

Abstract
Introduction:
As a consequence of their small size, high stability and high affinity, single domain antibody fragments (sdAb) are appealing targeting vectors for radiopharmaceutical development. With sdAbs binding to internalizing receptors like HER2, residualizing prosthetic agents can enhance tumor retention of radioiodine, which until now has been done with random labeling approaches. Herein we evaluate a site-specific strategy utilizing a radioiodinated, residualizing maleimido moiety and the anti-HER2 sdAb 5F7 bearing a GGC tail for conjugation.
Methods:
Maleimidoethyl 3-(guanidinomethyl)-5-iodobenzoate ([131I]MEGMB) and its N-succinimidyl ester analogue, iso-[125I]SGMIB, were labeled by halodestannylation and conjugated with 5F7GGC and 5F7, respectively. Radiochemical purity, immunoreactivity and binding affinity were determined. Paired-label experiments directly compared iso-[125I]SGMIB-5F7 and [131I]MEGMIB-5F7GGC with regard to internalization/residualization and affinity on HER2-expressing SKOV-3 ovarian carcinoma cells as well as biodistribution and metabolite distribution in athymic mice with subcutaneous SKOV-3 xenografts.
Results:
[131I]MEGMIB-5F7GGC had an immunoreactivity of 81.3% and Kd = 0.94 ± 0.27 nM. Internalization assays demonstrated high intracellular trapping for both conjugates, For example, at 1 h, intracellular retention was 50.30 ± 3.36% for [131I]MEGMIB-5F7GGC and 55.95 ± 3.27% for iso-[125I]SGMIB-5F7, while higher retention was seen for iso-[125I]SGMIB-5F7 at later time points. Peak tumor uptake was similar for both conjugates (8.35 ± 2.66% ID/g and 8.43 ± 2.84% ID/g for iso-[125I]SGMIB-5F7 and [131I]MEGMIB-5F7GGC at 1 h, respectively); however, more rapid normal tissue clearance was seen for [131I]MEGMIB-5F7GGC, with a 2-fold higher tumor-to-kidney ratio and a 3-fold higher tumor-to liver ratio compared with co-injected iso-[125I]SGMIB-5F7. Consisted with this, generation of labeled catabolites in the kidneys was higher for [131I]MEGMIB-5F7GGC.
Conclusion:
[131I]MEGMIB-5F7GGC offers similar tumor targeting as iso-[125I]SGMIB-5F7 but with generally lower normal tissue uptake.
Advances in knowledge and implication for patient care:
The site specific nature of the [131I]MEGMIB reagent may facilitate clinical translation, particularly for sdAb with compromised affinity after random labeling.
Keywords: Site-specific labeling, single domain antibody fragment, nanobody, VHH, radioiodination
1. Introduction
The human epidermal growth factor receptor type 2 (HER2) is a 185 kDa cell surface protein that is overexpressed in about 20–25% of breast cancers and other malignancies including ovarian cancer [1–3]. High levels of HER2 expression are generally associated with aggressiveness and metastasis, leading to a poor prognosis. Hence, many HER2-targeted therapeutic agents have been developed and widely applied clinically, exemplified by the use of trastuzumab (Herceptin®, Genentech) to treat patients with HER2-expressing cancers. Although many patients respond well, others do not, leading to efforts to enhance efficacy by using this monoclonal antibody (mAb) as a delivery vehicle for targeted radionuclide therapy (TRT). For example, trastuzumab conjugates with β-particle emitting radionuclides such as 177Lu [4, 5], 90Y [6], 186Re [7], 188Re [8] and 131I [9] have been evaluated as potential agents for TRT. However, the large size of intact mAbs is not ideal for TRT because slow normal tissue clearance leads to maximum tolerated doses that are not sufficient for adequate tumor control.
Single domain antibody fragments (sdAbs), also referred to as nanobodies or VHH (variable domain of heavy chain-only antibody), are proteins that retain the molecular specificity of mAbs but have a molecular weight of 13–15 kDa, an order of magnitude smaller than intact mAbs. Several HER2-specific sdAbs have been shown to possess high receptor binding affinity, fast normal tissue clearance, and the capability of more homogeneous delivery to tumors than trastuzumab [10–14]. These properties make anti-HER2 sdAbs an attractive platform for developing TRT agents. Our own efforts have focused on 5F7, an sdAb that binds to the same epitope on Domain IV of the HER2 extracellular domain as trastuzumab [15]. With regard to radionuclide, the exceedingly high kidney uptake and retention observed with radiometals [14, 16] led us to explore radioiodines for this purpose. Moreover, an additional advantage of this strategy is that 131I is a true theranostic, and other radioiodines are available with properties suitable for PET imaging (124I), SPECT imaging (123I), and Auger electron TRT (123I).
Particularly for TRT applications, prolonging the residence time of the radionuclide in the tumor is of critical importance. Residualizing prosthetic agents for protein labeling are designed to facilitate the intracellular trapping of radiolabeled catabolites after receptor mediated internalization, a process that occurs extensively with 5F7 sdAb [17]. The rapid cellular internalization of this and other receptor targeted sdAbs can be an advantage for TRT provided that effective trapping of radioactivity can be achieved without concomitantly increased normal tissue activity levels particularly in the kidneys. Of the prosthetic agents evaluated to date, N-succinimidyl 3-guanidinomethyl-5-[131I]iodobenzoate (iso-[131I]SGMIB) provides many of the properties desired for TRT including enhanced tumor uptake, in vivo stability against dehalogenation, and rapid clearance from normal tissues including the kidneys [18]. Nonetheless, there are two areas for improvement of the iso-[131I]SGMIB-5F7 reagent. First, although renal activity levels for iso-[131I]SGMIB-5F7 are lower than those observed with other labeling methods [12, 17, 18], and other sdAbs labeled with radiometals [14, 16], the kidneys are still the likely dose limiting organ. Thus, a prosthetic agent with similar residualizing properties as iso-[131I]SGMIB but with lower kidney levels would be desirable. And second, the random nature of N-hydroxysuccinimido (NHS active ester) conjugation chemistry could result in the modification of any of the five lysine residues on 5F7 sdAb molecule including one in its CDR2 region, which could be involved in HER2 binding.
