

Abstract
BACKGROUND AND PURPOSE: Pelizaeus-Merzbacher's disease (PMD) is caused by mutations in the proteolipid protein (PLP) gene. Recent studies have shown that an increased PLP dosage, resulting from total duplication of the PLP gene, invariably causes the classic form of PMD. The purpose of this study was to compare the MR findings of PMD attributable to PLP duplication with those of PMD arising from a missense mutation.
METHODS: Seven patients with PMD, three with a PLP missense mutation in either exon 2 or 5 (patients 1–3), and four with PLP duplication (patient 4 having larger PLP duplication than patients 5–7) were clinically classified as having either the classic or connatal form of PMD. Cerebral MR images were obtained to analyze the presence of myelination and T1 and T2 shortening in the deep gray matter. Multiple MR studies were performed in six of the seven patients to analyze longitudinal changes.
RESULTS: Four patients (patients 1–4) were classified as having connatal PMD, whereas the other three (patients 5–7) were classified as having classic PMD. Myelination in the cerebral corticospinal tract, optic radiation, and corpus callosum was observed in three cases of classic PMD with PLP duplication. In patient 4, myelination extended to the internal capsule, corona radiata, and centrum semiovale over a 3-year period. No myelination was observed in three PMD cases with a PLP point mutation. T2 shortening in the deep gray matter was recognized in all patients with PMD.
CONCLUSION: The presence of myelination in the cerebral corticospinal tract with diffuse white matter hypomyelination on MR images could be a marker for PMD with PLP duplication. It is suggested that progression of myelination may be present in connatal PMD with large PLP duplication.
Pelizaeus-Merzbacher's disease (PMD) is a rare X-linked inherited disorder affecting myelination of the central nervous system (1, 2). Pathologically, PMD, in contrast to other leukodystrophies like metachromatic leukodystrophy, adrenoleukodystrophy, and multiple sclerosis, is a dysmyelinating rather than a demyelinating disorder. In demyelinating disorders, myelin is formed, deposited around axons, and then destroyed later. In dysmyelinating disorders such as PMD, normal myelination never occurs.
Based on the time of onset and the clinical severity, PMD has traditionally been divided into four categories: the classic, connatal, transitional, and adult forms (1, 2). The classic and connatal forms are the most common. Classic PMD has its onset during late infancy. Early symptoms include nystagmoid, dancing or trembling eye movements, and delayed motor development followed by involuntary movements and spasticity. The course is usually protracted and it is often misdiagnosed as cerebral palsy. Connatal PMD is a rarer and more severe variant that begins at birth or in early infancy and has a more severe clinical course. Abnormal nystagmoid eye movements, extrapyramidal hyperkinesia, spasticity, optic atrophy, and seizures also occur during the early stage.
The basic defect in PMD patients is a defect in the proteolipid protein (PLP) gene. PLP is the most abundant protein in central nervous system myelin, accounting for approximately 50% of the total myelin protein (3). Traditionally, PLP and an isoform DM20, produced by alternative splicing of exon 3B, have been presumed to play a structural role in compact myelin, but more recent studies have suggested an additional role in glial cell development (4). A wide range of mutations in the PLP gene can result in PMD. The deletion of the 18q 22.3-qter region, which includes the myelin basic–protein gene, can also cause a variable myelin defect (5).
Hodes et al (6) reviewed 21 different missense mutations of the PLP gene, mostly located in exons 3 and 4. Missense mutations impair the cellular transport of PLP proteins to the membrane. The resulting accumulation of PLP in the endoplasmic reticulum finally leads to oligodendrocytic cell death. Mutations that inhibit the transport of both PLP and DM20 are found in patients with connatal PMD, whereas patients with classic PMD carry mutations that impair the transport of PLP only (7). Nonsense mutations, splice-site mutations, and small deletions have also been described (8). Sequential analysis of the entire PLP coding region, however, has failed to reveal mutations in approximately 80% of the patients with PMD.
