

Abstract
Inflammatory bowel disease (IBD) is a complex disorder that imposes a growing health burden. Multiple genetic associations have been identified in IBD, but the mechanisms underlying many of these associations are poorly understood. Animal models are needed to bridge this gap, but conventional laboratory mouse strains lack the genetic diversity of human populations. To more accurately model human genetic diversity, we utilized a panel of chromosome (Chr) substitution strains, carrying chromosomes from the wild-derived and genetically divergent PWD/PhJ (PWD) strain on the commonly used C57BL/6J (B6) background, as well as their parental B6 and PWD strains. Two models of IBD were used, TNBS- and DSS-induced colitis. Compared with B6 mice, PWD mice were highly susceptible to TNBS-induced colitis, but resistant to DSS-induced colitis. Using consomic mice, we identified several PWD-derived loci that exhibited profound effects on IBD susceptibility. The most pronounced of these were loci on Chr1 and Chr2, which yielded high susceptibility in both IBD models, each acting at distinct phases of the disease. Leveraging transcriptomic data from B6 and PWD immune cells, together with a machine learning approach incorporating human IBD genetic associations, we identified lead candidate genes, including Itga4, Pip4k2a, Lcn10, Lgmn, and Gpr65.
Introduction
Crohn’s Disease and Ulcerative Colitis, and microscopic colitis, the two major forms of inflammatory bowel disease (IBD), are common causes of chronic illness, and affect a large and varied patient population. Patients with less severe forms of IBD can manage their symptoms with a combination of steroids and immune modulators; however, patients with more severe forms of the disease have limited medical options and often require surgical resections of affected bowel 1. A better understanding of disease etiology could provide improved therapeutic, diagnostic, and preventative strategies.
Recent evidence implicates both genetic and environmental factors in IBD pathogenesis, whereby a combination of these factors leads to alterations in the gut epithelial barrier function, allowing for translocation of luminal antigens (e.g. from gut bacteria) and subsequent immune activation and initiation of inflammatory processes 2. With regard to the genetic contribution to IBD, the existence of multiple genetic loci influencing disease susceptibility was first clearly demonstrated in early animal studies using standard inbred laboratory strains of mice 3–5. More recently, the explosion in human GWAS has identified >200 genes associated with various forms of IBD 6–9. The most prominent of these associations include missense variants in ATG16L1, NOD2, and IL23R, genes involved in autophagy, and innate, and adaptive immune responses, respectively 10. Nonetheless, we are only beginning to understand the functional consequences of genetic variation contributing to IBD, and GWAS explain only a minor proportion of the total heritability of IBD 11. Additionally, the GWAS for most, if not all, immune-mediated diseases so far have defined the genetic basis of disease susceptibility only, and have not clearly delineated the genetics of disease progression, pathophysiology, and other critical sub-phenotypes. The mechanisms underlying newly identified genetic associations, and the interactions of genetics with the environment, such as the gut microbiome, are only beginning to be understood. Little is understood about these complex bidirectional interactions, yet this knowledge will be critical for the development of more effective personalized therapeutic and preventative modalities. Such studies are difficult to carry out in humans, and relevant animal models are needed to bridge this gap in our knowledge.
The laboratory mouse provides an indispensable, powerful, and genetically tractable tool to understand the basic mechanisms of human disease, and to establish causality for genetic or environmental factors. However, widely used knockout mice are rarely representative of natural genetic variation, while commonly used laboratory mouse strains lack the genetic diversity present in the human population. These problems can be overcome by the introduction of so-called wild-derived inbred strains of mice into the experimental design. Compared with conventional inbred laboratory mice such as, e.g., C57BL/6 and BALB/c, these mice have much greater genetic divergence, thus more closely recapitulating the natural evolutionarily selected genetic divergence of human populations 12–14. Perhaps more importantly, these mice have not undergone extended artificial selection in the laboratory, and thus genetically resemble their wild counterparts that encounter selective pressure from infectious and/or commensal microorganisms, which is a particularly important point when studying immune-mediated disease. Among these wild-derived strains are PWD/PhJ (PWD) and PWK/PhJ mice, trapped in the Czech Republic and established as inbred strains by the Forejt group 15. Importantly, this group also generated a panel of chromosome (Chr) substitution (consomic) strains by introgressing each PWD chromosome one at a time onto on the commonly used C57BL/6J (B6) background (notated as B6-ChrPWD) 16.
We have recently employed this model to identify genetic loci that control susceptibility to experimental autoimmune encephalomyelitis (EAE) 17, 18, the principal autoimmune model of multiple sclerosis (MS), an immune-mediated disease with similar genetic burden to IBD 19–21. Subsequently, we applied this approach to model and identify gene-environment interactions in EAE/MS 22. Importantly, we have shown that baseline immune cell transcriptomes differ profoundly between B6 and PWD mice, with significant enrichment of not only MS-signature genes, but also of human IBD-susceptibility genes 17, suggesting that PWD mice carry many naturally occurring genetic variants that regulate susceptibility to IBD.
Here, we have employed a panel of select B6-ChrPWD consomic strains, as well as the B6 and PWD parental strains, across two widely used chemically induced models of IBD: dextran sulfate sodium (DSS)- and 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis 23. We demonstrate that the two parental strains differ dramatically in their susceptibility to each of these models. Additionally, we demonstrate that multiple PWD-derived loci exert powerful bidirectional effects that are specific to each model of IBD, with partial genetic overlap between experimental IBD and EAE susceptibility, similar to the shared genetic burden of IBD and MS 21.
Results
PWD mice are resistant to DSS-induced colitis compared with B6 mice
To determine the susceptibility of PWD mice to IBD, we first employed the widely used DSS-induced colitis model. DSS disrupts the integrity of the intestinal epithelium, causing subsequent acute intestinal inflammation that most closely resembles human ulcerative colitis 23. To test the susceptibility of B6 and PWD mice to DSS-induced colitis, DSS (5%) was administered in drinking water for 5 days, then discontinued, and weight loss and disease activity were monitored as described in Materials and Methods. Compared with B6 mice, PWD mice showed more rapid weight loss initially, but after DSS discontinuation, their weight stabilized, while B6 continued to lose weight (Fig. 1A). Similarly, PWD mice exhibited a faster recovery as measured by disease activity score, a measure of disease severity (Fig. 1B). As an additional quantitative indicator of colitis severity, we measured the levels of fecal lipocalin 2 (LCN2), a highly sensitive, dynamic, and non-invasive biomarker of intestinal inflammation 24. As expected, beginning around day 5 post-DSS initiation, LCN2 production increased greatly in both strains, but PWD mice showed strikingly lower levels of fecal LCN2 and faster recovery by day 10, while the inflammation in B6 mice persisted (Fig. 1C). Consistent with this, semi-quantitative evaluation of histological damage and inflammation of the mucosa performed at day 10 post DSS induction revealed reduced levels of mucosal layer damage and inflammation in PWD mice compared with B6 (Fig. 1D). These results suggest that PWD mice are better able to resolve DSS-induced intestinal inflammation compared with B6 mice.
Figure 1. PWD mice are resistant to DSS-induced colitis.
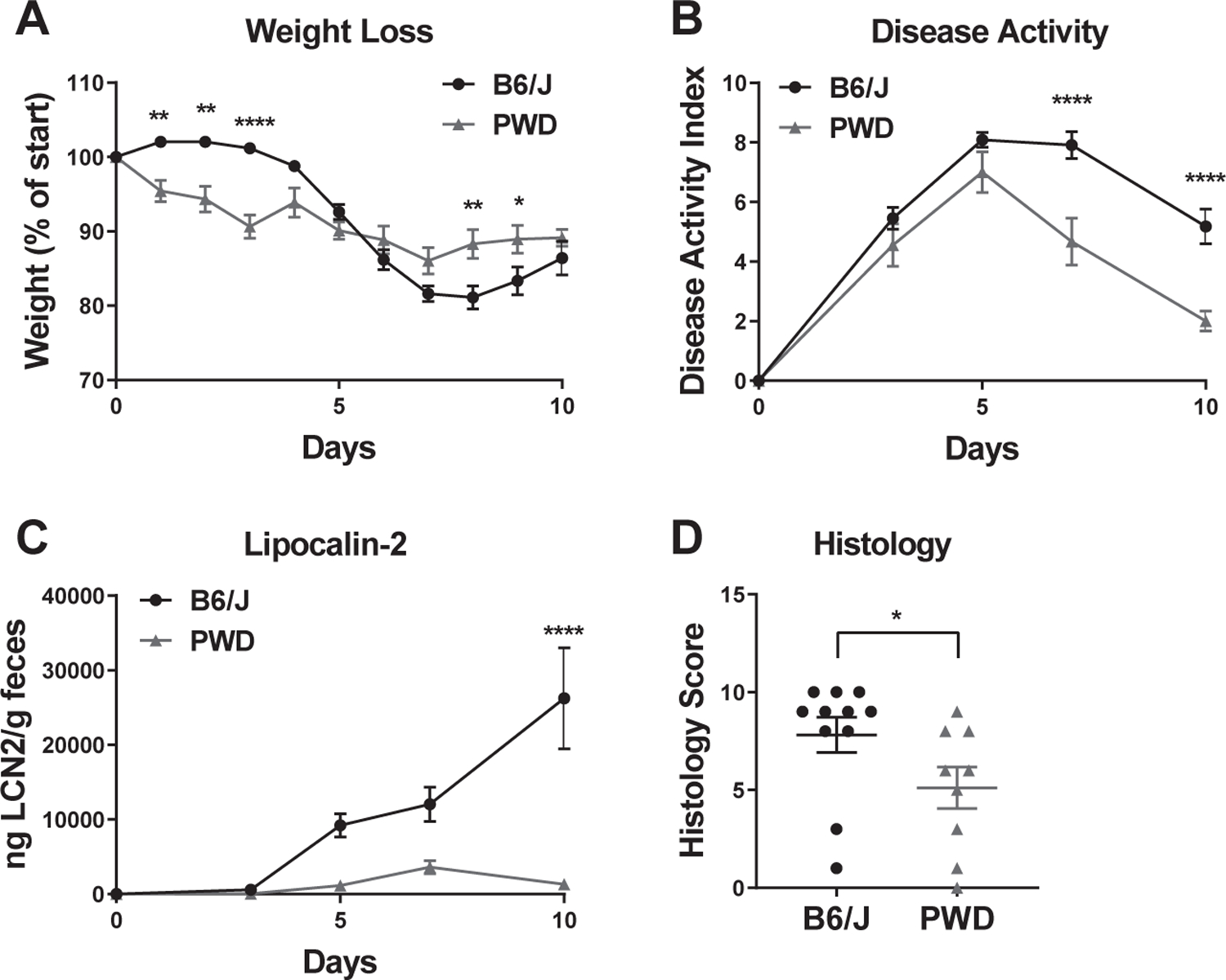
A 5% solution of DSS was administered to B6 and PWD mice in drinking water from days 0 – 5. Weight loss (A), disease activity index (B), were monitored as described in Materials and Methods. Disease activity index was calculated based on weight loss, stool consistency, and presence of blood in stool. Fecal LCN2 levels (C) were measured by ELISA. (D) Animals were euthanized on day 10, colons were fixed and processed for H&E sections, followed by semi-quantitative evaluation, as described in the Materials and Methods. Significance of differences between B6 and PWD were determined by two-way ANOVA with Holm-Sidak post-hoc comparisons in (A–C), and by Mann Whitney test in (D), and are indicated as follows: *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001. Numbers of animals studied of each sex are provided in Table 1. These studies were performed in parallel with those in Fig. 3.