Site-specific radiolabeling offers a solution to the second issue because it allows for precise chemical modification of an sdAb that can be designed to avoid damaging modification to amino acids involved in receptor recognition. Moreover, site-specific labeling also can provide a single product after radiolabeling, a major advantage for future clinical translation. Site-specific conjugation methods based on the thiol group on cysteine residues have become dominant in antibody drug conjugate (ADC) development [19–23]. Conjugation via Michael addition of thiol to maleimide is one of the most common methods for cysteine based site-specific conjugation, and its application in sdAbs has been reported previously [24–27].
In this study, we developed a novel residualizing prosthetic agent, maleimidoethyl 3-(guanidinomethyl)-5-iodobenzoate (MEGMB) for site-specific radioiodination of 5F7GGC, a previously described version of 5F7 bearing a C-terminal GGC tail [15]. It was designed to contain the iso-SGMIB motif but with the NHS ester replaced by a maleimido moiety for thiol conjugation. Radioiodinated MEGMIB, obtained by the radioiododestannylation of a tin precursor, was conjugated to sdAb 5F7GGC to provide [131I]MEGMIB-5F7GGC (Figure 1). The binding affinity, cellular internalization and tumor targeting capacity of [131I]MEGMIB-5F7GGC were evaluated in HER2-expressing preclinical models in direct comparison to iso-[125I]SGMIB-5F7. In this way, we could directly compare the characteristics of an sdAb subjected to NHS random labeling and site-specific maleimido labeling in paired label format.
Figure 1.
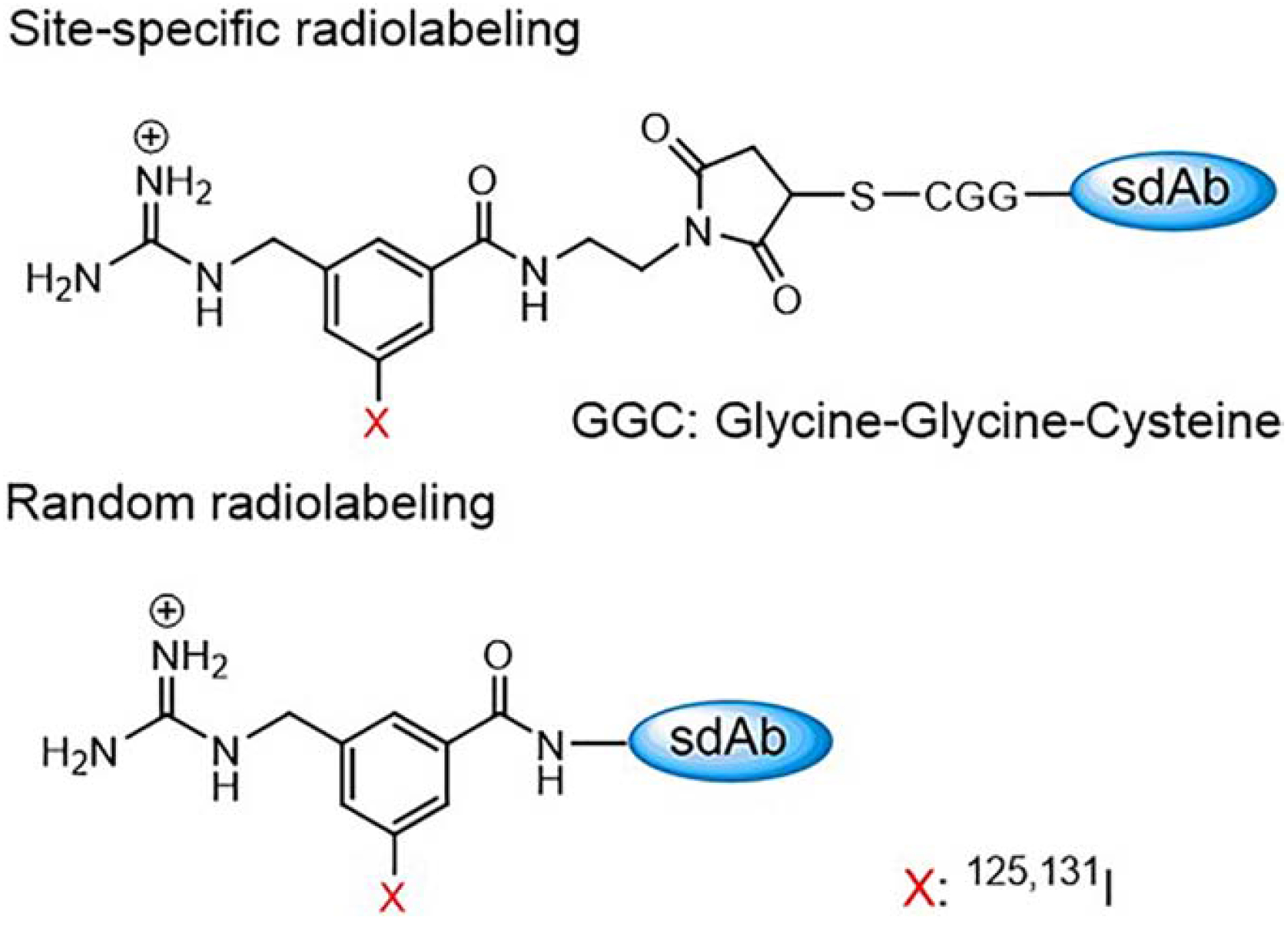
Site-specific radiolabeling of the anti-Her2 sdAb 5F7GGC on the cysteine tail and the random radiolabeling of sdAb 5F7 on the lysine residues.