Recent studies have shown that an increased PLP dosage, resulting from total duplication of the PLP gene, also causes PMD (8, 9). PLP over-expression caused by duplication of the gene also leads to glial cell degeneration. Duplication of the PLP gene has been shown to be a major cause of PMD (8, 9). PMD patients with PLP duplication invariably have the classic form with a relatively mild clinical course (8–10).
MR imaging is a useful method for assessing the dysmyelination of the cerebral white matter in PMD. MR imaging can show a hypomyelination pattern; ie, reversal of the white matter signal intensity on T1- and T2-weighted images (11). Nonetheless, there has been no report of a correlation between the genotype and MR imaging. The purpose of this study was to examine the MR findings of PMD attributable to PLP duplication compared with those arising from a missense PLP mutation.
Methods
Seven male patients with PMD, aged 2 to 17 years and from seven unrelated Japanese families, were involved in this study. The patients came from two hospitals in the city of Chiba. Genetic analysis of their PLP genes, obtained with previously reported methods (12), revealed missense point mutations in exon 2 (Leu45[CTA] → Arg[CGA]) in patient 1, exon 5 (Val209[GTT] → Asp[GAT]) in patient 2, and exon 5 (Pro210[CCA] → Leu[CTA]) in patient 3 (13). The proton MR spectroscopic findings in patients 1 and 3 were reported previously (14). These missense point mutations impaired the transport of both PLP and DM20. PLP gene duplications were identified in the other four patients (patients 4–7) by using an interphase fluourescence in situ hybridization (FISH) assay (9). To determine the size of the duplication and appropriate locations of the DNA rearrangement breakpoints, phage P1 artificial chromosomes (PAC) clones were used as FISH probes for breakpoint mapping (9). The size of the duplication in patient 4 was much larger than that found in the other three patients.
The patients were classified into the classic or connatal form based on the time of disease onset and the clinical severity (1). MR imaging was performed with a 0.5-T superconducting magnet in six patients (the exception being patient 6), or a 1.5-T superconducting magnet in five patients (patients 1, 3, 4, 5, and 6). At least two scans were obtained in 6 patients (patients 1–6). Axial or coronal T2-weighted images or both (2000/80/2 [TR/TE/excitations] for 0.5-T, 3000–4000/100/2 for 1.5-T) and T1-weighted images (500/30/2, TI =700 for 0.5 T; 500/30/2 for 1.5-T) were obtained in all seven patients. The parameters were as follows: matrix size, 224 × 160 for 0.5-T, 256 × 256 or 256 × 192 for 1.5-T; field of view, 25 cm; slice thickness, 7 mm for 0.5-T, 6 mm for 1.5-T; and slice gap, 2 mm. The images were examined for signs of myelination (comparison of T1 and T2 shortening within the cortex) in parts of the brain; that is, the cerebral corticospinal tract (the internal capsule, corona radiata, centrum semiovale, pre- or postcental cortex, and subcortical white matter), optic radiation, corpus callosum, and cerebellar white matter. On the basis of the signal intensity, myelination was graded as “absent,” “present,” and “intermediate.” Absent myelination indicated a higher signal on T2-weighted and a lower signal on T1-weighted images in the cerebral cortex. Present myelination indicated a lower-to-equal signal on T2- and a higher signal on T1-weighted images of the cerebral cortex. Intermediate myelination showed a signal between that of absent and present myelination. When myelination was present, the “tigroid” appearance of preserved myelin was also evaluated.
T1 and T2 shortening within the cortex was evaluated in the deep gray matter regions; ie, the thalamus, striatum, and globus pallidus. Cerebral and cerebellar atrophy was also examined. Cerebral CT was performed at least once in all patients to identify low-density white matter areas and to rule out calcification, which could also shorten the T1 and T2 values.
