

Abstract
Summary: We present the CT, MR, and autopsy findings of central brain herniation in a 9-year-old boy undergoing treatment for diabetic ketoacidosis (DKA). Severe cerebral edema resulting in central brain herniation is an uncommon complication of the treatment of DKA but carries with it high morbidity and mortality. Radiologic imaging and autopsy findings in this case revealed striking infarctions of central brain structures.
Central or transtentorial herniation of the diencephalon is a distinct pattern of supratentorial brain shift that may occur as a result of diffuse cerebral edema or in response to bilateral supratentorial intracranial expansive processes (1–3). Some degree of generalized cerebral edema is a common complication of the treatment of severe diabetic ketoacidosis (DKA) (1, 4). Severe cerebral edema resulting in central brain herniation is uncommon in this condition but carries with it high mortality and morbidity (1, 4–6). Early recognition and treatment of cerebral edema in DKA is vital for patient management (1, 6). To our knowledge, little has been described in the radiologic literature regarding central herniation in DKA. We report the striking radiographic and autopsy findings of central brain herniation secondary to DKA, including previously unreported infarctions of anterior basilar cerebral structures.
Case Report
A previously healthy, 9-year-old boy presented to an outside hospital complaining of respiratory distress, abdominal pain, and malaise. Laboratory analysis was significant for serum glucose of 1,056, and the patient was diagnosed with DKA. The patient was treated with subcutaneous and intravenous insulin and oral and intravenous fluid resuscitation. Subsequently, the patient became acutely unresponsive and required transfer to the pediatric intensive care unit at our institution. On admission, the patient had fixed and dilated pupils and a Glasgow coma scale of 5. CT scans of the head taken when the patient was admitted to the hospital showed diffuse cerebral edema with effacement of sulci and basal cisterns (Fig 1A). Owing to probable intracranial pressure elevation, intravenous fluids were restricted initially while the patient was treated with a continuous insulin drip. Mannitol was administered prior to placement of a ventriculostomy catheter for intracranial pressure monitoring 1 day after admission.
fig 1.
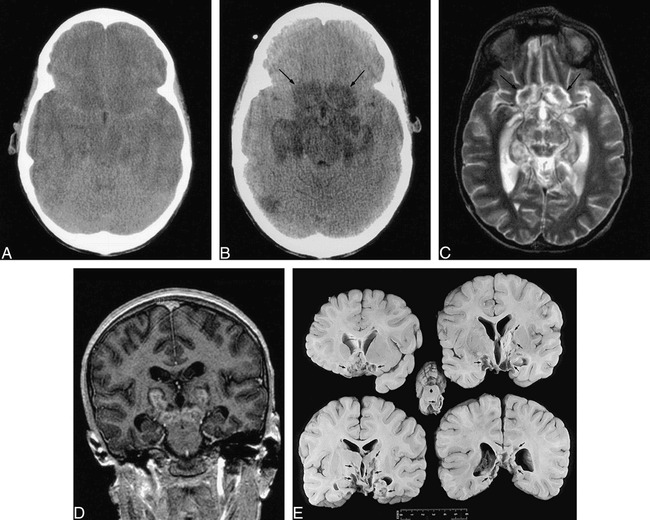
Nine-year-old boy with diffuse cerebral edema and central herniation secondary to treatment of DKA.
A, Axial noncontrast CT scan shows diffuse cerebral edema with effacement of sulci and basal cisterns.
B, Axial noncontrast CT scan obtained 2 days after A shows marked low-density infarcts in the gyrus recti and medial orbital gyri (arrows), globus pallidi, hippocampi/parahippocampal gyri, hypothalamus, midbrain, and posterior right temporal lobe.
C–D, Axial noncontrast T2-weighted (4000/105/1) (C) and coronal postcontrast T1-weighted SPGR (14.4/3.7/1) (D) MR images obtained 24 days after A show cavitary infarcts in the gyrus recti and medial orbital gyri (arrows), medial temporal lobes, midbrain and thalami. Enhancement is present within the thalamic and midbrain lesions. There is diffuse cerebral atrophy.
E, Coronal gross pathologic sections through cerebrum and axial section through midbrain show remote infarctions involving the basilar cerebral structures, basal ganglia, thalami, hippocampi and midbrain (arrows).
A follow-up CT scan performed 2 days after admission demonstrated marked low-density changes in the thalami, hypothalamus, anterior perforated substance, subfrontal area, midbrain, globus pallidi, hippocampi/parahippocampal gyri, posterior cingulate gyri, right posterior temporal lobe, as well as the left occipital lobe and splenium of the corpus callosum (Fig 1B). The findings were interpreted as infarction in the central portion of the brain.
An EEG obtained after the head CT scan showed diffuse slow-wave activity. Follow-up EEGs indicated slight improvement. Eight days after admission, the patient developed seizure activity and was treated with anticonvulsants. A follow-up CT scan performed at that time revealed bilateral hippocampal/parahippocampal and basal ganglia high densities consistent with hemorrhage. There was interval decrease also in cerebral edema and evolution of other previously described central brain infarctions. An MR scan obtained 24 days after admission demonstrated diffuse brain atrophy in addition to the lesions demonstrated on previous CT scans (Fig 1C–D). There was pathologic enhancement in many of the lesions (Fig 1D). MR venograms and MR angiograms were normal.