2. Materials and methods
2.1. General
The reagents used in this work were purchased from Sigma-Aldrich except as noted below. Sodium [125I]iodide (629 GBq)/mg) and sodium [131I]iodide (185 GBq/mg) each in 0.1 M NaOH were obtained from Perkin Elmer Life and Analytical Sciences (Boston, MA). The synthesis iso-[125/131I]SGMIB and conjugation of this prosthetic agent to 5F7 sdAb were performed as reported previously [17, 18]. The anti-HER2 mAb, trastuzumab (Roche/Genentech), was obtained from the Duke University Medical Center Pharmacy. Normal phase column chromatography was performed with the Biotage Isolera chromatography system (Charlotte, NC) using their prepacked columns. High performance liquid chromatography (HPLC) was conducted using two Agilent 1260 Infinity systems equipped with a 1260 Infinity Multiple Wavelength Detector (Santa Clara, CA). For monitoring radioactivity, one system was connected to a Dual Scan-RAM flow activity detector/TLC scanner and the other to a Flow-RAM detector (Lablogic, Tampa, Florida); both the HPLC and the gamma detectors were controlled by LabLogic Laura software. Hydrophobic Interaction Chromatography (HIC) HPLC for the separation of sdAb 5F7 conjugates utilized a TSKgel Butyl-NPR 2.5 μm particle-size, 4.6 mm ID × 10 cm column (Tosoh Bioscience, Montgomeryville, PA) eluted as follows: Mobile Phase A, 25 mM sodium phosphate buffer and 1.5 M ammonium sulfate (pH=7.0); Mobile phase B, 25 mM sodium phosphate buffer and 2-propanol (80% v/v sodium phosphate buffer and 20% v/v 2-propanol) (pH=7.0). The column was eluted with 0 to 100% B in 20 min at a flow rate of 0.5 mL/min. A size-exclusion HPLC system was used for the purification and identification of 5F7GGC sdAb conjugate. For this, an Agilent PL Multisolvent 20 column was eluted with pure Milli-Q® water as the mobile phase. Disposable PD-10 desalting columns for gel filtration were purchased from GE Healthcare (Piscataway, NJ). Radioactivity levels were measured using a CRC-7 dose calibrator (Capintec, Pittsburgh, PA) for higher activities, and either an LKB 1282 (Wallac, Finland) or a Perkin Elmer Wizard II (Shelton, CT) automated gamma counter for lower activities. Proton NMR spectra were obtained on a 400 MHz spectrometer (Varian/Agilent; Inova), and chemical shifts are reported in δ units using the residual solvent peak as a reference. Mass spectra were obtained using an Advion (Ithaca, NY) ExpressionL CMS LC-MS System attached to an Agilent 1260 infinity HPLC, like the system described above. This mass spectrometer has the capability of determining molecular weights of compounds directly from TLC plates (Plate Express), and by ESI, atmospheric pressure chemical ionization (APCI), and atmospheric solids analysis probe (ASAP). High resolution mass spectra were obtained by flow injection into an Agilent 6224 TOF LC/MS system under positive ion mode. Protein mass spectra were obtained using a Bruker Autoflex Speed LRF MALDI-TOF system.
2.2. Anti-HER2 single domain antibody fragments
Production, purification and characterization details for anti-HER2 sdAb 5F7GGC have been reported [15] as have those for the 5F7 construct without the GGC tail [18]. The anti-HER2 sdAb 5F7 and 5F7GGC, obtained as a gift from Dr. Hilde Revets (formerly of Ablynx NV Ghent, Belgium), were selected from phage libraries derived from llamas immunized with SKBR-3 human breast carcinoma cells [15]. The sdAb 5F7 lacking the GGC tail was present exclusively as a monomer while the sdAb 5F7GGC was present as a mixture of dimer and monomer (~30% dimer).
2.3. Synthesis of intermediates and final unlabeled prosthetic agents
2.3.1. Synthesis of 2-(trimethylsilyl)ethyl 3-bromo-5-methylbenzoate (1).
A mixture of 3-bromo-5-methylbenzoic acid (5.0 g, 1 eq, 23 mmol), 2-(trimethylsilyl)ethan-1-ol (3.0 g, 1.1 eq, 26 mmol), and 4-N,N-dimethylaminopyridine (DMAP) (0.28 g, 0.1 eq, 2.3 mmol) in dichloromethane (100 mL) was stirred at 20°C for 5 min. The condensing agent, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (5.8 g, 1.3 eq, 30 mmol) was added and the mixture was stirred at 25°C for 17 h. The reaction mixture was partitioned between water and dichloromethane. The dichloromethane layer was separated, dried over anhydrous MgSO4 and the solvent was evaporated. The resultant crude mixture was purified by normal phase chromatography using a 50 g BIOTAGE SNAP ULTRA column and 9:1 hexanes:EtOAc as the mobile phase to afford 2-(trimethylsilyl)ethyl 3-bromo-5-methylbenzoate (5.5 g, 75%) as an oil: 1H NMR (CDCl3): δ 0.08 (s, 9H), 1.11 (t, 2H), 2.35 (s, 3H), 4.39 (t, 2H), 7.47 (s, 1H), 7.67 (s, 1H), 7.87 (s, 1H). HRMS Calcd for C13H19BrO2Si (M + Na)+: 337.0230. Found: 337.0223.