Results
The clinical data and MR findings in each patient are presented in the following Table. Four patients (all three patients with a PLP point mutation and one with large PLP duplication [patients 1–4]) were classified as having connatal PMD, whereas the other three patients with PLP duplication (patients 5–7) were identified as having classic PMD. Nystagmus began at birth or soon after birth in cases of connatal PMD, and after 3 weeks in classic PMD. Patient 5 could crawl, and patients 6 and 7 could walk with assistance. On the other hand, the patients with connatal PMD never acquired head control.
Clinical and MR findings for all seven patients

Myelination in the cerebral corticospinal tract (the internal capsule, corona radiata, centrum semiovale, pre- or postcentral cortex, and subcortical white matter), optic radiation, and corpus callosum was observed in three cases of classic PMD with PLP duplication (patients 5–7, Fig 1). High signal intensity on T1-weighted images was more prominent than was low signal intensity on T2-weighted images. In a case of connatal PMD with large PLP duplication (patient 4), myelination was recognized only in the posterior limb of the internal capsule at age 1 year 9 months (Fig 2A–B). Myelination, however, extended into the internal capsule, corona radiata, and centrum semiovale 3 years later (Fig 2C–D). On the other hand, no myelination was observed in the three PMD cases with PLP point mutations (patients 1–3, Fig 3). No progression of myelination was observed in six patients, the exception being patient 4. A tigroid pattern of myelination was not observed in any patient.
fig 1.
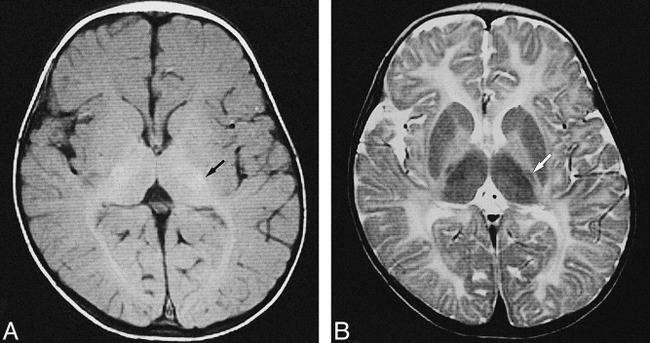
MR images of patient 6 with PMD arising from PLP duplication (classic form) obtained when the patient was 3 years old.
A, Transverse T1-weighted image shows high signal intensity in the posterior limb of the internal capsule (arrow) and optic radiation, representing myelination. T1 shortening was also recognized in the bilateral thalamus.
B, Transverse T2-weighted image shows abnormal high signal intensity in the diffuse cerebral white matter, representing hypomyelination, and low signal in the posterior limb of the internal capsule (white arrow), representing myelination. Low signal intensity was recognized in the bilateral striatum and thalamus.
fig 2.
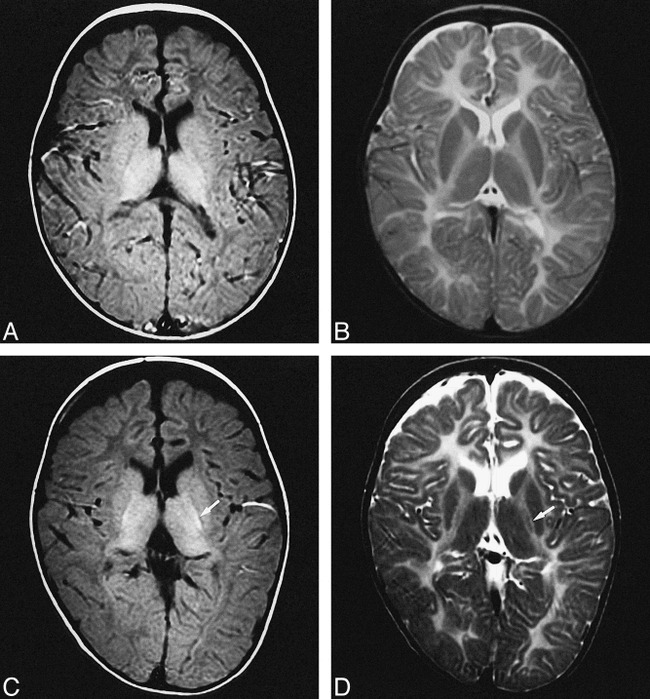
MR images of patient 4 with PMD attributed to PLP duplication (connatal form).