PWD mice are highly susceptible to TNBS-induced colitis compared with B6 mice
DSS-induced colitis recapitulates some, but not all aspects of human IBD. We employed another commonly used chemical model of IBD, TNBS-induced colitis, which models different aspects of IBD pathogenesis. TNBS is a reactive hapten that initiates a Th1/Th17-driven acute inflammatory response against intestinal and/or microbial antigens, which results in intestinal inflammation that mimics many immunologic and histopathologic aspects of human IBD, most closely resembling Crohn’s disease 23. Age and sex-matched B6 and PWD mice received an intracolonic injection of 200 mg/kg TNBS in 50% ethanol, a dose which induced mild-moderate clinical signs of colitis and weight loss in B6 mice (Fig. 2A and B). In contrast, PWD exhibited rapid and irreversible weight loss, signs of severe colitis, and 100% lethality within 6 days, all of which were significantly greater compared with B6 mice (Fig. 2A–C). These changes were not observed in vehicle (ethanol) injected control B6 and PWD animals (data not shown).
Figure 2. PWD mice are highly susceptible to TNBS-induced colitis.
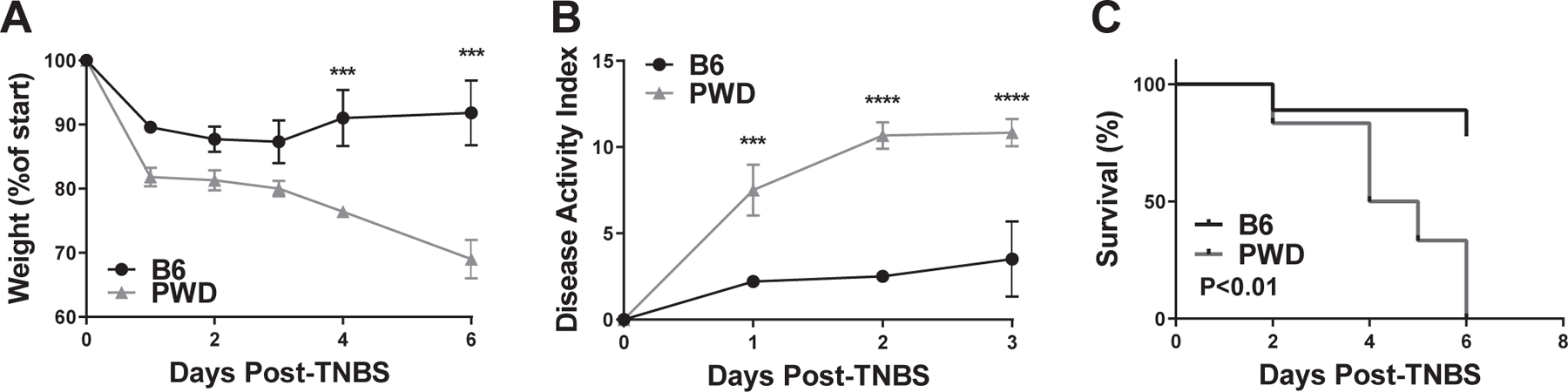
B6 and PWD mice received an intracolonic enema of 200 mg/kg TNBS in 50% ethanol, followed by monitoring of weight loss (A), disease activity index (B), and survival (C). Significance of differences were determined by two-way ANOVA with Holm-Sidak post-hoc comparisons in (A) and (B), and by Mantel-Cox test in (C), and are indicated as follows: *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001. Numbers of animals studied were as follows: B6 (N=7, 4 males and 3 females) and PWD (N=6, 3 males and 3 females). These mice represent an independent cohort from those studied in Fig. 4.
Taken together, these data demonstrate that compared with B6, PWD mice exhibit highly divergent responses in two different models of experimental colitis, strongly suggesting the existence of genetic variants differentially controlling susceptibility to various aspects of IBD.
PWD-derived loci regulate susceptibility to DSS-induced colitis
In order to identify PWD loci controlling IBD phenotypes, we studied a panel of select B6-ChrPWD consomic strains. There are 27 B6-ChrPWD strains in all, one strain for each Chr, with the exception of Chrs 10, 11, and X, each of which is represented by 3 sub-consomic strains carrying reciprocal portions (~1/3) of the respective Chrs 16. Strain names are abbreviated to include only the chromosome number that is derived from PWD, e.g., B6-Chr1PWD is abbreviated as Chr1PWD. From this panel, we selected the following eight strains on the basis of their susceptibility to EAE, an animal model of MS, since MS is an immune-mediated disease with a partially overlapping genetic burden compared to IBD 21. We selected Chr1PWD, Chr6PWD, Chr11.1PWD, Chr11.3PWD, and Chr16PWD mice, which are more susceptible to EAE compared with B6, and Chr2PWD, Chr10.3PWD, Chr15PWD mice, which are more resistant to EAE compared with B6 18, 22. Additionally, given the prominent role of MHC in immune-mediated diseases, including IBD 25, 26, we included two Chr17PWD consomic strains. The first, designated Chr17SPWD (“S” for “short”) was the original strain obtained from Jackson laboratories that we studied for EAE susceptibility 18, which we previously discovered unexpectedly carries the H-2/MHC-containing interval derived from B6 22 (genotyping shown in Fig. S1A). The second, designated Chr17FPWD (“F” for “full”), was provided directly by the Forejt laboratory. This strain was genotyped to confirm that the entire Chr17 (including H-2) is of PWD-origin (Fig. S1B). We have also confirmed that Chr17SPWD mice indeed carry the B6 MHC class II antigen, using allele-specific antibodies and flow cytometry (data not shown).
Colitis was induced by administration of DSS in the drinking water, as described above. No mortality was observed in B6 mice, and minimal mortality was seen in consomic strains, with the dramatic exception of Chr2PWD mice, 89% of which succumbed by day 10 post-induction (Fig. 3A). Accordingly, Chr2PWD mice exhibited very rapid weight loss and exacerbated disease activity scores, compared with B6 mice (Fig. 3B–D). Compared with B6 mice, Chr1PWD and Chr17SPWD mice also exhibited more severe colitis, as indicated by more severe weight loss and/or exacerbated disease activity (Fig. 3B–D). In contrast, compared with B6, Chr11.1PWD mice exhibited reduced weight loss, and disease activity. Chr10.3PWD, Chr11.3PWD, Chr15PWD, Chr16PWD and surprisingly, Chr17FPWD, exhibited similar colitis severity parameters compared with B6 (Fig. 3B–D). LCN2 measurements and histological analysis were carried out for select strains exhibiting significant or suggestive differences in weight loss and/or disease activity, although samples for Chr2PWD mice were not available due to early mortality and severe disease. LCN2 levels and histological condition largely followed disease activity measurements, with elevated LCN2 levels and/or more severe histological damage observed in Chr1PWD, Chr2PWD, and Chr17SPWD mice, with a much earlier and acute onset for Chr2PWD compared with the other two strains (Fig. 3E and F). Chr16PWD mice exhibited histologic evidence of modestly exacerbated colitis (Fig. 3F) in the absence of significant differences for other colitis indicators (Fig. 3A–E). A summary of all quantitative colitis parameters across all of the consomic strains is provided in Table 1, and additional kinetics of weight loss, disease activity, and LCN2 levels are shown in (Fig. S2). Taken together, these data illustrate that PWD loci exert potent bidirectional effects on multiple aspects of DSS-induced colitis severity.
Figure 3. PWD loci regulate susceptibility to DSS-induced colitis.