2.3.2. Synthesis of 2-(trimethylsilyl)ethyl 3-bromo-5-(hydroxymethyl)benzoate (2).
Azobisisobutyronitrile (AIBN) (0.26 g, 0.1 eq, 1.6 mmol) was added to a refluxing mixture of 1 (5.0 g, 1 eq, 16 mmol), N-bromosuccinimide (NBS) (4.2 g, 1.5 eq, 24 mmol) in 1,2-dichloroethane (75 mL). The reaction was allowed to proceed for 2 h with TLC monitoring of its progress; extra NBS was added as needed. Solvent was evaporated and ether (100 mL) was added to the resultant oil. The precipitate that formed was filtered off and ether from the filtrate was evaporated to obtain an oil. This crude oil and sodium acetate (5.2 g, 4.0 eq, 63 mmol) were taken in DMF (50 mL) and the resultant heterogeneous mixture was stirred at 60°C for 16 h. The reaction mixture was cooled to 20°C and partitioned between ethyl acetate and water. The ethyl acetate layer was separated, dried over MgSO4, and concentrated to give an oil. It was dissolved in methanol (100 mL) and after cooling the solution to 0–5°C, gaseous ammonia was bubbled through it for 3 min. The sealed reaction vessel was fitted with an argon balloon, allowed to warm to 20°C and then stirred for 16 h. Methanol was evaporated and the resultant residue was subjected to silica gel chromatography using a 50 g BIOTAGE SNAP ULTRA column and 5:1 hexanes:EtOAc as the mobile phase to afford 2-(trimethylsilyl)ethyl 3-bromo-5-(hydroxymethyl)benzoate (2.7 g, 51%) as an off-white solid: 1H NMR (CDCl3): δ 0.08 (s, 9H), 1.14 (t, 2H), 4.41 (t, 2H), 4.74 (s, 2H), 7.72 (s, 1H), 7.94 (s, 1H), 8.08 (s, 1H). HRMS Calcd for C13H19BrO3Si (M + Na)+ : 353.0179. Found: 353.0173.
2.3.3. Synthesis of 2-(trimethylsilyl)ethyl 3-(hydroxymethyl)-5-(trimethylstannyl)benzoate (3).
A mixture of 2 (1.2 g, 1 eq, 3.6 mmol), and hexamethylditin (3.6 g, 2.3 mL, 3.0 eq, 11 mmol) in dioxane (50 mL) was heated at 100°C for 15 min. Bis(triphenylphosphine)palladium(II) chloride (0.25 g, 0.1 eq, 0.36 mmol) was added and the mixture was stirred for 1 h at 100°C; progress of the reaction was monitored by TLC (4:1 hexanes:EtOAc, Rf of 2 = 0.5, Rf of 3 = 0.6). The reaction mixture was poured on ice and the crude product was extracted with EtOAc. The black-colored ethyl acetate extract containing some particulates was filtered through a Celite bed and the filter cake was washed with EtOAc. The filtrate was dried over MgSO4 and the ethyl acetate was evaporated. The resultant crude oil was purified using a 25 g SNAP Ultra Biotage column and 5:1 hexanes:EtOAc as the mobile phase to give 2-(trimethylsilyl)ethyl 3-(hydroxymethyl)-5-(trimethylstannyl)benzoate (1.2 g, 80%) as a colorless oil: 1H NMR (CDCl3): δ 0.08 (s, 9H), 0.29 (s, 9H), 1.14 (t, 2H), 4.41 (t, 2H), 4.74 (s, 2H), 7.54 (s, 1H), 7.95 (s, 1H), 8.03 (s, 1H). HRMS Calcd for C16H28O3Si[120Sn] (M + Na)+: 439.0722. Found: 439.0722.
2.3.4. Synthesis of 2-(trimethylsilyl)ethyl 3-((2,3-bis(tert-butoxycarbonyl)guanidino)-methyl)-5-(trimethylstannyl)benzoate (4).
Diisopropyl azodicarboxylate (DIAD) (0.80 g, 0.77 mL, 1.5 eq, 4.0 mmol) was added to a cooled (0–5°C) solution of 3 (1.1 g, 1 eq, 2.6 mmol), 1,3-bis(tertbutoxycarbonyl)guanidine (0.76 g, 1.1 eq, 2.9 mmol), and triphenylphosphine (1.0 g, 1.5 eq, 4.0 mmol) in tetrahydrofuran (50 mL). The reaction was allowed to proceed at 20°C for 16 h. The reaction mixture was concentrated to dryness and the resultant material purified using a 50 g Biotage SNAP ULTRA column and 5:1 hexanes:EtOAc as the mobile phase to yield 2-(trimethylsilyl)ethyl (E)-3-((2,3-bis(tert-butoxycarbonyl)-guanidino)methyl)-5-(trimethylstannyl)benzoate (1.3 g, 75%) as a white foam: 1H NMR (CDCl3): δ 0.08 (s, 9H), 0.30 (s, 9H), 1.12 (t, 2H), 1.39 (s, 9H), 1.49 (s, 9H), 2.04 (s, 1H), 4.41 (t, 2H), 5.22 (s, 2H), 7.72 (s, 1H), 7.90 (s, 1H), 8.03 (s, 1H). HRMS Calcd for C27H47N3O6Si[120Sn] (M + H)+: 658.2329. Found: 658.2340.