A and B, T1- and T2-weighted images obtained when the patient was 1 year 9 months old.
C and D, T1- and T2-weighted images obtained when the patient was 5 years old. Myelination extends into the internal capsule over a 3-year period (white arrow).
fig 3.
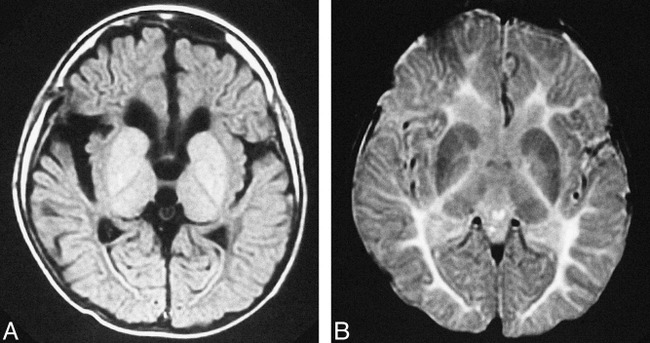
MR images of patient 2 with PMD attributed to a PLP point mutation (connatal form) at the age of 13.
A and B, Transverse T1- and T2-weighted images show no myelination in the cerebral white matter, including the posterior limb of the internal capsule. T1 and T2 shortening was recognized in the bilateral striatum, globus pallidus, and thalamus.
T1 and T2 shortening in the deep gray matter (ie, striatum, globus pallidus, and thalamus) was, to some degree, recognized in all patients in this study. Low signal intensity on T2-weighted images was more significant than high signal intensity on T1-weighted images. The longitudinal MR study showed progressive T2 shortening in the basal ganglionic regions.
Cerebral and cerebellar atrophy was present in connatal PMD. The atrophy was not present on initial scanning in patients 1 and 3 at the ages of 9 and 10 months. In the other four patients, no atrophy was observed.
Calcification was absent in all patients. Also, CT showed slightly low-density areas in the cerebral and cerebellar white matter in all patients; however, differences could not be seen between PMD with PLP point mutations and PMD with PLP duplication.
Discussion
From the MR perspective, postnatal brain development consists primarily of changes in signal intensity secondary to the process of myelination (15). Brain myelination on MR images is shown to occur at different rates and at different times on T1-weighted images compared with those found on T2-weighted images. For example, at birth, high signal intensity on T1-weighted images is observed in the cerebellar peduncle and the posterior limb of the internal capsule, which shows low signal intensity on T2-weighted images during the interval between birth and 2 months after birth. Changes in white matter maturation are seen best on T1-weighted images during the first 6 to 8 months of life, and on T2-weighted images between 6 and 18 months. The exact reason for these differences has not been determined. It, however, is known that T1 shortening correlates temporally with the increased interaction of the hydrogen molecules of water binding to the member components of myelin as well as with the increases in cholesterol and glycolipids that accompany the formation of myelin from oligodendrocytes. T2 shortening correlates temporally with the tightening of the myelin spiral around the axon; that is, the maturation of the myelin sheath (15, 16). The seven PMD patients in this study were older than 9 months and therefore should have had an essentially adult pattern, at least on T1-weighted images.
Three patients with classic PMD with PLP duplication (patients 5–7) exhibited myelination on MR imaging in the cerebral corticospinal tract, optic radiation, and corpus callosum. In fact, pathologic analysis showed a relatively good state of myelination within the internal capsule (10) in cases of classic PMD. The T1 shortening in these regions was more prominent than was T2 shortening. This immature myelination pattern, also seen in the neonatal period (15), might reflect the pathologic findings in classic PMD; that is, the myelin sheath in the myelin islets is more or less damaged and shows varicose and bandlike distension, thinning, and pallor (10).
