P, Moore PK, Zhu YZ, H2S biosynthesis and catabolism: new insights from molecular studies, Cell. Mol. Life Sci 74 (2017) 1391–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mani S, Li H, Untereiner A, Wu L, Yang G, Austin RC, Dickhout JG, Lhoták Š, Meng QH, Wang R, Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis, Circulation 127 (2013) 2523–2534. [DOI] [PubMed] [Google Scholar]
- [16].Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N, Mice deficient in cystathionine beta-synthase: Animal models for mild and severe homocyst(e)inemia, Proc. Natl. Acad. Sci. U.S.A 92 (1995) 1585–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miles EW, Kraus JP, Cystathionine beta-synthase: Structure, function, regulation, and location of homocystinuria-causing mutations, J. Biol. Chem 279 (2004) 29871–29874. [DOI] [PubMed] [Google Scholar]
- [18].Kimura Y, Koike S, Shibuya N, Lefer D, Ogasawara Y, Kimura H, 3-mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (cys-ssh and gssh) together with signaling molecules H2S2, H2S3 and H2S, Sci. Rep 7 (2017) 10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yadav PK, Yamada K, Chiku T, Koutmos M, Banerjee R, Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase, J. Biol. Chem 288 (2013) 20002–20013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nagahara N, Multiple role of 3-mercaptopyruvate sulfurtransferase: antioxidative function, H2S and polysulfide production and possible SOx production, Br. J. Pharmacol 175 (2018) 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H, Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide, J. Biochem 146 (2009) 623–626. [DOI] [PubMed] [Google Scholar]
- [22].Tomita M, Nagahara N, Ito T, Expression of 3-mercaptopyruvate sulfurtransferase in the mouse, Molecules 21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr., Gojon G Jr., Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ, H₂s protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase, Circulation 127 (2013) 1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bucci M, Vellecco V, Cantalupo A, Brancaleone V, Zhou Z, Evangelista S, Calderone V, Papapetropoulos A, Cirino G, Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ace inhibition, Cardiovasc. Res 102 (2014) 138–147. [DOI] [PubMed] [Google Scholar]
- [25].Nagahara N, Nagano M, Ito T, Shimamura K, Akimoto T, Suzuki H, Antioxidant enzyme, 3-mercaptopyruvate sulfurtransferase-knockout mice exhibit increased anxiety-like behaviors: a model for human mercaptolactate-cysteine disulfiduria, Sci. Rep 3 (2013) 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Peleli M, Zollbrecht C, Montenegro MF, Hezel M, Zhong J, Persson EG, Holmdahl R, Weitzberg E, Lundberg JO, Carlström M, Enhanced xor activity in enos-deficient mice: Effects on the nitrate-nitrite-no pathway and ros homeostasis, Free Radic. Biol. Med 99 (2016) 472–484. [DOI] [PubMed] [Google Scholar]
- [27].Bibli SI, Zhou Z, Zukunft S, Fisslthaler B, Andreadou I, Szabo C, Brouckaert P, Fleming I, Papapetropoulos A, Tyrosine phosphorylation of enos regulates myocardial survival after an ischaemic insult: role of pyk2, Cardiovasc. Res 113 (2017) 926–937. [DOI] [PubMed] [Google Scholar]
- [28].Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ, Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function, Proc. Natl. Acad. Sci. U.S.A 104 (2007) 15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Papathanasiou S, Rickelt S, Soriano ME, Schips TG, Maier HJ, Davos CH, Varela A, Kaklamanis L, Mann DL, Capetanaki Y, Tumor necrosis factor-α confers cardioprotection through ectopic expression of keratins k8 and k18, Nat. Med 21 (2015) 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li D, Wu J, Bai Y, Zhao X, Liu L, Isolation and culture of adult mouse cardiomyocytes for cell signaling and in vitro cardiac hypertrophy, J Vis Exp 21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Leisegang MS, Bibli SI, Günther S, Pflüger-Müller B, Oo JA, Höper C, Seredinski S, Yekelchyk M, Schmitz-Rixen T, Schürmann C, Hu J, Looso M, Sigala F, Boon RA, Fleming I, Brandes RP, Pleiotropic effects of laminar flow and statins depend on the krüppel-like factor-induced lncrna mantis, Eur. Heart J 40 (2019) 2523–2533. [DOI] [PubMed] [Google Scholar]
- [32].King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ, Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent, Proc. Natl. Acad. Sci. U.S.A 111 (2014) 3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bibli SI, Luck B, Zukunft S, Wittig J, Chen W, Xian M, Papapetropoulos A, Hu J, Fleming I, A selective and sensitive method for quantification of endogenous polysulfide production in biological samples, Redox Biol 18 (2018) 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Katsouda A, Szabo C, Papapetropoulos A, Reduced adipose tissue H2S in obesity, Pharmacol. Res 128 (2015) 190–199. [DOI] [PubMed] [Google Scholar]