2.3.5. Synthesis of N-succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzoate (Boc2-iso-SGMTB).
Compound 4 (1.2 g, 1 eq, 1.8 mmol) was dissolved in anhydrous tetrahydrofuran (50 mL) and the solution cooled to 0–5°C. A 1 M solution of tetra-n-butylammonium fluoride (TBAF) (0.53 g, 2.0 mL, 1.1 eq, 2.0 mmol) was added to the above and the mixture stirred at 25°C 16 h. Tetrahydrofuran was evaporated and the resultant oil dissolved in 75 mL of dichloromethane. N-hydroxysuccinimide (0.32 g, 1.5 eq, 2.7 mmol) and DMAP (22 mg, 0.1 eq, 0.18 mmol) were added and the solution was stirred at 25°C for about 5 min. EDC (0.53 g, 1.5 eq, 2.7 mmol) was added to the above and the mixture stirred at 25°C for 16 h under argon. The reaction mixture was partitioned between water and dichloromethane, and the organic layer dried over anhydrous MgSO4 and filtered. The volatiles were evaporated and the crude material chromatographed using a 25 g BIOTAGE SNAP ULTRA column and 1:1 hexanes:EtOAc as the mobile phase to afford Boc2-iso-SGMTB (781 mg, 65%) as a white foam: 1H NMR (CDCl3): δ 0.30 (s, 9H), 1.40 (s, 9H), 1.47 (s, 9H), 2.89 (s, 4H), 5.16 (s, 2H), 7.87 (s, 1H), 7.99 (s, 1H), 8.09 (s, 1H). HRMS Calcd for C26H39N4O8[120Sn] (M + H)+: 655.1790. Found: 655.1794.
2.3.6. Synthesis of N-succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-iodobenzoate (Boc2-iso-SGMIB).
A solution of Boc2-iso-SGMTB (222 mg, 1 eq, 340 μmol), potassium iodide (56 mg, 1 eq, 340 μmol) and N-chlorosuccinimide (136 mg, 3 eq, 1.02 mmol) in 10 mL DMF was stirred overnight at 20°C. DMF was evaporated under reduced pressure and the resultant crude material was taken in ethyl acetate (20 mL), washed with water (20 mL × 3), and dried over anhydrous MgSO4. The volatiles from the filtrate were evaporated and the residual material chromatographed using a 25 g BIOTAGE SNAP ULTRA column and 1:1 hexanes:EtOAc as the mobile phase to afford Boc2-iso-SGMIB (190 mg, 90%) as a white foam: 1H NMR (CDCl3): δ 1.45 (s, 9H), 1.50 (s, 9H), 2.91 (s, 4H), 5.14 (s, 2H), 8.07 (s, 1H), 8.13 (s, 1H), 8.35 (s, 1H). HRMS Calcd for C23H30IN4O8 (M + H)+: 617.1108. Found: 617.1100.
2.3.7. Synthesis of N-maleimidoethyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(trimethylstannyl)benzamide (Boc2-MEGMTB).
A solution of Boc2-iso-SGMTB (10 mg, 1 eq, 15.3 μmol), 2-maleimidoethylamine hydrochloride (3.2 mg, 1.2 eq, 18.4 μmol) and N, N-diisopropylethylamine (DIPEA, 3 mg, 4 μL, 1.5 eq, 23 μmol) in DMF (1 mL) was stirred at 20°C for 4 h. DMF was evaporated under reduced pressure and the residue taken in acetonitrile and subjected to reversed-phase HPLC purification. For this, an Agilent ZORBAX semi-prep C18 column was eluted at a flow rate of 4 mL/min with a mobile phase gradient consisting of 0.1% formic acid both in water (solvent A) and acetonitrile (solvent B); the proportion of B was increased linearly from 40% to 80% in 18 min. Lyophilization of HPLC fractions containing the product afforded 3.1 mg (30%) of Boc2-MEGMTB as a clear oil: 1H NMR (CDCl3): δ 0.3 (s, 9H), 1.49 (s, 18H), 3.63 (m, 2H), 3.81 (m, 2H), 5.15 (s, 2H), 6.73 (s, 2H), 7.60 (s, 1H), 7.73 (s, 1H), 7.79 (s, 1H). HRMS Calcd for C28H41N5O7[120Sn](M + H)+: 680.2101. Found: 680.2110.
2.3.8. Synthesis of N-maleimidoethyl)-3-(guanidinomethyl)-5-iodobenzamide (MEGMIB).
A solution of Boc2-iso-SGMIB (10 mg, 1 eq, 16 μmol), 2-maleimidoethylamine hydrochloride (3.43 mg, 1.2 eq, 19 μmol) and DIPEA (3.1 mg, 4.2 μL, 1.5 eq, 24 μmol) in DMF (1 mL) was stirred at 20°C for 4 h. DMF was evaporated under reduced pressure and the residual material was treated with trifluoroacetic acid (TFA, 1 mL). After 2 h, TFA was evaporated with a stream of argon, and the residue was reconstituted in acetonitrile. The crude product was purified using reversed-phase HPLC as above except the proportion of solvent B was increased from 5% to 15% in 15 min to afford MEGMIB (2 mg, 28%) as white powder: 1H NMR (MeOD-d4): δ 1.22 (s, 1H), 3.48 (m, 2H), 3.67 (m, 2H), 4.36 (s, 2H), 6.74 (s, 2H), 7.63 (s, 1H), 7.78 (s, 1H), 7.95 (s, 1H), 8.48 (s, 1H). HRMS Calcd for C15H16IN5O3(M + H)+: 442.0312. Found: 442.0321.
2.3.9. Synthesis of unlabeled iso-SGMIB-5F7 conjugate.
Deprotection of Boc2-iso-SGMIB with TFA was performed following a previously reported method [17, 18, 28, 29]. A solution of 5F7 sdAb in 0.1 M borate buffer, pH 8.5 (1.7 mL of 3 mg/mL; 5.1 mg, 0.4 μmol, 1 eq) was added to iso-SGMIB (500 μg, 1.2 μmol, 3 eq) in a half-dram glass vial. The vial was vortexed for 20 s and incubated at 30°C for 2 h. A 1:1 SGMIB-5F7 conjugate (i.e. 1 iso-SGMIB per 5F7 sdAb) was isolated from the mixture by HIC HPLC using a TSKgel Butyl-NPR column (see above for conditions).