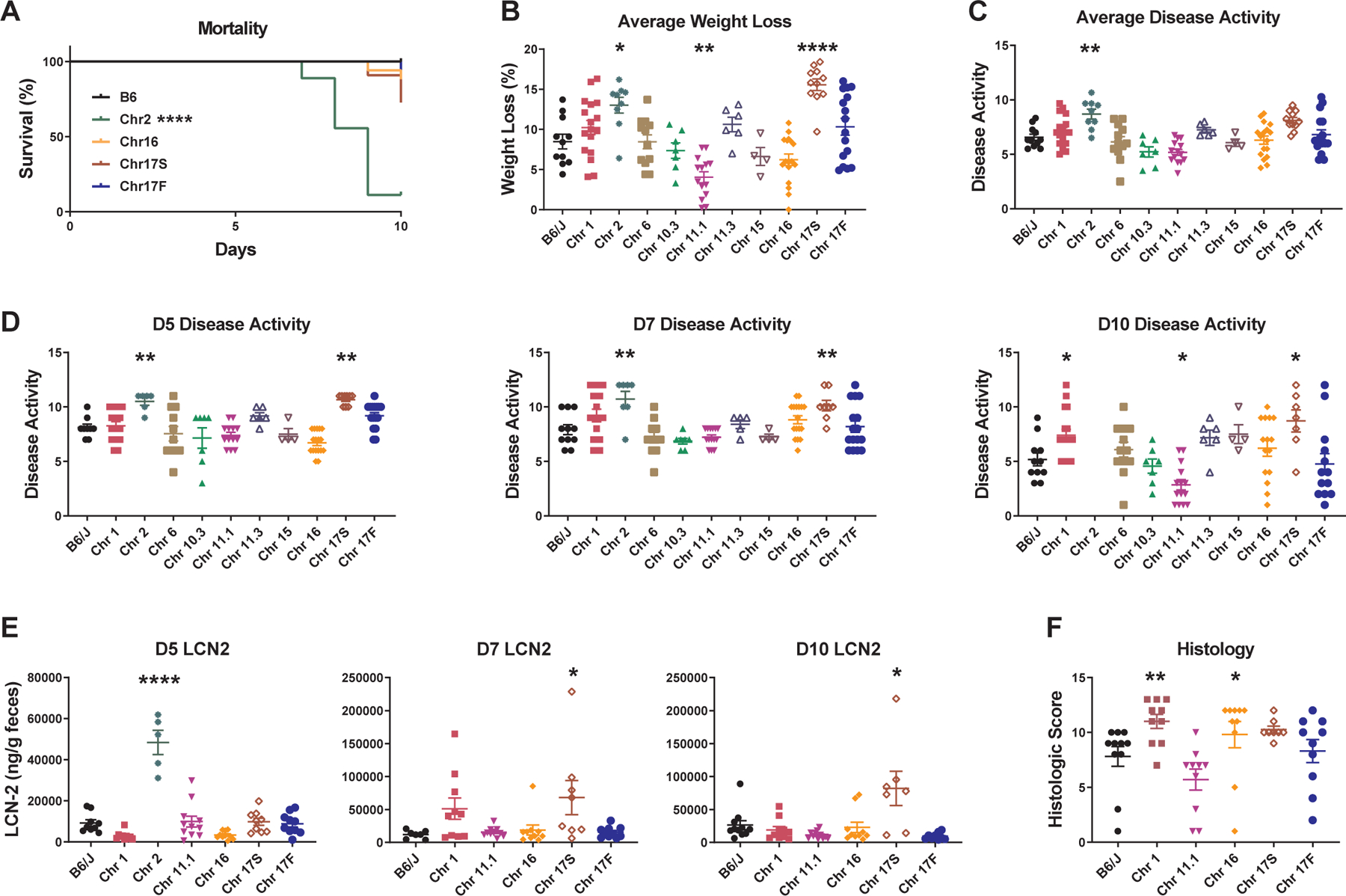
DSS colitis was induced and evaluated in B6.ChrPWD consomic strains and B6 controls as described in Fig. 1. (A) Survival is shown for B6 mice and any consomic strains that had less than 100% survival. (B) Average weight loss was calculated as the average % weight loss across all days that were measured. (C) Average disease activity was calculated as the average disease activity score across time points measured, excluding D0 (D3, D5, D7, D10). (D) Disease activity scores at the indicated time points. (E) LCN2 levels were measured by ELISA at the indicated time points. (F) Colonic tissue was collected from surviving animals on D10 and evaluated by semiquantitative histology as in Fig. 1. Significance of differences between each consomic strain and B6 controls was calculated using: Mantel-Cox test in (A); ordinary one-way ANOVA with Dunnet’s post-hoc comparisons in (B) (C), and (E); Kruskal-Wallis one-way ANOVA with Dunn’s post-hoc comparisons in (D), and (F), and are indicated as follows: and are indicated as follows: *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001. Numbers of animals studied of each sex are provided in Table 2.
Table 1. Summary of DSS-induced colitis severity parameters across the consomic panel.
Summary data from Fig. 1, Fig. 3 and Fig. 5. Comparisons with B6 controls are summarized as follows: “=”, not significantly different from B6; “↓” significantly lower than B6, “↑”, significantly higher than B6; “ref.”, B6 reference; “ND”, not done. Significance of differences is indicated using asterisks, as in Fig. 3. For measurements with comparisons across multiple time points (disease activity and LCN2), the measurement with highest significance (if any) at any single time point comparison is provided. *EAE susceptibility was determined in previous studies {Bearoff 2015; Bearoff 2016; Krementsov 2018}. Comparisons with B6 controls are summarized as follows: “=”, not significantly different from B6; “↓” significantly lower than B6, “↑”, significantly higher than B6; “ref.”, B6 reference; “ND”, not done.
| Strain | Female N | Male N | Mortality | Weight Loss | Disease Activity | LCN-2 | Histology | *EAE |
|---|---|---|---|---|---|---|---|---|
| B6 | 7 | 4 | ref. | ref. | ref. | ref. | ref. | ref. |
| PWD | 5 | 4 | = | ↓** | ↓**** | ↓**** | ↓* | ↓ |
| Chr1 | 10 | 7 | = | = | ↑* | = | ↑* | ↑ |
| Chr2 | 6 | 3 | ↑**** | ↑* | ↑** | ↑**** | ND | ↓ |
| Chr6 | 6 | 6 | = | = | = | ND | ND | ↑ |
| Chr10.3 | 4 | 3 | = | = | = | ND | ND | ↓ |
| Chr11.1 | 6 | 8 | = | ↓** | ↓* | = | = | ↑ |
| Chr11.3 | 6 | 0 | = | = | = | ND | ND | ↑ |
| Chr12 | 2 | 4 | = | ↑**** | ↑**** | ND | ND | = |
| Chr15 | 0 | 4 | = | = | = | ND | ND | ↓ |
| Chr16 | 5 | 12 | = | = | = | = | ↑* | ↑ |
| Chr17S | 5 | 6 | = | ↑**** | ↑** | ↑*** | = | = |
| Chr17F | 5 | 11 | = | = | = | = | = | ND |
PWD-derived loci regulate susceptibility to TNBS-induced colitis
In order to identify genes controlling severity across multiple models of IBD, we applied the TNBS-induced colitis model to a subset of six of the B6-ChrPWD consomic strains that had been studied in the DSS-induced colitis paradigm, namely: Chr1PWD, Chr2PWD, Chr11.1PWD, Chr16PWD, Chr17SPWDand Chr17FPWD, along with B6 controls. As in the DSS model, B6 and other consomic strains exhibited relatively low mortality, with the dramatic exception of Chr2PWD mice, which had 91% mortality (Fig. 4A) and rapid weight loss (Fig. 4B and Fig. S3). As in the DSS model, samples for LCN2 levels and histology from Chr2PWD mice were not available due to early mortality and severe disease. Additionally, compared with B6, Chr1PWD mice had increased disease activity (Fig. 4C and D), and greatly elevated LCN2 levels (Fig. 4E) particularly at later time points, indicating that these mice fail to resolve TNBS-induced intestinal inflammation. Chr17FPWD mice had significantly reduced weight loss, disease activity, and histologic damage scores (Fig. 4B, C, D, and F). The other strains exhibited minimal differences compared with B6 (Fig. 4). A summary of all quantitative colitis parameters across all of the consomic strains is provided in Table 2, and additional kinetics of weight loss, disease activity, and LCN2 levels are shown in (Fig. S3A–C). Taken together, these results suggest that PWD-derived loci on Chr1 and Chr2 potently exacerbate colitis, with effects on the chronic and acute phase, respectively.
Figure 4. PWD loci regulate susceptibility to TNBS-induced colitis.
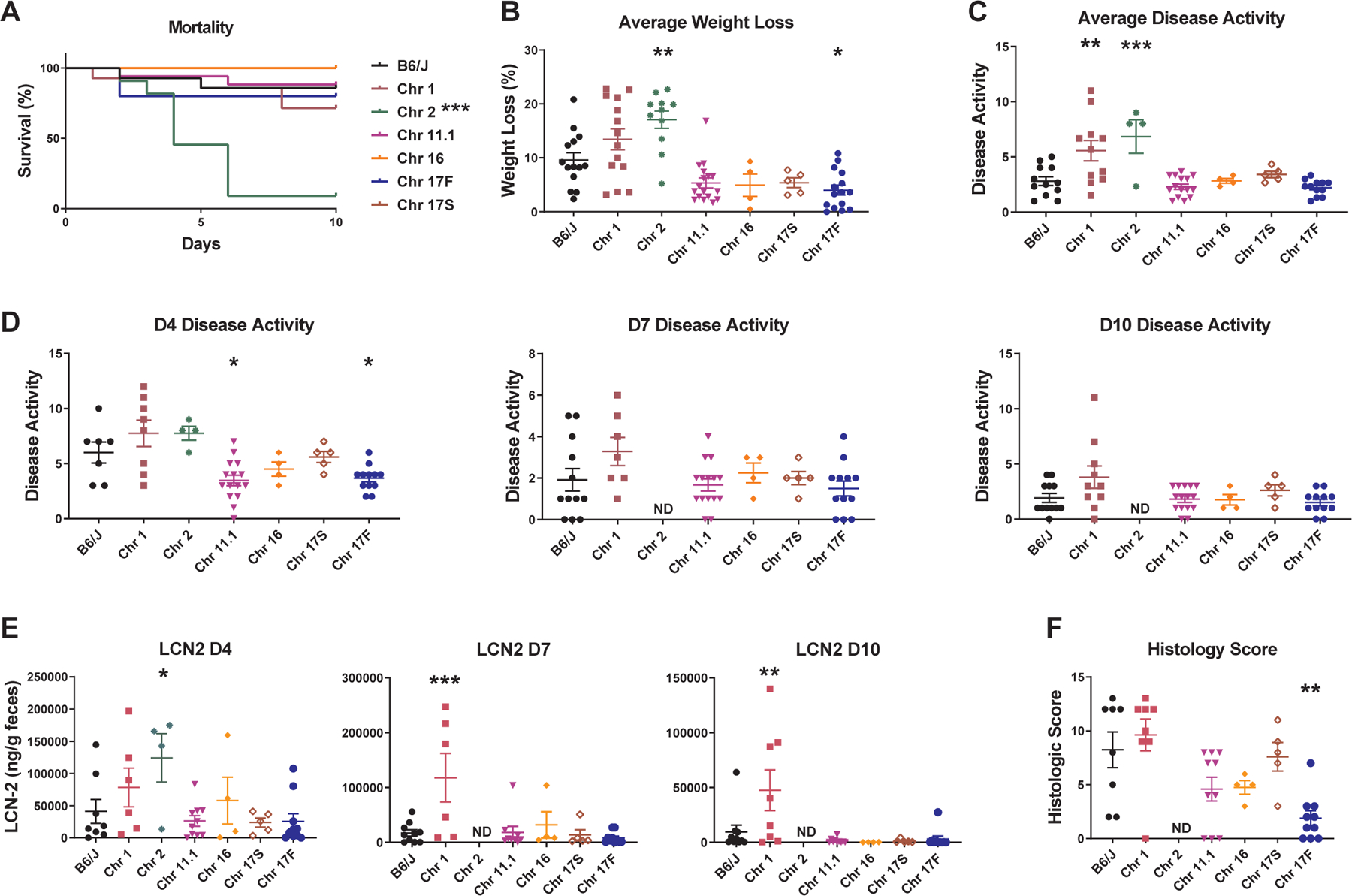
DSS colitis was induced and evaluated in B6.ChrPWD consomic strains and B6 controls as described in Fig. 2. (A) Survival is shown for all mice. (B) Average weight loss was calculated as the average % weight loss across all days that were measured. (C) Average disease activity was calculated as the average disease activity score across time points measured, excluding D0 (D4, D7, D10). (D) Disease activity scores at the indicated time points. (E) LCN2 levels were measured by ELISA at the indicated time points. (F) Colonic tissue was collected from surviving animals on D10 and evaluated by semi-quantitative histology as in Fig. 1. Significance of differences between each consomic strain and B6 controls was calculated using: Mantel-Cox test in (A); ordinary one-way ANOVA with Dunnet’s post-hoc comparisons in (B) (C), and (E); Kruskal-Wallis one-way ANOVA with Dunn’s post-hoc comparisons in (D), and (F), and are indicated as follows: and are indicated as follows: *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001. Numbers of animals studied of each sex are provided in Table 2.