Patient 4 (connatal PMD with large PLP duplication) presented interesting neuroradiologic and clinical findings. Nezu et al (17) and van der Knaap and Valk (18) previously reported the absence of any progression of myelination in PMD. They suggested an arrest of myelination occurred before or soon after birth in PMD. MR imaging in patient 4, however, revealed longitudinal progression of myelination in the internal capsule, corona radiata, and centrum semiovale over a 3-year period. Myelination in the optic radiation and corpus callosum, which was present in the other three classic PMD cases with PLP duplication, was absent on follow-up MR imaging. The state of myelination in patient 4 was, therefore, between that of classic PMD with PLP duplication and connatal PMD with a PLP point mutation. From the clinical point of view, more than 40 PMD patients with PLP duplication had classic PMD with a relatively mild clinical course (19); however, patient 4 was classified as having connatal PMD with severe clinical manifestations, although he had PLP duplication.
We recently reported on 13 PMD patients with PLP duplication, who commonly had classic PMD (9). Among them, two exceptionally severe cases (clinically connatal PMD) carried large duplications, suggesting that either the size of the duplicated genomic region or the location of the breakpoint may affect the clinical severity. A duplicated large fragment may contain other genes as well as PLP. The duplication may affect these genes, resulting in the phenotypic variation and delayed myelination that was seen on MR images in patient 4.
On the other hand, myelination was totally absent on MR images in three cases of connatal PMD with a PLP point mutation. This finding coincides with the pathologic findings in connatal PMD; that is, dysmyelination spreads diffusely over all parts of the brain and is so intense that, in large areas, not a single myelinated fiber is to be found. Accordingly, the presence of myelination in the corticospinal tract with diffuse white matter hypomyelination on MR images could be a marker for PMD with PLP duplication.
In the literature, descriptions of the pathologic features of classic PMD (10) have reported that the central white matter shows a patchy distribution of dysmyelination with preserved myelin islands. These findings have no relation to such neuroanatomic structures as fiber pathways (including the corticospinal tract and optic radiation) and systems, but are frequently found perivascularly, sometimes as zones of preserved myelin along stretches of blood vessels, thus giving rise to the characteristic “tigroid” appearance. Caro and Marks (20) reported MR images of patients with classic PMD, who exhibited a homogeneous low intensity on T1-weighted images, and heterogeneous high signal intensity with small scattered areas of normal signal on T2-weighted images in the white matter, especially in the convexities of the brain. They interpreted the MR findings as reflecting the tigroid pattern of myelination. Nevertheless, the absence of T1 shortening that would be caused by the myelination remained unexplained. The tigroid pattern of myelination not seen in the four patients studied by Wang et al (21) also was not seen in the present patients. To determine whether or not the “tigroid” pattern of myelination can be detected on MR images, high-resolution MR imaging should be performed in patients with PMD.
All seven of our patients showed low signal intensity on T2-weighted images and high signal intensity on T1-weighted images in the thalamus and putamen. The low signal intensity on T2-weighted images was more prominent than was the high signal intensity on T1-weighted images. These findings are similar to those found in the normal infantile pattern, which is believed to be secondary to myelination of the axons within the deep nuclei (15). Pathologic examination (1), however, revealed that all central nervous system myelin sheaths, including those of the deep gray matter regions, were involved, even in cases of classic PMD. Therefore, other mechanisms should be considered for the T2 and T1 shortening in the deep gray matter in PMD cases.