2.3.10. Synthesis of unlabeled MEGMIB-5F7GGC conjugate.
The 5F7GGC sdAb, consisting of both dimer and monomer, was first reduced to convert the dimer to a monomer. For this, the buffer was exchanged to a reducing buffer (0.2 M NH4OAc, pH = 6.3, 5 mM EDTA) using a Vivaspin concentrator (Vivaspin® 500, 5 kDa molecular weight cut off (MWCO), GE Healthcare). The reducing buffer as well as the reaction mixture was degassed and purged with argon at each step to ensure a non-oxidizing environment. Gel-immobilized tris(2-carboxyethyl) phosphine hydrochloride (Thermo Scientific, Rockford, IL) was used to reduce the intermolecular disulfide bond formed between the C-terminal cysteines on 5F7GGC using a protocol provided by the manufacturer. Briefly, equal volumes of a TCEP gel slurry (8 μmol/mL; 0.2 mL) and 5F7GCC (see below) were added to a 1.5 mL centrifuge tube that was centrifuged at 1000 × g for 1 min. The supernatant was removed and the reducing buffer (500 μL) was added to the gel, the mixture vortexed for 1 min, and then the tube was centrifuged at 1000 × g for 1 min. This process was repeated four times. After the final removal of the supernatant from the TCEP gel slurry, 5F7GGC (2 mg, 1 eq, 0.153 μmol, in 1 mL reducing buffer) was added. The reduction was carried out by shaking the tube at 37°C on a thermo-shaker for 2 h. The tube was then centrifuged at 1000 × g for 1 min, and the supernatant containing monomeric 5F7GGC was removed and immediately added to a glass vial containing MEGMIB (203 μg, 3 eq, 0.459 μmol) and incubated at 37°C for 2 h. The MEGMIB-5F7GGC conjugate was purified using size-exclusion HPLC using an Agilent PL Multisolvent 20 column.
2.3.11. Quality control of sdAb and conjugates.
The molecular weights of 5F7, 5F7GGC, MEGMIB-5F7GGC and iso-SGMIB-5F7 were determined by MALDI-TOF mass spectrometry. The binding affinity (Kd) of the sdAbs and their conjugates for the HER2-Fc protein was measured by surface plasmon resonance using the Biacore system (Duke Human Vaccine Institute Shared Resource).
2.4. Cell culture conditions
Reagents for cell culture were obtained from Thermo Fisher Scientific (Waltham, MA), except where noted. SKOV-3 human ovarian carcinoma cells were obtained from the Duke University Cell Culture Facility. SKOV-3 cells were grown in McCoy’s 5A medium, containing 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were cultured at 37°C in a 5% CO2 humidified incubator.
2.5. Radiochemistry
Conjugation of [131I]MEGMIB to monomeric/reduced 5F7GGC was performed adapting a previously reported method [18, 30]. Briefly, a methanolic solution of N-chlorosuccinimide (NCS) (0.2 mg/mL) and 1% (v/v) acetic acid (100 μL), and sodium [131I]iodide (1–5 μL, 37–185 MBq) were added to a half-dram glass vial containing Boc2-MEGMTB (50 μg, 0.07 μmol). The vial was vortexed for a short time and the reaction was allowed to proceed at 20°C for 15 min. The volatiles were evaporated under a stream of argon and the residual activity reconstituted in 50% acetonitrile in water (100 μL). This solution was injected onto a reversed-phase HPLC column eluted a flow rate of 2 mL/min with a gradient consisting of 0.1% TFA in water (solvent A) and 0.1% TFA in acetonitrile (solvent B); proportion of B was linearly increased from 40% to 80% over 8 min. The HPLC fractions containing the product (tR = 4.5 min) were pooled and most of the acetonitrile from it was removed using a stream of argon. The product Boc2-[131I]MEGMIB was extracted with 2 × 1 mL of ethyl acetate and transferred to an one-dram glass vial. Ethyl acetate was evaporated with argon, TFA (100 μL) added and the deprotection of Boc2-[131I]MEGMIB was allowed to proceed at 20° C for 10 min. Subsequently, TFA was removed under a stream of argon, followed by co-evaporation with ethyl acetate (100 μL × 3). Monomeric sdAb 5F7GGC, freshly obtained as described above, added to the above vial containing [131I]MEGMIB. The conjugation was carried out at 37°C for 45 min. The labeled sdAb was isolated by gel filtration over a PD-10 column using PBS as the mobile phase. Fractions containing the [131I]MEGMIB-5F7GGC were pooled and used for further studies.
2.6. Quality control of the [131I]MEGMIB-5F7GGC
The radiochemical purity of labeled sdAb was evaluated by SDS-PAGE/phosphor imaging and size-exclusion HPLC. The immunoreactive fraction was determined by the Lindmo method [31] following a reported procedure [32]. HER2 binding affinity of [131I]MEGMIB-5F7GGC was determined using SKOV-3 cells by the saturation binding assay as described before [28, 29]. Nonspecific binding was determined in parallel assays by co-incubating cells with 100-fold molar excess of trastuzumab.
2.7. In vitro stability study
The in vitro stability of both iso-[125I]SGMIB-5F7 and [131I]MEGMIB-5F7GGC conjugates was investigated by incubating them individually in 1 mL of PBS, fetal bovine serum (FBS) or human serum (HS) at 37°C. The radiochemical purity was checked by performing SDS-PAGE/phosphor imaging of 1 μL aliquots at 24 and 48 h.