Table 2. Summary of TNBS-induced colitis severity parameters across the consomic panel.
Summary data from Fig. 2 and Fig. 4. Comparisons with B6 controls are summarized as follows: “=”, not significantly different from B6; “↓” significantly lower than B6, “↑”, significantly higher than B6; “ref.”, B6 reference; “ND”, not done. Significance of differences is indicated using asterisks, as in Fig. 4. For measurements with comparisons across multiple time points (disease activity and LCN2), the measurement with highest significance (if any) at any single time point comparison is provided. *EAE susceptibility was determined in previous studies {Bearoff 2015; Bearoff 2016; Krementsov 2018}. Comparisons with B6 controls are summarized as follows: “=”, not significantly different from B6; “↓” significantly lower than B6, “↑”, significantly higher than B6; “ref.”, B6 reference; “ND”, not done.
| Strain | Female N | Male N | Mortality | Weight Loss | Disease Activity | LCN-2 | Histology | *EAE |
|---|---|---|---|---|---|---|---|---|
| B6 | 8 | 6 | ref. | ref. | ref. | ref. | ref. | ref. |
| PWD | 3 | 3 | ↑** | ↑** | ↑**** | ND | ND | ↓ |
| Chr1 | 6 | 8 | = | = | ↑** | ↑*** | = | ↑ |
| Chr2 | 6 | 5 | ↑**** | ↑** | ↑**** | ↑* | ND | ↓ |
| Chr11.1 | 8 | 8 | = | = | ↓* | = | = | ↑ |
| Chr16 | 0 | 4 | = | = | = | = | = | ↑ |
| Chr17S | 0 | 5 | = | = | = | = | = | = |
| Chr17F | 4 | 8 | = | ↓* | ↓* | = | ↓** | ND |
We next compared susceptibility of B6-ChrPWD consomic strains across three different models of inflammatory disease: the TNBS- and DSS-induced colitis models, and the EAE; the latter using our published data 17, 18, 22. Chr1PWD and Chr2PWD strains stand out as having significant phenotypes across all three models. Interestingly, the direction of effect is consistent for Chr1PWD mice, which exhibit more severe disease across all three models. Thus, this chromosome likely carries PWD alleles that enhance inflammation across multiple tissues. In contrast, while Chr2PWD mice have extremely high susceptibility in both colitis models, they are resistant to EAE. Thus, it is likely that PWD alleles on this chromosome impact gut inflammation specifically, and may affect the baseline state of the gut to an inflammation-predisposed state. We tested this idea by measuring baseline LCN2 levels prior to induction of colitis and found that indeed Chr2PWD mice exhibit elevated LCN2 levels in the absence of overt inflammatory stimuli (Fig. S3D), although a histopathological analysis did not reveal any overt signs of inflammation or barrier disruption (data not shown). Taken together, our results suggest that novel PWD-derived alleles on Chr1 and Chr2 regulate gut inflammation, likely by distinct mechanisms.
IBD in the B6.ChrPWD consomic model recapitulates Collaborative Cross genetics
A key recent development for mouse genetics has been the introduction of the Collaborative Cross (CC) systems genetics resource. The CC strains are derived from an eight-way intercross between classic and wild-derived strains (including B6 and PWK/PhJ (PWK); the latter a very close relative of the PWD strain 27). De Villena and colleagues recently identified a CC strain, CC011/Unc, with a high penetrance of spontaneous IBD 28. Subsequent backcross mapping by B6 identified four loci, with the major locus (Ccc1) being mostly PWK-derived, on distal Chr12 28. Our B6.ChrPWD consomic model yields an opportunity to support or refute the candidacy of the Ccc1 locus for IBD susceptibility. To test whether PWK-homologous PWD-derived loci on Chr12 could regulate IBD susceptibility, we tested the susceptibility of Chr12PWD consomic mice to experimental colitis. Of note, the Chr12PWD consomic strain was not part of the panel in our first study (Fig. 3), which had only included strains with significant EAE phenotypes, which Chr12PWD lacked (Tables 1–3). Unlike CC011/Unc, Chr12PWD consomic mice in our facility did not exhibit spontaneous colitis, suggesting that epistatic interactions between Ccc1 and additional loci in CC011/Unc mice are required for spontaneous IBD, a notion supported by the findings of De Villena and colleagues 28. Therefore, DSS treatment in Chr12PWD consomic mice and matched set of B6 control mice was employed to bypass the requirement for those epistatic interactions. Compared with B6 controls, Chr12PWD consomic mice demonstrated significantly higher weight loss and disease severity index, as well as a trend towards higher mortality (Fig. 5A–C). These results suggest that PWK-homologous PWD loci on Chr12 promote experimental IBD, supporting the candidacy of the Ccc1 locus. These results also demonstrate the utility of the B6.ChrPWD consomic model as a parallel and complementary physical mapping approach to the more complex and expensive CC approaches.
Table 3. Comparison between B6.ChrPWD consomic strain susceptibility across two models of experimental IBD and a model of MS.
Susceptibility to TNBS or DSS colitis is indicated as different from B6 if at least two parameters in Table 1 or Table 2 were significantly different from B6, and consistent in direction of change. *EAE susceptibility was determined in previous studies {Bearoff 2015; Bearoff 2016; Krementsov 2018}. Comparisons with B6 controls are summarized as follows: “=”, not significantly different from B6; “↓” significantly lower than B6, “↑”, significantly higher than B6; “ref.”, B6 reference; “ND”, not done.
| Strain | DSS | TNBS | *EAE |
|---|---|---|---|
| B6 | ref. | ref. | ref. |
| PWD | ↓ | ↑ | ↓ |
| Chr1 | ↑ | ↑ | ↑ |
| Chr2 | ↑ | ↑ | ↓ |
| Chr6 | = | ND | ↑ |
| Chr10.3 | = | ND | ↓ |
| Chr11.1 | ↓ | = | ↑ |
| Chr11.3 | = | ND | ↑ |
| Chr12 | ↑ | ND | = |
| Chr15 | = | ND | ↓ |
| Chr16 | = | = | ↑ |
| Chr17S | ↑ | = | = |
| Chr17F | = | ↓ | ND |
Figure 5. B6.Chr12PWD mice are highly susceptible to DSS-induced colitis.
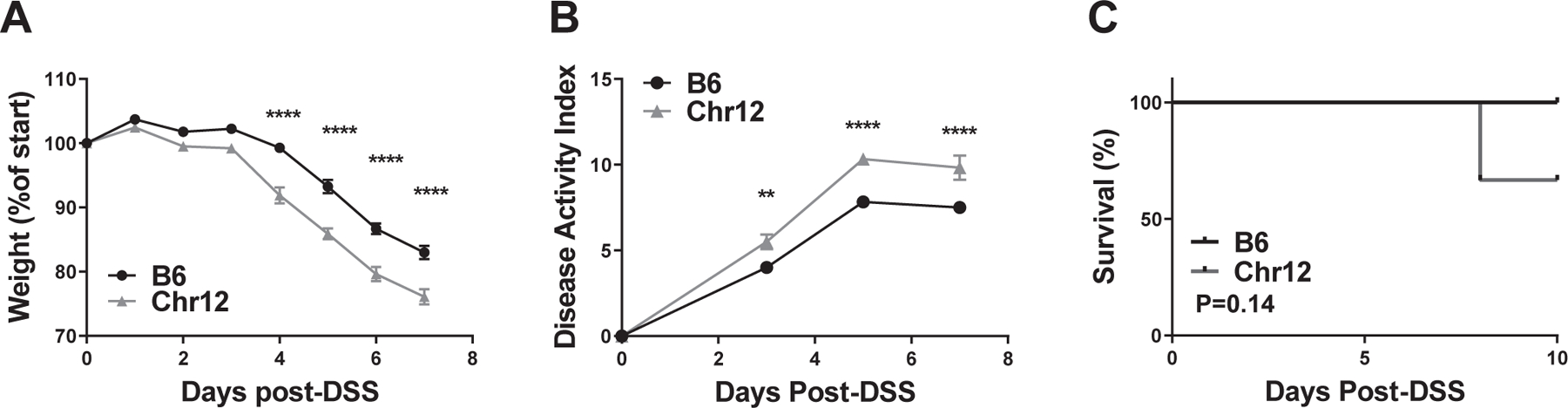
A 5% solution of DSS was administered in drinking water as in Fig. 2, followed by monitoring of weight loss (A), disease activity index (B), and survival (C). Weights and disease activity are shown up until day 7, since after this point data for some mice would be missing due to mortality. Significance of differences were determined by two-way ANOVA with Holm-Sidak post-hoc comparisons in (A) and (B), and by Mantel-Cox test in (C), and are indicated as follows: **, P<0.01; ***, P<0.0001. Numbers of animals studied were as follows: B6 (N=6, 3 males and 3 females), Chr12PWD (N=6, 4 males and 3 females). The B6 animals in this figure represent an independent cohort from those in Fig. 2, with colitis induced simultaneously with Chr12PWD mice.