Prominent T2 shortening in the deep gray and white matter was recently reported in children with certain pathologic states, such as ischemic anoxic brain injury and cerebral infarction (22, 23). This abnormal T2 shortening may either arise from iron deposition caused by to interruption of axonal projections from the basal ganglia and thalamus to the cortex or may be attributed directly to free radicals produced during cell injury. These regions exhibited slightly higher signal intensity on T1-weighted images (24); however, this finding was often ambiguous. To examine the presence or absence of iron deposition in PMD cases, gradient acquisition images should be obtained.
Another possible mechanism for T2 shortening in the deep gray matter is maturational changes of the neuronal network (25). Maturational changes of neurons, particularly increasing synaptic density, may reduce the amount of free water in the brain, resulting in shortening of the T2 value (26). In PMD, axonal processes appear normal, and the neuronal architecture is unchanged pathologically. Proton MR spectroscopy revealed a normal N-acetylaspartate/creatine ratio (N-acetylaspartate is believed to be only present in neurons and is regarded as a neuronal marker) in two of our patients with PMD that was attributed to a PLP point mutation (14). Accordingly, normal neuronal maturation in the deep gray matter may cause signal changes without myelination.
Conclusion
Finally, the presence of myelination in the cerebral corticospinal tract, with diffuse white matter hypomyelination observed on MR images, could be a marker for PMD with PLP duplication and may help to establish a clinical prognosis. It is suggested that progression of myelination may occur in connatal PMD with large PLP duplication.
Acknowledgments
We wish to thank Fuminori Morita, Yoshitada Nakano, and Kenichiro Okumura for their excellent technical support.
Footnotes
Address reprint requests to Jun-ichi Takanashi, Department of Pediatrics, Faculty of Medicine, Chiba University, 1–8–1 Inohana, Chuo-ku, Chiba-shi, Chiba 260–8677, Japan.
References
- 1.Seitelberger F, Pelizaeus-Merzbacher disease. In: Vinken PJ, Bruyn GW, eds. Handbook of Clinical Neurology. Vol 10. Leukodystrophies and Poliodystrophies. Amsterdam: Elsevier; 1970:150–202
- 2.Zoghbi HY, Ballabio A, Pelizaeus-Merzbacher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. 7th ed. New York: McGraw-Hill; 1995:4581–4585
- 3.Aubourg P, The leukodystrophies: a window to myelin. Nature Genet 1993;5:105-106 [DOI] [PubMed] [Google Scholar]
- 4.Griffiths IR, Montague P, Dickinson P, The proteolipid protein gene. Neuropath Appl Neurobiol 1995;21:85-96 [DOI] [PubMed] [Google Scholar]
- 5.van der Knaap MS, Valk J, 18q-syndrome. In: van der Knaap MS, Valk J, eds. Magnetic Resonance of Myelin, Myelination, and Myelin Disorders. 2nd ed. Berlin: Springer-Verlag; 1995:190–191
- 6.Hodes ME, Pratt VM, Dlouhy SR, Genetics of Pelizaeus-Merzbacher disease. Dev Neurosci 1993;15:383-394 [DOI] [PubMed] [Google Scholar]
- 7.Gow A, Lazzarini RA, A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat Genet 1996;13:422-428 [DOI] [PubMed] [Google Scholar]
- 8.Sistermans EA, de Coo RFM, De Wijs IJ, Van Oost BA, Duplication of the proteolipid protein gene is the major cause of Pelizaeus-Merzbacher disease. Neurology 1998;50:1749-1754 [DOI] [PubMed] [Google Scholar]