The patient was discharged to a rehabilitation facility 33 days after admission with a discharge diagnosis of insulin-dependent diabetes mellitus presenting as DKA and cerebral infarction. His Glasgow coma scale on discharge had improved to 9. One year later the patient died from respiratory complications. Postmortem examination was performed and showed remote, multifocal, bilateral infarctions involving basilar cerebral structures, the basal ganglia, thalami, hippocampi and midbrain in distributions of the anterior choroidal, anterior cerebral, and posterior cerebral arteries (Fig 1E).
Discussion
Brain herniation may result from a multitude of pathologic processes that increase intracerebral mass (7). Types of brain herniation include subfalcine, transalar, tonsillar, and transtentorial (7, 8). Bilateral transtentorial herniation, also known as central herniation, may occur as the ultimate outcome of unilateral transtentorial herniation or as a complication of bilateral supratentorial intracranial expansive processes (often acute traumatic or vascular lesions) (2, 9). Central herniation is the end result of downward displacement of the cerebral hemispheres and the basal nuclei compressing and displacing the diencephalon and the midbrain rostrocaudally through the tentorial notch (3). There may be herniation also of both temporal unci and parahippocampal gyri as well as the lingual gyrus and isthmus of the gyrus fornicatus into the tentorial hiatus (2, 7, 8). The brain stem is compressed transversely and will appear elongated on its anteroposterior axis on axial imaging. The basal cisterns may be obliterated completely, especially the perimesencephalic (interpeduncular, quadrigeminal, ambient, and crural) and suprasellar cisterns (2, 8). There may be downward shifting of the basilar artery and pineal gland (10).
Central transtentorial herniation must be distinguished from temporal or uncal transtentorial herniation (2, 3). This latter type of herniation characteristically occurs when expanding lesions arising in the temporal fossa or temporal lobe shift the inner, basal edge of the uncus and hippocampal gyrus toward the midline, across the incisural edge of the tentorium. This may result in flattening and displacement of the midbrain.
Clinically, the earliest signs of uncal herniation signify dysfunction in structures outside the brain parenchyma (eg, cranial nerve III) whereas the first signs of central herniation are diencephalic dysfunction. In fact, Plum and Posner elegantly described two distinct clinical syndromes—the central syndrome of rostrocaudal deterioration and the syndrome of uncal herniation and lateral brain stem compression (3). Both of these syndromes are recognizably distinct in their course but both merge into a similar picture when pathologic changes extend to involve the midbrain level or below.
The clinical manifestations of central herniation will depend upon the degree of compression or displacement of anatomic structures. The first evidence of central herniation is usually a change in alertness or behavior secondary to diencephalic dysfunction followed by respiratory, ocular, and motor signs (3). As the syndrome progresses, uncal and parahippocampal herniation may result in compression of the third nerve and tectum, leading to unilateral or bilateral third-nerve palsy, failure of upward gaze, and loss of the pupillary light reflex. When the herniation is of sufficient severity, obstruction of the aqueduct may occur leading to increased intraventricular pressure and varying degrees of hydrocephalus and papilledema (7, 8). Infarction in territories supplied by the anterior choroidal, posterior cerebral, thalamoperforate, and superior cerebellar arteries resulting from vascular compression at the tentorial notch have been described well (4, 9, 11). Furthermore, focal infarction may occur in the mamillary bodies and anterior lobe of the pituitary gland secondary to decreased blood flow through the hypothalamo-hypophyseal portal vessels (9). With midbrain displacement and compression, there may be ischemia of corticospinal and corticobulbar pathways. With continued midbrain descent through the incisura, there may be mechanical shearing of the pontine and mesencephalic perforating vessels, resulting in Duret's hemorrhages (7). Compression of the respiratory and cardiac centers of the reticular substance leads to irreversible coma or death (7).
In our case, the patient's sudden change in mental status and lack of unilateral localizing findings were consistent clinically with the syndrome of central herniation (3). The diffuse edema noted on the initial CT scan, with secondary obliteration of the basal cisterns and elongation of the brain stem, was compatible radiographically with central herniation (2). A follow-up CT scan revealed marked low attenuation consistent with infarction or edema involving vascular territories of the anterior choroidal, anterior cerebral, posterior cerebral, and thalamoperforate arteries. Of particular interest is the sharply defined low-density lesion present within the subfrontal region involving the gyrus recti, medial orbital gyri, and subcallosal gyrus. To our knowledge, infarcts in these regions secondary to central herniation have never been described previously. The vascular supply to these structures emanates from the anterior cerebral arteries (ACAs) via the olfactory artery, subcallosal artery, orbito-frontal artery, recurrent artery of Heubner, and ACA perforators (12). It is likely, therefore, that occlusion of the proximal branches of the ACAs occurred secondary to mechanical compression against the planum sphenoidale or stretching by the downward shift of the central brain structures or both (3). Alternatively, the low density present within the subfrontal region may reflect venous congestion or pressure necrosis with secondary edema. This has been described within the midbrain as it descends through the incisura (7). Regardless of etiology, the low-density changes revealed on CT scans within the subfrontal region progressed to necrosis and cavitary change on MR imaging and autopsy. In fact, widespread cavitation and necrosis were noted also at autopsy within the vascular distributions of the anterior choroidal, posterior cerebral, and thalamoperforate arteries.