Identification of candidate genes regulating experimental IBD susceptibility
In order to identify candidate PWD alleles regulating IBD susceptibility in Chr1PWD and Chr2PWD mice, we undertook a multipronged approach (Fig. 6A). We leveraged our published transcriptional profiles across five different types of B6 and PWD immune cells 17, reasoning that IBD is an immune cell-mediated disease, and that differential expression of genes between B6 and PWD is likely to result in functional differences. Five different immune cells types were profiled: CD11b+CD11c+ antigen presenting cells (APCs), CD19+ B cells (Bcells), TCRβ+CD4+CD25− effector T cells (CD4), TCRβ+CD8+ T cells (CD8), and TCRβ+CD4+CD25+ regulatory T cells (Treg) (see Methods and 17). For the first approach (IBD GWAS 1:1 candidates), to increase the relevance to human IBD, we filtered genes differentially expressed between B6 and PWD in a given cell type by published IBD GWAS candidate loci. We selected the largest IBD GWAS meta-analysis to date, which had identified a total of 241 associated loci, which implicate 619 candidate genes based on proximity and linkage disequilibrium to the marker SNPs 9, (Table S1). The size of this gene set is also optimal for our machine learning algorithm 29, below. This analysis identified 8 genes on Chr1, including known immune regulators Cd28, Ptprc, and Slamf8, as well as 10 genes on Chr2, including Cd40, Il2ra, and Itga4 (Fig. 6C).
Figure 6. Identification and prioritization of novel gene candidates associated with IBD susceptibility.
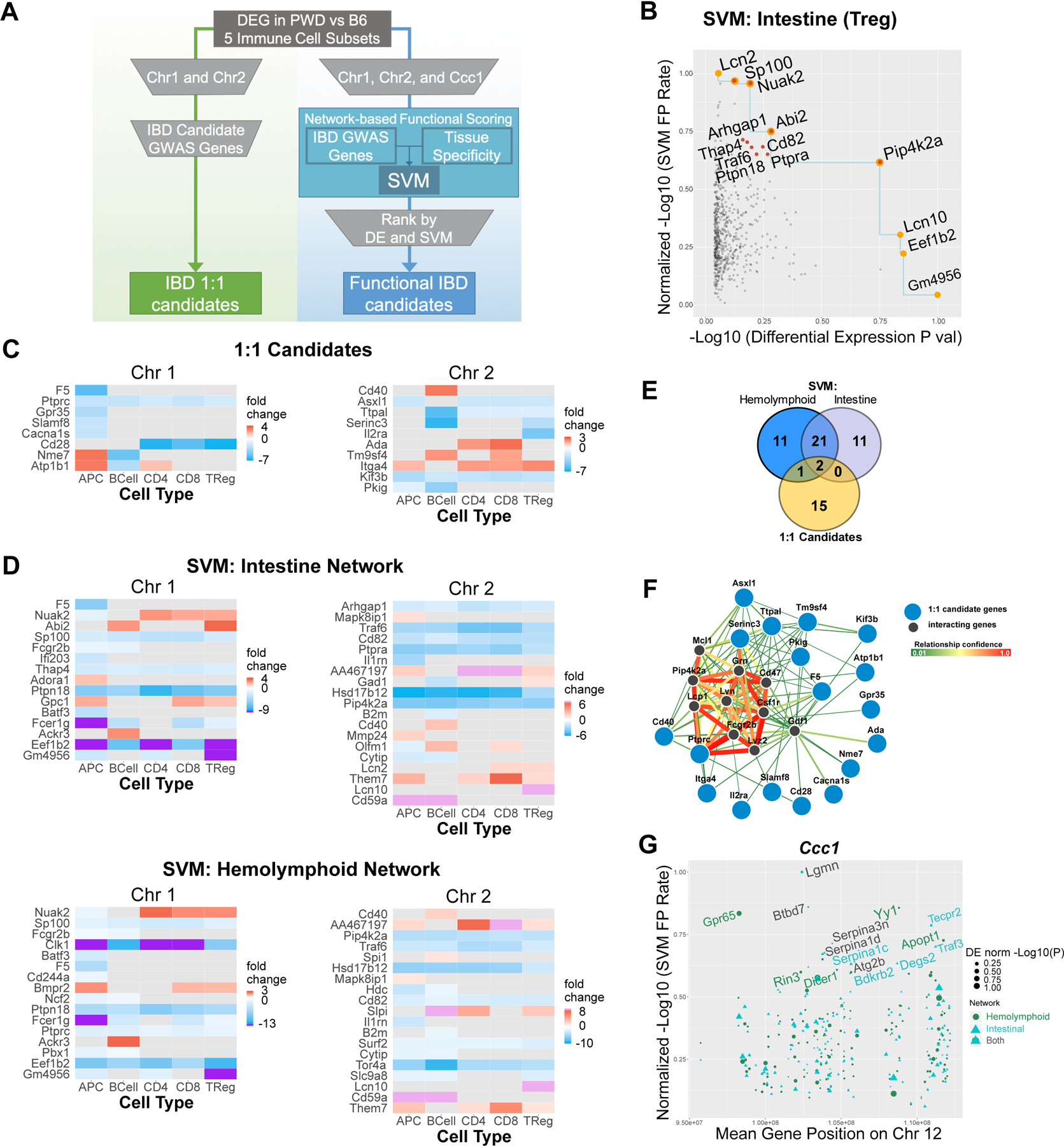
(A) Schematic illustrating the two distinct workflows that were used to identify and prioritize candidate genes. IBD 1:1 candidates were identified by the overlap of highly differentially expressed genes in B6 vs. PWD immune cells and IBD GWAS candidate genes (see C). Scores from tissue-specific “Intestine” and “Hemolymphoid” network SVMs that were trained with IBD GWAS genes were integrated with differential expression to identify functional IBD candidates (see B, D–F). (B) SVM using the intenstine network was integrated with the differentially expressed gene dataset from PWD and B6 Tregs. Significantly differentially expressed genes from chromosome 1 and 2 are plotted by normalized −log10(p-value) on the x-axis and normalized SVM −log10(false positive rate) (FPR)SVM on the y-axis. FPRSVM and p-values were normalized by their maximum value (see Methods). Orange points denote genes located on the Pareto front, represented by the blue line. Red points denote the top ten ranked genes based on the final score, which combines the count of the number of genes that have both a lower −log10(p-value) and −log10(FPR). Orange and red indicates top ranked genes that also fall on the Pareto front. SVMFPR vs. differential expression plots for each of the five immune cell subtypes from the intestine and hemolymphoid network SVMs can be found in Fig. S4. Heatmaps of differential gene expression between PWD and B6 immune cell types (log2(fold change PWD/B6)) for the top candidate genes identified by the 1:1 IBD candidate approach (C) or the SVM approach (D). Purple and pink denote extreme differential expression values, and gray denotes genes whose differential expression was not significant (false discovery rate <0.05). (E) Overlap between genes identified using IBD 1:1 candidate approach and the intestine and hemolymphoid networks SVMs. (F) Functional connectivity of the top 1:1 IBD candidate genes on Chr1 and Chr2 with 10 most functionally connected predicted interactor genes was visualized using the FNTM tool. (G) Genes from the Ccc1 locus are plotted by their genomic location on the x-axis and normalized −log10(FPRSVM) on the y-axis. Each gene has a score from the intestine and hemolymphoid network SVM, and the size of each point indicates normalized significance of differential expression (“DE norm log10(P)”). The names of top ten genes (ranked by SVM score) from each network are annotated.
The approach above is limited by the assumption that PWD alleles of orthologous IBD GWAS genes are themselves regulating susceptibility to experimental IBD in our models. An alternative possibility is that PWD alleles in genes that are functionally related to IBD GWAS genes are responsible. To address the latter possibility, we utilized an agnostic machine learning approach to rank associated genes by functional relatedness to IBD GWAS genes, as we recently published 30. Briefly, we used support vector machine (SVM) classifiers together with the Functional Networks of Tissues in Mouse (FNTM) database 31 to infer the functional connectivity among IBD GWAS genes, i.e. to identify a functional sub-network containing these genes. We trained two SVM classifiers, one with the intestinal system network and another with the hemolymphoid system network, reasoning that IBD risk alleles could influence either gut physiology per se or the immune system more broadly. We then applied the SVM classifier to score genes on mouse Chr1 and Chr2 by their functional association to IBD GWAS genes in either network (Table S2). To further enrich our functional predictions using direct functional data from B6 and PWD immune cells, we used the significance of differential expression between B6 and PWD immune cells (see above) as a second ranking metric, together with the SVM score (Fig. 6B and Fig. S4). The blue line in Fig. 6B indicates the Pareto front of the gene set in this space, and highlights the tradeoff between functional scores and differential expression as candidate ranking metrics. Some highly differentially expressed genes are not strongly functionally associated with IBD GWAS candidates and vice versa (e.g., Lcn2 vs. Lcn10 in Fig. 6B). An ideal candidate gene is a strong candidate on both measures, i.e. located in the top right corner of the Fig. 6B and Fig. S4 (e.g., Pip4k2a in Fig. 6B). To obtain a final, integrated functional score, we ranked every candidate gene by the number of genes on both Chr1 and Chr2 in this space for which the candidate is superior on both measures, i.e. the number of genes for which the candidate is both more strongly differentially expressed and more functionally associated to IBD GWAS genes (see Methods for details; integrated scores are provided in Table S3). The top ten genes by this ranking cluster near the Pareto front toward the upper right corner or the plot (highlighted by red dots in Fig. 6B and Fig. S4). To visualize the expression patterns for these top-ranked genes, we generated differential expression heat maps for the genes that were top-ten candidates in any of the five cell types for both tissues, or genes located on the Pareto front (Fig. 6D). Overall, this approach prioritized a total of 46 unique genes (Table S4), with only three of these genes (Cd40, F5, and Ptprc) overlapping with the 1:1 candidates identified above (see Fig. 6E), suggesting that this approach has identified novel candidate genes with higher functional connectivity compared with the 1:1 approach. Interestingly, this approach identified Lcn2, a known biomarker in colitis used in our studies above, as highly functionally related to the IBD phenotype (Fig. 6B), but with marginal differential expression significance. As a more balanced candidate, this approach also identified the related Lcn10 gene, which not only has a high functional connectivity to IBD, but is also highly differentially expressed between PWD and B6 specifically in T regulatory (Treg) cells (Fig. 6C, D and Fig. S4). A visual example of functional connectivity between 1:1 IBD candidates on Chr1 and Chr2 and highly functionally connected genes (inferred using the FNTM network), including Pip4k2, is shown in Fig. 6F.