- 9.Inoue K, Osaka H, Imaizumi K, et al. PLP duplication causing Pelizaeus-Merzbacher disease: phenotype manifestation and molecular mechanism. Ann Neurol 1999;45:624-632 [PubMed] [Google Scholar]
- 10.Seitelberger F, Neuropathology and genetics of Pelizaeus-Merzbacher disease. Brain Pathol 1995;5:267-273 [DOI] [PubMed] [Google Scholar]
- 11.Barkovich AJ, Toxic and metabolic brain disorders. In: Barkovich AJ, ed. Pediatric Neuroimaging. 2nd ed. New York: Raven Press; 1995:55–105
- 12.Osaka H, Kawanishi C, Inoue K, et al. Novel nonsense proteolipid protein gene mutation as a cause of X-linked spastic paraplegia in twin males. Biochem Biophys Res Commun 1995;24:835-841 [DOI] [PubMed] [Google Scholar]
- 13.Inoue K, Osaka H, Kawanishi C, et al. Mutations in the proteolipid protein gene in Japanese families with Pelizaeus-Merzbacher disease. Neurology 1997;48:283-285 [DOI] [PubMed] [Google Scholar]
- 14.Takanashi J, Sugita K, Ishii I, Osaka H, Niimi H, Evaluation of Pelizaeus-Merzbacher disease with proton magnetic resonance spectroscopy. AJNR Am J Neuroradiol 1997;18:533-535 [PMC free article] [PubMed] [Google Scholar]
- 15.Barkovich AJ, Normal development of the neonatal and infant brain, skull, and spine. In: Barkovich AJ, ed. Pediatric Neuroimaging. 2nd ed. New York: Raven Press; 1995:9–54
- 16.van der Knaap MS, Valk J, Myelination and retarded myelination. In: van der Knaap MS, Valk J, eds. Magnetic Resonance of Myelin, Myelination, and Myelin Disorders. 2nd ed. Berlin: Springer-Verlag; 1995:31–52
- 17.Nezu A, Kimura S, Takeshita S, Osaka H, Kimura K, Inoue K, An MRI and MRS study of Pelizaeus-Merzbacher disease. Pediatr Neurol 1998;18:334-337 [DOI] [PubMed] [Google Scholar]
- 18.van der Knaap MS, Valk J, The reflection of histology in MR imaging of Pelizaeus-Merzbacher disease. AJNR Am J Neuroradiol 1989;10:99-103 [PMC free article] [PubMed] [Google Scholar]
- 19.Cailloux F, Giraud G, Courtois V, et al. Proteolipid protein gene involvement in leukodystrophies suggesting a dysmyelinating process. Eur J Paediatric Neurol 1997;1:A49-A50 [Google Scholar]
- 20.Caro PA, Marks HG, Magnetic resonance imaging and computed tomography in Pelizaeus-Merzbacher disease. Magn Reson Imaging 1990;8:791-796 [DOI] [PubMed] [Google Scholar]
- 21.Wang P-J, Young C, Liu H-M, Chang Y-C, Shen Y-Z, Neurophysiologic studies and MRI in Pelizaeus-Merzbacher disease: comparison of classic and connatal forms. Pediatr Neurol 1995;12:47-53 [DOI] [PubMed] [Google Scholar]
- 22.Aoki S, Okada Y, Nishimura K, et al. Normal deposition of brain iron in childhood and adolescence: MR imaging at 1.5 T. Radiology 1989;172:381-385 [DOI] [PubMed] [Google Scholar]
- 23.Takanashi J, Sugita K, Tanabe Y, Ito C, Date H, Niimi H, T2 shortening in childhood moyamoya disease. Neuroradiology 1996;38:S169-S173 [DOI] [PubMed] [Google Scholar]
- 24.Drayer B, Burger P, Darwin R, Reiderer S, Herfkens R, Johnson GA, MRI of brain iron. AJR Am J Roentogenol 1986;147:103-110 [DOI] [PubMed] [Google Scholar]
- 25.Barkovich AJ, MR of the normal neonatal brain: assessment of deep structures. AJNR Am J Neuroradiol 1998;19:1397-1403 [PMC free article] [PubMed] [Google Scholar]
- 26.Korogi Y, Takahashi M, Sumi M, et al. MR signal intensity of the perirolandic cortex in the neonate and infant. Neuroradiology 1996;38:578-584 [DOI] [PubMed] [Google Scholar]