2.8. Determination of internalization in vitro
Internalization and cellular retention of [131I]MEGMIB-5F7GGC, in tandem with iso-[125I]SGMIB-5F7, in SKOV-3 cells was assessed in a paired-label format using procedures reported for similar molecules [18, 33]. In these assays, nonspecific uptake was assessed in parallel experiments by co-incubating cells with 100-fold molar excess of trastuzumab.
2.9. Biodistribution
All experiments involving animals were performed under a protocol approved by the Duke University IACUC. Subcutaneous SKOV-3 xenografts were established by inoculating 10-week old female athymic mice (~25 g), obtained from a colony maintained by the Duke University Division of Laboratory Animal Resources, in the shoulder with 5 × 106 SKOV-3 cells in 50% matrigel (Corning Inc., NY) in the above medium (100 μL). Biodistribution studies were performed 6–8 weeks later, when tumors reached 350 – 500 mm3 in size. The biodistribution of [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7 was determined in a paired-label format in groups of five mice injected via the tail vein with 0.30–0.37 MBq of each labeled conjugate. Each mouse received a total of 2–6 μg of sdAb (5F7 and 5F7GGC combined, based on measured specific activities). At 1 h, 4 h and 24 h after the tracer administration, blood and urine were collected and the mice were killed by an overdose of isofluorane. Tumor and other tissues were harvested, blot-dried, weighed and counted along with the injection standards for 125I and 131I activity using an automated gamma counter. From these counts, the percentage of the injected dose (%ID) per organ, per gram of tissue (%ID/g) and tumor-to-normal tissue ratios were calculated.
2.10. Catabolism of labeled conjugates in athymic mice bearing SKOV-3 xenografts
The in vivo catabolism of [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7 was investigated in paired-label format. A group of five mice was injected intravenously with 0.30–0.37 MBq each of [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7 (a total of 2–6 μg of sdAb). At 1 h, urine samples were collected, and the mice were euthanized. The kidneys and tumor from each mouse were harvested, weighed and counted on the automated gamma counter. Urine from each mouse was diluted to 1 mL with PBS, loaded onto a 0.22 μm Spin-X® centrifuge tube filter (Corning Costar), washed with PBS to precondition, and centrifuged at 14,000 rpm for 30 min. The filtrate and the filter were counted on the automated gamma counter. The kidneys and tumor from each mouse were washed with deionized water to remove any adherent blood and each transferred to a glass tube. Cell lysis buffer (RIPA cell lysis buffer, 500 μL × 2), along with protease inhibitor (Halt™ Phosphatase Inhibitor Cocktail, Thermo Fisher), was added to each tube, and the tissue was homogenized using a hand-held homogenizer. The mixture was centrifuged at 14,000 rpm for 20 min and the supernatants separated. The supernatants were decanted, counted on the automated gamma counter and loaded onto a preconditioned 0.22 μm SpinX column, and centrifuged at 14,000 rpm for 30 min. The filtrate and the filter were counted on the automated gamma counter. In each case, 200 μL of the filtrate was loaded onto either a 5-kDa or a 30-kDa MWCO Vivaspin® filter and centrifuged at 14,000 rpm for 40 min. Both the filtrates and the upper compartments of the Vivaspin® filters were counted for 125I and 131I. The activity present in the filtrates and the filter was calculated as a percentage of the total radioactivity loaded onto the Vivaspin® filter.
2.11. Statistical analyses
Data are presented as mean ± standard deviation. Differences in the behavior of co-incubated (in vitro) or co-administered (in vivo) labeled conjugates were analyzed for statistical significance using a paired two-tailed Student t-test in GraphPad Prism 8. Differences with a P value < 0.05 were considered statistically significant.
3. Results
3.1. Chemistry
Previously, Boc2-iso-SGMTB was synthesized using 3-(methoxycarbonyl)-5-nitrobenzoic acid as the starting material, in six steps with an overall yield of 4% [17, 18, 28, 29]. In order to reduce synthesis steps and improve the overall yield, a modified synthesis starting with 3-bromo-5-methylbenzoic acid was developed (Scheme 1). With this new approach, it was possible to synthesize Boc2-iso-SGMTB in 5 steps with an overall yield of 15%. The starting material was first converted to trimethylsilylethyl ester 1 in 75% yield. Benzylic bromination of 1 was performed with NBS, and the resultant benzyl bromide was converted to benzyl alcohol 2 via sequential acetylation and ammonolysis in situ in an overall yield of 80%. Compound 2 was subjected to palladium-mediated stannylation using hexamethylditin to afford 3 in 75% yield. Compound 4, synthesized from 3 in 75% yield via the Mitsunobu reaction, was deprotected using TBAF, and the crude product re-esterified to afford Boc2-iso-SGMTB in 65% yield. Conjugation of Boc2-iso-SGMTB with 2-maleimidoethylamine gave the required tin precursor, Boc2-MEGMTB, in 40% yield. Iodination of Boc2-iso-SGMTB with KI and NCS in DMF rendered Boc2-iso-SGMIB in 90% yield. Conjugation of this with 2-maleimidoethylamine and removal of Boc groups from the resultant product by treatment with TFA afforded the cold standard MEGMIB in 31% yield for the two steps. NMR and mass spectral data for all compounds are consistent with their structures.
Scheme 1.