Additionally, given that our experiments in Chr12PWD mice (see Fig. 5) supported the candidacy of the Ccc1 locus identified in CC mice, we took advantage of the mapping performed by De Villena and colleagues in their study, which narrowed this interval to 94.8–112.3 Mb on Chr12. Using the network-based functional scoring approach described above, we ranked candidate genes in this interval, identifying Lgmn as the top candidate identified by both network SVMs, and Gpr65 by the hemolymphoid network SVM, with both candidates showing robust differential expression between B6 and PWD (Fig. 6G and Table S2). Taken together, we have used a machine learning approach to augment our own and others’ physical mapping data, providing a manageable list of plausible candidate genes to be pursued in follow-up studies.
Discussion
In this study, we have utilized the genetic diversity of wild-derived PWD mice to map susceptibility loci across two different chemically induced models of IBD. Interestingly, the parental B6 and PWD strains exhibited divergent susceptibility across the two models, with PWD exhibiting low susceptibility to DSS- but high susceptibility to TNBS-induced colitis. These disparate phenotypes likely have to do with the differing nature of these two models, reviewed in detail elsewhere 23, 32. Briefly, DSS treatment models an intestinal epithelial barrier defect, followed by innate immune activation, whereas the hapten TNBS induces an adaptive T cell response against bacterial or self-antigens 32. Additionally, in terms of histopathology, localization, and tissue layers involved, the DSS and TNBS models more closely resemble ulcerative colitis and Crohn’s disease, respectively 23. While there is a large genetic overlap between these two types of IBD in humans, many susceptibility genes are unique to each disease 8, thus it is possible that the differential response of the PWD mice across the two different models reflects disease subtype-specific genes. This intriguing possibility will be addressed in future studies.
Two of our B6.ChrPWD strains, Chr1PWD and Chr2PWD, exhibited markedly enhanced susceptibility across both colitis models. Chr2PWD mice rapidly developed acute inflammation and exhibited very high mortality. It appears that this strain carries alleles that predispose it to intestinal inflammation, possibly due to a genetic defect in intestinal barrier function, a notion that is supported by increased baseline inflammation in this strain (Fig. S3D). In contrast, Chr1PWD mice exhibit enhanced inflammation at later time points, suggestive of chronic inflammation. We hypothesize that this phenotype is likely due to a genetically controlled state of heightened immune responsiveness, which is also consistent with the finding that Chr1PWD mice have markedly enhanced severity of EAE 18, 22. The latter is also consistent with the well-documented genetic overlap between IBD and other immune-mediated diseases such as MS 21. Somewhat surprisingly, while parental PWD mice were resistant to DSS-induced colitis, only one of the consomic strains studied (Chr11.1PWD) showed any evidence of resistance in this model. We suspect that either: 1) the remaining consomic strains not included in our study can capture the DSS resistance loci in parental PWD mice, or 2) that the resistance phenotype in PWD mice is dependent on epistasis between loci on multiple chromosomes.
Augmenting our physical mapping data, we used 2 different gene prioritization approaches to identify plausible candidate genes in our loci of interest. Using the simpler 1:1 IBD GWAS candidate approach, we identified seventeen genes of interest, including Slamf8, Cd40, and Itga4 (see Fig. 6C). With regard to the former gene, a non-synonymous SNP in SLAMF8 was recently associated with IBD susceptibility 9. CD40/Cd40 encodes a costimulatory molecule expressed on the surface of antigen presenting cells, whose neutralization elicits spontaneous colitis in mice 33, similar to how neutralization of costimulatory molecule CTLA-4 results in IBD in humans treated with checkpoint inhibitors for cancer immunotherapy 34. With regard to ITGA4/Itga4, this gene encodes integrin alpha 4, which is known to regulate lymphocyte entry into inflamed gut and CNS tissues, and its blockade by the drug natalizumab is therapeutic in Crohn’s disease and MS 35, 36. Strikingly, recent human studies demonstrate that the IBD risk allele at ITGA4 results in differential expression in activated immune cells 9, similar to our observations in B6 and PWD immune cells, where the PWD Itga4 allele shows higher expression in all immune cell types except B cells (Fig. 6C).
Using the SVM-based prioritization approach, we identified 46 genes of interest as highly functionally related the IBD phenotype and highly differentially expressed in PWD vs. B6 immune cell types. Some of the top ranked genes across multiple cell types included Pip4k2a, Ptpra, and Traf6, all exhibiting strongly downregulated expression in PWD across all 5 cell types. Pip4k2a encodes one of the many enzymes involved in the interconversion of phosphoinositides, cellular membrane lipids involved in a variety of processes, including a key role in inflammation 37, although a role for this particular isoform of this enzyme in inflammation has not yet been established. Ptpra encodes a member of the protein tyrosine phosphatases, which are well documented regulators of T cell receptor signaling 38. Ptpra was recently described to promote inflammation in a model of rheumatoid arthritis 39. Traf6 encodes TRAF6, a key adaptor molecule involved in innate immune signaling in response to bacterial signals 40, consistent with a potential role for this gene in modulating the response against commensal or pathogenic bacteria driving intestinal inflammation. Future studies will address the functional role of the candidates prioritized by our approaches.
There are two important limitations to our candidate gene approach. The first is that it integrates our published gene expression data, which was microarray-based, and thus subject to SNP variation in PWD transcripts that can affect microarray probe binding, since the probes were designed using a B6 reference genome 41. This particularly applies to genes that exhibit apparent downregulation in PWD, and such candidate genes will require additional preliminary validation, e.g. by qRT-PCR, prior to further analysis, although we note that functional connectivity as detected by our SVM algorithm is likely to rule out many such spurious hits. A future possible approach will be to “de-SNP” the data by removing any SNP-containing probes, as recently described for the Affymetrix platforms 41. A second limitation has to do with the input IBD GWAS gene dataset, in which many genes are implicated across a given locus simply by proximity and linkage disequilibrium, resulting in a number of likely false positives. As fine mapping continues to improve GWAS resolution and accuracy, our analyses can incorporate these refined gene lists and improve our prediction accuracy. However, because our SVM approach scores GWAS hits according to how well they cohere with other hits within a functional network, we expect spurious positional candidates to be de-prioritized, while multiple relevant genes in a locus regulated by an intergenic SNP (e.g. a topologically associating domain) can all receive high scores.
The MHC locus in humans regulates susceptibility to multiple autoimmune or inflammatory diseases in humans, including IBD 25, with prominent epistatic interactions 26. In this light, our mapping results on mouse Chr17 are complex, but not unexpected. Chr17SPWD mice (which carry the B6 H-2 (MHC) haplotype, but most of their Chr17 is still PWD-derived; see Fig. S1A) are more susceptible to DSS-, but not TNBS-induced colitis compared with B6 mice. In contrast, Chr17FPWD mice (which carry Chr17 that is fully PWD-derived; see Fig. S1A) are more resistant to TNBS- but not DSS-induced colitis compared with B6 mice. These results suggest the existence of multiple interacting alleles regulating colitis susceptibility on Chr17. Additionally, the stronger potential effect of the PWD-derived H-2 locus in Chr17FPWD mice on TNBS- but not DSS-induced colitis susceptibility may be related to the nature of the model, with TNBS hapten effects being more dependent on an adaptive immune response 23, and thus more dependent on antigen presentation by MHC, which is highly likely to be impacted by polymorphisms in the H-2 locus in Ch17FPWD mice. Lastly, we note that Chr17FPWD mice were imported from a different vivarium, and likely harbor a distinct microbiome, which may interact with the host genetic background to yield differential IBD susceptibility. Future studies can address these possibilities in more detail.