DKA is a relatively common acute complication of insulin-dependent diabetes mellitus and, although usually treated without significant sequelae, it is the most common cause of death in children with type I diabetes (4, 5). Most of these deaths are secondary to CNS disease, primarily cerebral edema (4, 5). Some degree of cerebral edema appears to be a universal consequence during treatment of severe DKA; however, only rarely is the edema severe enough to cause brain stem herniation (1). Brain stem herniation, or more precisely, central herniation in DKA is an ominous sign with high morbidity and mortality (4–6). The cause of cerebral edema in patients treated for DKA is uncertain (1, 4). Various factors have been considered, including the rate of fluid administration, rate of glucose reduction, integrity of the blood-brain barrier, rate of acid-base correction, and osmolar disequilibrium between vascular and extravascular brain compartments (1, 4, 6).
Although the development of central brain herniation after initiation of treatment for DKA is unpredictable, it typically becomes evident 6 to 13 hours after treatment has begun (4, 6). Clinical signs include sudden deterioration of consciousness, absent brain stem responses, and eventual respiratory arrest (4–6). Early, aggressive treatment of cerebral edema with mannitol and fluid restriction may reverse the life-threatening herniation (5, 6). This requires meticulous monitoring of the patient's neurologic state and familiarity with the appearance of brain edema and central herniation on CT scans or MR imaging.
Acknowledgments
The authors wish to thank Belinda De Libero and Margaret Kowaluk for their secretarial and photographic support in the preparation of this case report.
Footnotes
Address reprint requests to David A. Shrier, MD, Department of Radiology, PO Box 648, Neuroradiology Division, University of Rochester Medical Center, 601 Elmwood Avenue, Rochester, NY 14642.
References
- 1.Duck SC, Wyatt DT, Factors associated with brain herniation in the treatment of diabetic ketoacidosis. J Pediatr 1988;113:10-14 [DOI] [PubMed] [Google Scholar]
- 2.Nguyen JP, Djindjian M, Brugières P, Badiane S, Melon E, Poirier J, Anatomy-computerized tomography correlations in transtentorial brain herniation. J Neuroradiol 1989;16:181-196 [PubMed] [Google Scholar]
- 3.Plum F, Posner JB, The Diagnosis of Stupor and Coma. Contemporary Neurology Series-Edition 3. Philadelphia: F.A. Davis; 1980:96–112 [PubMed]
- 4.Eskander EN, Weller SJ, Frim DM, Hydrocephalus requiring urgent external ventricular drainage in a patient with diabetic ketoacidosis and cerebral edema: case report. Neurosurgery 1997;40:836-839 [DOI] [PubMed] [Google Scholar]
- 5.Rogers B, Sills I, Cohen M, Seidel FG, Diabetic ketoacidosis. Neurologic collapse during treatment followed by severe developmental morbidity. Clin Pediatr 1990;29:451-456 [DOI] [PubMed] [Google Scholar]
- 6.Kalis NN, Van Der Merwe P-L, Schoeman JF, Smith RML, Cerebral oedema with coning in diabetic keto-acidosis. S Afr Med J 1991;79:727-731 [PubMed] [Google Scholar]
- 7.Laine FJ, Shedden AI, Dunn MM, Ghatak NR, Acquired intracranial herniations: MR imaging findings. AJR Am J Roentgenol 1995;165:967-973 [DOI] [PubMed] [Google Scholar]
- 8.Stovring J, Descending tentorial herniation: findings on computed tomography. Neuroradiology 1977;14:101-105 [DOI] [PubMed] [Google Scholar]
- 9.Graham DI, Lantos PL, eds Greenfields Neuropathology. Sixth Edition. New York: Oxford University Press ;1997:177
- 10.Hahn FJ, Gurney J, CT signs of central descending transtentorial herniation. (Correspondence). AJNR Am J Neuroradiol 1985;6:844-845 [PMC free article] [PubMed] [Google Scholar]
- 11.Endo M, Ichikawa F, Miyasaka Y, Yada K, Ohwada T, Capsular and thalamic infarction caused by tentorial herniation subsequent to head trauma. Neuroradiology 1991;33:296-299 [DOI] [PubMed] [Google Scholar]
- 12.Marinkoviæ S, Gibo H, Brigante L, Milisavljeviæ M, Donzelli R, Arteries of the brain and spinal cord. In: De Angelis, ed. Anatomic Features and Clinical Significance. Avellino, Italy: 1997; 179–189