- [35].Peleli M, Flacker P, Zhuge Z, Gomez C, Wheelock CE, Persson AEG, Carlstrom M, Renal denervation attenuates hypertension and renal dysfunction in a model of cardiovascular and renal disease, which is associated with reduced nadph and xanthine oxidase activity, Redox Biol 13 (2017) 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Siragusa M, Thöle J, Bibli SI, Luck B, Loot AE, de Silva K, Wittig I, Heidler J, Stingl H, Randriamboavonjy V, Kohlstedt K, Brüne B, Weigert A, Fisslthaler B, Fleming I, Nitric oxide maintains endothelial redox homeostasis through pkm2 inhibition, EMBO J 38 (2019) e100938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid P, Giannogonas A, Cantalupo S, Dhayade KP, Karalis R, Wang R, Cirino Feil G, cGMP-dependent protein kinase contributes to hydrogen sulfide-stimulated vasorelaxation, PLoS ONE 7 (2012) e53319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nagahara N, Tanaka M, Tanaka Y, Ito T, Novel characterization of antioxidant enzyme, 3-mercaptopyruvate sulfurtransferase-knockout mice: Overexpression of the evolutionarily-related enzyme rhodanese, Antioxidants (Basel) 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hanaoka K, Sasakura K, Suwanai Y, Toma-Fukai S, Shimamoto K, Takano Y, Shibuya N, Terai T, Komatsu T, Ueno T, Ogasawara Y, Tsuchiya Y, Watanabe Y, Kimura H, Wang C, Uchiyama M, Kojima H, Okabe T, Urano Y, Shimizu T, Nagano T, Discovery and mechanistic characterization of selective inhibitors of H2S-producing enzyme: 3-mercaptopyruvate sulfurtransferase (3MST) targeting active-site cysteine persulfide, Sci. Rep 7 (2017) 40227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tullio F, Angotti C, Perrelli MG, Penna C, Pagliaro P, Redox balance and cardioprotection, Basic Res. Cardiol 108 (2013) 392. [DOI] [PubMed] [Google Scholar]
- [41].Nagahara N, Sreeja VG, Li Q, Shimizu T, Tsuchiya T, Fujii-Kuriyama YA, A point mutation in a silencer module reduces the promoter activity for the human mercaptopyruvate sulfurtransferase, BBA 1680 (2004) 176–184. [DOI] [PubMed] [Google Scholar]
- [42].Kimura Y, Toyofuku Y, Koike S, Shibuya N, Nagahara N, Lefer DJ, Ogasawara Y, Kimura H, Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain, Sci. Rep 5 (2015) 14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Szabo C, Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: mechanisms and implications, Am. J. Physiol. Cell Physiol 312 (2017) C3–C15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bibli SI, Yang G, Zhou Z, Wang R, Topouzis S, Papapetropoulos A, Role of cGMP in hydrogen sulfide signaling, Nitric Oxide 46 (2015) 7–13. [DOI] [PubMed] [Google Scholar]
- [45].Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I, Martin E, Szabo C, Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation, Proc. Natl. Acad. Sci. U.S.A 109 (2012) 9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Coletta C, Módis K, Szczesny B, Brunyánszki A, Oláh G, Rios EC, Yanagi K, Ahmad A, Papapetropoulos A, Szabo C, Regulation of vascular tone, angiogenesis and cellular bioenergetics by the 3-mercaptopyruvate sulfurtransferase/H2S pathway: functional impairment by hyperglycemia and restoration by dl-α-lipoic acid, Mol. Med 21 (2015) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wedmann R, Bertlein S, Macinkovic I, Böltz S, Miljkovic JL, Muñoz LE, Herrmann M, Filipovic M, Working with “H2S”: facts and apparent artifacts, Nitric Oxide 41 (2014) 85–96. [DOI] [PubMed] [Google Scholar]
- [48].Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L, Wang R, Hydrogen sulfide protects against cellular senescence via s-sulfhydration of keap1 and activation of NRF2, Antioxid. Redox Signal 18 (2013) 1906–1919. [DOI] [PubMed] [Google Scholar]
- [49].Zivanovic J, Kouroussis E, Kohl JB, Adhikari B, Bursac B, Schott-Roux S, Petrovic D, Miljkovic JL, Thomas-Lopez D, Jung Y, Miler M, Mitchell S, Milosevic V, Gomes JE, Benha M, Gonzalez-Zorn B, Ivanovic-Burmazovic I, Torregrossa R, Mitchell JR, Whiteman M, Schwarz G, Snyder SH, Paul BD, Carroll KS, Filipovic MR, Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-sulfhydration, Cell Metab 30 (2019) 1152–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dhingra R, Vasan RS, Age as a risk factor, Med. Clin. North Am 96 (2012) 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]