Our work adds to an already extensive literature on genetic mapping of experimental IBD determinants in inbred mice, although the majority of this work has been performed in classic inbred strains. Early studies using spontaneous colitis in IL-10 deficient mice and crosses between the B6 and C3H/HeJBir (C3H) classic inbred strains have mapped numerous loci, although the identity of the candidate genes remains unclear 42. Similar linkage studies with classical inbred strains have been done in the DSS model (B6 ˣ C3H cross) and the TNBS model (B6 ˣ SJL/J), with similar results (reviewed in 43). The studies are subject to the aforementioned limitations of genetic diversity inherent to conventional laboratory strains. To our knowledge, there is only one study to date that utilized genetics of wild-derived mice to map IBD loci. De Villena and colleagues utilized the Collaborative Cross resource to identify a partially inbred CC strain, CC011/Unc, with very high penetrance of spontaneous IBD 28. Subsequent backcross mapping by B6 identified four loci, one of them (Ccc1) mostly PWK-derived, on Chr12, and another (Ccc3) WSB/EiJ-derived locus on Chr1 28. We utilized the B6.Chr12PWD consomic strain to support their association mapping data by physical mapping, supporting a role for PWD/PWK alleles at the Ccc1 locus in promoting IBD susceptibility. Additionally, our SVM prioritization approaches identified several plausible candidate genes in this locus, including Lgmn and Gpr65. The former gene encodes the cysteine protease legumain, which is highly expressed by dendritic cells, and plays a key role for antigen processing and presentation 44, including self-antigens 45. GPR65/Gpr65 encodes a lysosomal pH-sensing G protein-coupled receptor that may be involved in clearance of intracellular bacteria, with a missense variant in this gene associated with increased IBD risk 46. Interestingly, a SNP query (using the Mouse Phenome Database) revealed the PWK allele of Gpr65 results in two missense variants compared with B6. Our study illustrates the potential utility of the PWD-consomic mice as a complementary resource to the CC that can be used for physical mapping and/or direct editing of candidate alleles identified in CC mapping populations, on a uniform (B6) background. On the other hand, future mapping studies of DSS- and TNBS-induced colitis susceptibility loci using the CC resource would provide a logical extension of our current study, and could provide the mapping resolution necessary to identify the candidate genes responsible for the profound and divergent phenotypes described herein.
IBD, like most chronic diseases, has a complex and a multifactorial etiology. Our findings demonstrate that this complexity, particularly the genetic etiology, can be modeled and dissected using mouse models, where environmental and genetic variables can be precisely controlled and manipulated. In this regard, incorporation of genetic diversity from wild-derived mice is instrumental in trying to approximate the immense genetic diversity present in human population. Future approaches will continue to identify novel genetic elements controlling not only IBD susceptibility/incidence, but also various aspects of disease progression and pathology, something that is still lacking in human genetic studies. The results from such animal studies will be integrated with emerging genetic and epidemiologic data from human IBD to generate a comprehensive picture of the complexities underlying IBD etiology, aiming to provide future avenues for therapeutic and prophylactic interventions.
Materials and Methods
Animals
C57BL/6J (B6), PWD/PhJ (PWD), and B6.ChrPWD consomic mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA), then bred and housed in a single room within the vivarium at the Larner College of Medicine at the University of Vermont for five or more generations. The experimental procedures used in this study were approved by the Animal Care and Use Committee of the University of Vermont.
To ensure their correct identity and to enhance rigor and reproducibility of these studies, B6.ChrPWD consomic mice were subjected to genome-wide SNP genotyping using DartMouse genotyping services (Dartmouth College, NH, USA), as previously described by us 22. All mice used in this study were of the expected genotypes, with the following exception. Chr17SPWD mice were found to carry a homozygous B6-derived interval between 30 and 45 Mb on Chr17, encompassing H-2. Chr17FPWD mice with the full PWD-derived Chr17 were generously provided by Dr. Jiri Forejt (Institute of Molecular Genetics of the ASCR, Czech Republic) and housed in the vivarium at UVM for two or more generations prior to experimentation.
Induction and evaluation of colitis
Colitis was induced in age- and sex-matched (when practical, see below) male and female B6, PWD, and B6.ChrPWD consomic mice between 8 and 14 weeks of age, essentially as previously described by us 47. Comparable numbers of males and females were used when available (see Tables 1 and 2), and data for both sexes were pooled. For the DSS-induced colitis model, mice received a 5% solution of DSS (MP Biomedicals, USA) in their drinking water for 5 days, which was replaced by regular drinking water for the subsequent 5 days. Alternatively, for the TNBS-induced colitis model, mice received intracolonic enemas containing 200mg/kg TNBS in 50% ethanol in 50–100 μL. The TNBS dose was weight-adjusted.
The mice were monitored daily for weight loss, stool consistency, and the presence of blood in the stool (either grossly visible or by hemoccult test), which together was used to calculate classic colitis disease activity score 48. Note that we did not always succeed in collection of fecal samples from each individual mouse, thus scores for those mice lacking fecal samples were not determined for a given day of collection. All mice that survived through day 10 of the study were euthanized, their colons were measured for length and thickness, examined at the macroscopic level for evidence of ulceration and adhesion, and assigned a macroscopic score on the basis of this criteria, as previously described. With the exception of histological scoring (see below), the investigators were not blinded to the identity of the consomic strains, although no pre-conceived notion about strain-specific outcomes was present at the start of the experiments. For all comparisons that used statistical tests that made the assumption of equal variance, the standard deviation did not differ significantly (P>0.05) by group, with the exception of LCN2 measurements, for which the standard deviation did differ significantly (P<0.05).
For histological evaluation of colitis severity, mice were euthanized at 4 weeks of age, colons were removed, prepared using the “Swiss roll” technique, fixed overnight in formalin, followed by 70% ethanol. Tissues were paraffin-embedded, sectioned, and stained with H&E at the UVM Medical Center histology laboratory. Histological damage scoring was performed on the basis of a semi-quantitative scoring system, as previously described by our laboratory 49, 50. The following features were considered and scored as follows: extent of destruction of normal mucosal architecture (0 = normal; 3 = maximal damage), presence and degree of cellular infiltration (0 = normal; 3 = maximal infiltration), extent of muscle thickening (0 = normal; 3 = maximal thickness), presence or absence of crypt abscesses (0 = absent; 1 = present) and the presence or absence of goblet cell mucus (0 = absent; 1 = present). Scoring was done by trained lab personnel, blinded to the identity of the samples.
In keeping with the NIH mandate on inclusion of sex as a biological variable in preclinical studies, we attempted to include male and female mice from each strain, when they were available. While some modest differences in weight loss between males and females were observed, these did not consistently extend to other parameters, and more importantly, differ by strain (data not shown), and hence male and female data were pooled to maximize the statistical power to detect genotype effects. In some cases when cohorts consisting of only a single sex were available for initial studies, additional cohorts to balance the sexes were included only in the case when a significant difference or a trend in disease severity was observed, thereby minimizing unnecessary animal use. Hence some strains studied included only a single sex (Chr11.3PWD and Chr15PWD for DSS, and Chr16PWD and Chr17SPWD for TNBS). Sample sizes (N/strain) for experiments were chosen based on pilot experiments in B6 mice and practical considerations such as breeding performance and age-matched cohort availability. No randomization was used, since untreated controls were not part of the experimental design and all mice were treated with DSS or TNBS.
Fecal LCN2 measurements
Fecal LCN2 levels were determined using a commercially available ELISA kit (R&D Systems, cat# DY1857), as follows. Fecal pellets were collected on ice and stored at −80°C. Pellets weighed and PBS with 0.1% Tween-20 was added to achieve 100 mg feces/ml. Silicon carbide beads (BioSpec Products, Bartlesville, OK, USA) were added to enhance homogenization. Samples were homogenized by vortexing for 10 min at 4°C, insoluble material was pelleted by centrifugation, and supernatants were used for ELISA at serial dilutions ranging from 1:10 to 1:20,000.
Candidate gene prioritization and bioinformatics
For candidate gene prioritization, the following two datasets were used. The first was a previously published transcriptomic analysis of differential gene expression between B6 and PWD mice across five different cell types: CD4 T cells (CD45+NK1.1−CD19−TCR+CD4+CD25-); CD8 T cells (CD45+NK1.1−CD19−TCR+CD8+); Treg cells (CD45+NK1.1−CD19–TCR+CD4+CD25+); APCs (CD45+NK1.1–CD19–TCR–CD11b+CD11c+), obtained by fluorescence activated cell sorting from the spleen and lymph nodes, with RNA expression measured by microarray (Illumina Bead Array), as previously described by us 17. The second was a list of 619 candidate genes from IBD GWAS 9.
To generate quantitative scores for functional association with IBD, the IBD GWAS data set and the Functional Networks of Tissues in Mouse (FNTM) were used to train support vector machine classifiers (SVMs) 31. The “Intestine” and “Hemolymphoid” tissue-specific functional genomic networks from FNTM were chosen for training. In these networks, nodes are genes that are connected by weighted links, whose values are continuous between 0 and 1 encoding predicted relatedness. Genes that are functionally relevant to IBD are expected to be functionally related in the FNTM networks to IDB GWAS genes, as the latter have an established association to the disease. The intestine and hemolymphoid SVMs were used to identify sub-networks of genes that are tightly interconnected to IBD GWAS genes in the respective network. Specifically, every gene in the genome was represented by a vector of network weights to each of the 619 IBD GWAS genes. Under the hypothesis that the IBD GWAS genes are more functionally related to each other than they are to random genes, the SVM was trained to distinguish the IBD GWAS genes from an equal-sized random set of genes from the genome. The model then scores how functionally similar any gene in the genome is to the IBD GWAS genes based on its functional connections. In order to average over the stochasticity in the choice of random background genes, we trained 100 SVMs per network and computed a final score by averaging the estimated false positive rate (FPR) from each model.
The trained SVMs were used to score each positional candidate gene on Chr1, Chr2, and the Ccc1 locus. The final output of this analysis for each gene is a pair of FPR values, one for each tissue network, that encodes how strongly related a gene is to the IBD GWAS genes. All machine learning was carried out as previously described 30.
To identify genes that had simultaneously high functional scores and differential expression in immune cell subtypes between B6 and PWD mice, –log10(FPRi) vs. –log10(pi) were plotted together, where FPRi and pi are the SVM FPR and differential expression p-value for the ith gene. Plots combining Chr 1 and 2 were generated for each immune cell type for both networks. For visualization, all FPR and p-values are normalized by their smallest value so that the highest −log10(FPRi) and −log10(pi) values equal one. The Pareto front marks the tradeoff between the two axes, and genes that lie toward the top-right corner are both strongly functionally related to IBD GWAS genes and differentially expressed. A combined final gene score, Si, is defined as follows:
A gene’s final score is, therefore, the count of the number of other genes that have both a higher (worse) FPR and differential expression p-value. A final gene score was computed for every gene on Chr 1 and Chr 2 for each tissue network and each immune cell type, yielding a total of ten scores. Genes that were either in the top ten list for at least one cell type or on the Pareto front (or both) were ranked highly. Heat maps of differential expression fold change were plotted for top-ranked genes and were ordered by their maximum final score across all cell types.
A plot integrating SVM FPR values, genomic location, and the maximum differential expression p-value across all cell types was generated for genes on the Ccc1 locus with both networks overlaid on the plot. The ten genes with the highest SVM scores from each network were selectively identified.
Supplementary Material
Funding and acknowledgements
This work was supported by the following grants: NIH/NINDS R01 NS097596, NIH/NIAID R21 AI145306, and VCIID COBRE Pilot Project award to DNK (supported by NIH/NIGMS grant P30 GM118228; PI: Ralph Budd); NIH NLM R21 LM012615 to ALT and JMM; and NIH DK113800 to GMM.
Footnotes
Code availability
All R code used for the SVM analysis can be accessed at https://github.com/MahoneyLabGroup/IBD.
The authors declare no conflict of interest.
References
- 1.Engel MA, Neurath MF. New pathophysiological insights and modern treatment of IBD. Journal of gastroenterology 2010; 45(6): 571–83. [DOI] [PubMed] [Google Scholar]
- 2.Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol 2014; 14(5): 329–42. [DOI] [PubMed] [Google Scholar]
- 3.Farmer MA, Sundberg JP, Bristol IJ, Churchill GA, Li R, Elson CO et al. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc Natl Acad Sci U S A 2001; 98(24): 13820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ermann J, Garrett WS, Kuchroo J, Rourida K, Glickman JN, Bleich A et al. Severity of innate immune-mediated colitis is controlled by the cytokine deficiency-induced colitis susceptibility-1 (Cdcs1) locus. Proc Natl Acad Sci U S A 2011; 108(17): 7137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sundberg JP, Elson CO, Bedigian H, Birkenmeier EH. Spontaneous, heritable colitis in a new substrain of C3H/HeJ mice. Gastroenterology 1994; 107(6): 1726–35. [DOI] [PubMed] [Google Scholar]
- 6.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet 2010; 42(12): 1118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet 2011; 43(3): 246–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491(7422): 119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017; 49(2): 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGovern DP, Kugathasan S, Cho JH. Genetics of Inflammatory Bowel Diseases. Gastroenterology 2015; 149(5): 1163–1176 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon H, Trier Moller F, Andersen V, Harbord M. Heritability in inflammatory bowel disease: from the first twin study to genome-wide association studies. Inflammatory bowel diseases 2015; 21(6): 1428–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H, Bell TA, Churchill GA, Pardo-Manuel de Villena F. On the subspecific origin of the laboratory mouse. Nat Genet 2007; 39(9): 1100–7. [DOI] [PubMed] [Google Scholar]
- 13.Yang H, Wang JR, Didion JP, Buus RJ, Bell TA, Welsh CE et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet 2011; 43(7): 648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011; 477(7364): 289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gregorova S, Forejt J. PWD/Ph and PWK/Ph inbred mouse strains of Mus m. musculus subspecies--a valuable resource of phenotypic variations and genomic polymorphisms. Folia biologica 2000; 46(1): 31–41. [DOI] [PubMed] [Google Scholar]
- 16.Gregorova S, Divina P, Storchova R, Trachtulec Z, Fotopulosova V, Svenson KL et al. Mouse consomic strains: Exploiting genetic divergence between Mus m musculus and Mus m domesticus subspecies. Genome Res 2008; 18(3): 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bearoff F, Del Rio R, Case LK, Dragon JA, Nguyen-Vu T, Lin CY et al. Natural genetic variation profoundly regulates gene expression in immune cells and dictates susceptibility to CNS autoimmunity. Genes and immunity 2016; 17(7): 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bearoff F, Case LK, Krementsov DN, Wall EH, Saligrama N, Blankenhorn EP et al. Identification of Genetic Determinants of the Sexual Dimorphism in CNS Autoimmunity. PLoS One 2015; 10(2): e0117993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blankenhorn EP, Butterfield R, Case LK, Wall EH, del Rio R, Diehl SA et al. Genetics of experimental allergic encephalomyelitis supports the role of T helper cells in multiple sclerosis pathogenesis. Ann Neurol 2011; 70(6): 887–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baranzini SE, Oksenberg JR. The Genetics of Multiple Sclerosis: From 0 to 200 in 50 Years. Trends Genet 2017; 33(12): 960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cotsapas C, Voight BF, Rossin E, Lage K, Neale BM, Wallace C et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet 2011; 7(8): e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krementsov DN, Asarian L, Fang Q, McGill MM, Teuscher C. Sex-Specific Gene-by-Vitamin D Interactions Regulate Susceptibility to Central Nervous System Autoimmunity. Front Immunol 2018; 9: 1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc 2007; 2(3): 541–6. [DOI] [PubMed] [Google Scholar]
- 24.Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One 2012; 7(9): e44328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goyette P, Boucher G, Mallon D, Ellinghaus E, Jostins L, Huang H et al. High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat Genet 2015; 47(2): 172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Wei Z, Cardinale CJ, Gusareva ES, Van Steen K, Sleiman P et al. Multiple Epistasis Interactions Within MHC Are Associated With Ulcerative Colitis. Front Genet 2019; 10: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 2004; 36(11): 1133–7. [DOI] [PubMed] [Google Scholar]
- 28.Rogala AR, Morgan AP, Christensen AM, Gooch TJ, Bell TA, Miller DR et al. The Collaborative Cross as a resource for modeling human disease: CC011/Unc, a new mouse model for spontaneous colitis. Mamm Genome 2014; 25(3–4): 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan Y, Gorenshteyn D, Burmeister M, Wong AK, Schimenti JC, Handel MA et al. Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput Biol 2012; 8(9): e1002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyler AL, Raza A, Krementsov DN, Case LK, Huang R, Ma RZ et al. Network-Based Functional Prediction Augments Genetic Association To Predict Candidate Genes for Histamine Hypersensitivity in Mice. G3 (Bethesda) 2019; 9(12): 4223–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goya J, Wong AK, Yao V, Krishnan A, Homilius M, Troyanskaya OG. FNTM: a server for predicting functional networks of tissues in mouse. Nucleic acids research 2015; 43(W1): W182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valatas V, Bamias G, Kolios G. Experimental colitis models: Insights into the pathogenesis of inflammatory bowel disease and translational issues. Eur J Pharmacol 2015; 759: 253–64. [DOI] [PubMed] [Google Scholar]
- 33.Joyce-Shaikh B, Cua DJ, Bauché D. Induction and Analysis of Anti-CD40-induced Colitis in Mice. Bio-protocol 2019; 9(3): e3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coutzac C, Adam J, Soularue E, Collins M, Racine A, Mussini C et al. Colon Immune-Related Adverse Events: Anti-CTLA-4 and Anti-PD-1 Blockade Induce Distinct Immunopathological Entities. J Crohns Colitis 2017; 11(10): 1238–1246. [DOI] [PubMed] [Google Scholar]
- 35.Pagnini C, Arseneau KO, Cominelli F. Natalizumab in the treatment of Crohn’s disease patients. Expert Opin Biol Ther 2017; 17(11): 1433–1438. [DOI] [PubMed] [Google Scholar]
- 36.Kalincik T, Brown JWL, Robertson N, Willis M, Scolding N, Rice CM et al. Treatment effectiveness of alemtuzumab compared with natalizumab, fingolimod, and interferon beta in relapsing-remitting multiple sclerosis: a cohort study. Lancet Neurol 2017; 16(4): 271–281. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki T, Takasuga S, Sasaki J, Kofuji S, Eguchi S, Yamazaki M et al. Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res 2009; 48(6): 307–43. [DOI] [PubMed] [Google Scholar]
- 38.Stanford SM, Rapini N, Bottini N. Regulation of TCR signalling by tyrosine phosphatases: from immune homeostasis to autoimmunity. Immunology 2012; 137(1): 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanford SM, Svensson MN, Sacchetti C, Pilo CA, Wu DJ, Kiosses WB et al. Receptor Protein Tyrosine Phosphatase alpha-Mediated Enhancement of Rheumatoid Synovial Fibroblast Signaling and Promotion of Arthritis in Mice. Arthritis Rheumatol 2016; 68(2): 359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dainichi T, Matsumoto R, Mostafa A, Kabashima K. Immune Control by TRAF6-Mediated Pathways of Epithelial Cells in the EIME (Epithelial Immune Microenvironment). Front Immunol 2019; 10: 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quigley D Equalizer reduces SNP bias in Affymetrix microarrays. BMC Bioinformatics 2015; 16: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keubler LM, Buettner M, Hager C, Bleich A. A Multihit Model: Colitis Lessons from the Interleukin-10-deficient Mouse. Inflammatory bowel diseases 2015; 21(8): 1967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buettner M, Bleich A. Mapping colitis susceptibility in mouse models: distal chromosome 3 contains major loci related to Cdcs1. Physiol Genomics 2013; 45(20): 925–30. [DOI] [PubMed] [Google Scholar]
- 44.Dall E, Brandstetter H. Structure and function of legumain in health and disease. Biochimie 2016; 122: 126–50. [DOI] [PubMed] [Google Scholar]
- 45.Manoury B, Mazzeo D, Fugger L, Viner N, Ponsford M, Streeter H et al. Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol 2002; 3(2): 169–74. [DOI] [PubMed] [Google Scholar]
- 46.Lassen KG, McKenzie CI, Mari M, Murano T, Begun J, Baxt LA et al. Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity 2016; 44(6): 1392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spohn SN, Bianco F, Scott RB, Keenan CM, Linton AA, O’Neill CH et al. Protective Actions of Epithelial 5-Hydroxytryptamine 4 Receptors in Normal and Inflamed Colon. Gastroenterology 2016; 151(5): 933–944 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Laboratory investigation; a journal of technical methods and pathology 1993; 69(2): 238–49. [PubMed] [Google Scholar]
- 49.Linden DR, Chen JX, Gershon MD, Sharkey KA, Mawe GM. Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol 2003; 285(1): G207–16. [DOI] [PubMed] [Google Scholar]
- 50.Raza A, Crothers JW, McGill MM, Mawe GM, Teuscher C, Krementsov DN. Anti-inflammatory roles of p38alpha MAPK in macrophages are context dependent and require IL-10. J Leukoc Biol 2017; 102(5): 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.