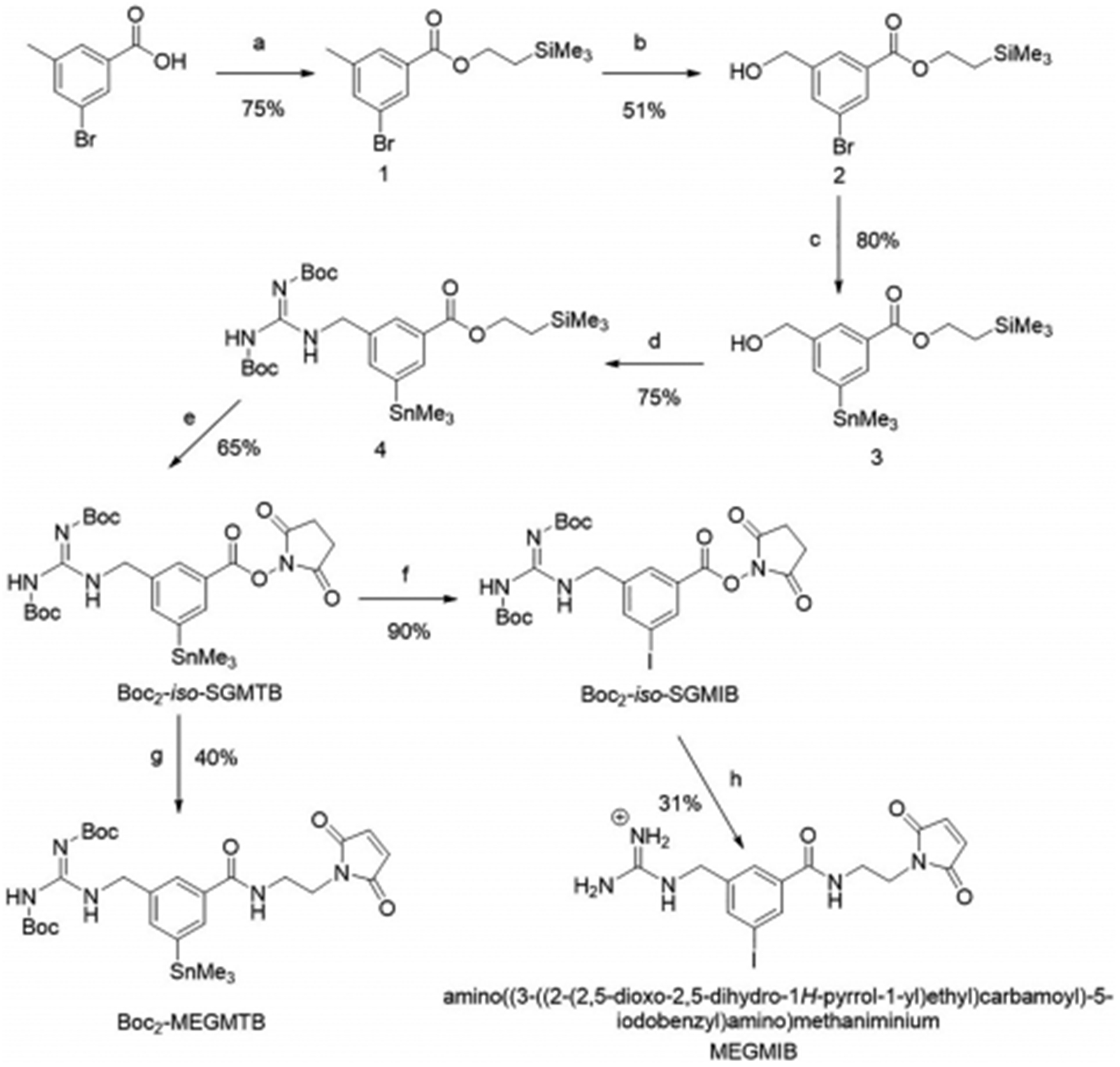
Synthesis of Boc2-iso-SGMTB, Boc2-MEGMTB tin precursor and the cold standard MEGMIB. a) 2-(trimethylsilyl)ethan-1-ol, DMAP, EDC, DCM; b) i: NBS, AIBN, DCE, ii: NaOAc, DMF, iii: NH3, MeOH; c) (Ph3P)2PdCl2, hexametheylditin, 1,4-dioxane; d) 1,3-bis(tert-butoxycarbonyl)guanidine, TPP, DIAD, THF; e) i: TBAF, THF, ii: 1-hydroxypyrrolidine-2,5-dione, EDC, DMAP, DCM; f) N-chlorosuccinimide, KI, DMF; g) 2-maleimidoethylamine hydrochloride, DIPEA, DMF; h) i: 2-maleimidoethylamine hydrochloride, DIPEA, DMF, ii: TFA.
3.2. Conjugation of unlabeled prosthetic agents to sdAb 5F7 and 5F7GGC
When three equivalents of iso-SGMIB were used for the synthesis of iso-SGMIB-5F7, conjugates with 1, 2 and 3 iso-SGMIB moieties per sdAb were produced. The 1:1 conjugate from this mixture was isolated by hydrophobic interaction chromatography (HIC) and the purity of the iso-SGMIB-5F7 conjugate thus obtained, determined by analytical size-exclusion HPLC, was >95%. Its molecular weight, determined by MALDI mass analysis, was 13118.5 Da consistent with the calculated value of 13117 Da. The binding affinity of iso-SGMIB-5F7 to HER2-Fc measured by surface plasmon resonance was 0.21 nM, and that determined in parallel for unmodified 5F7 was 0.22 nM. Size-exclusion HPLC indicated that 5FGGC was completely converted to the monomeric form after reduction with TCEP (Figure 2A). Using the same HPLC system, it was possible to separate MEGMIB-5F7GGC conjugate from both monomeric and dimeric 5F7GGC with a purity of >95% (Figure 2A). Its molecular weight was 13468.0 Da compared with a calculated value of 13470.0 Da. The HER2-binding affinity, determined by SPR as above, was 0.2 nM for the MEGMIB-5F7GGC conjugate.
Figure 2.
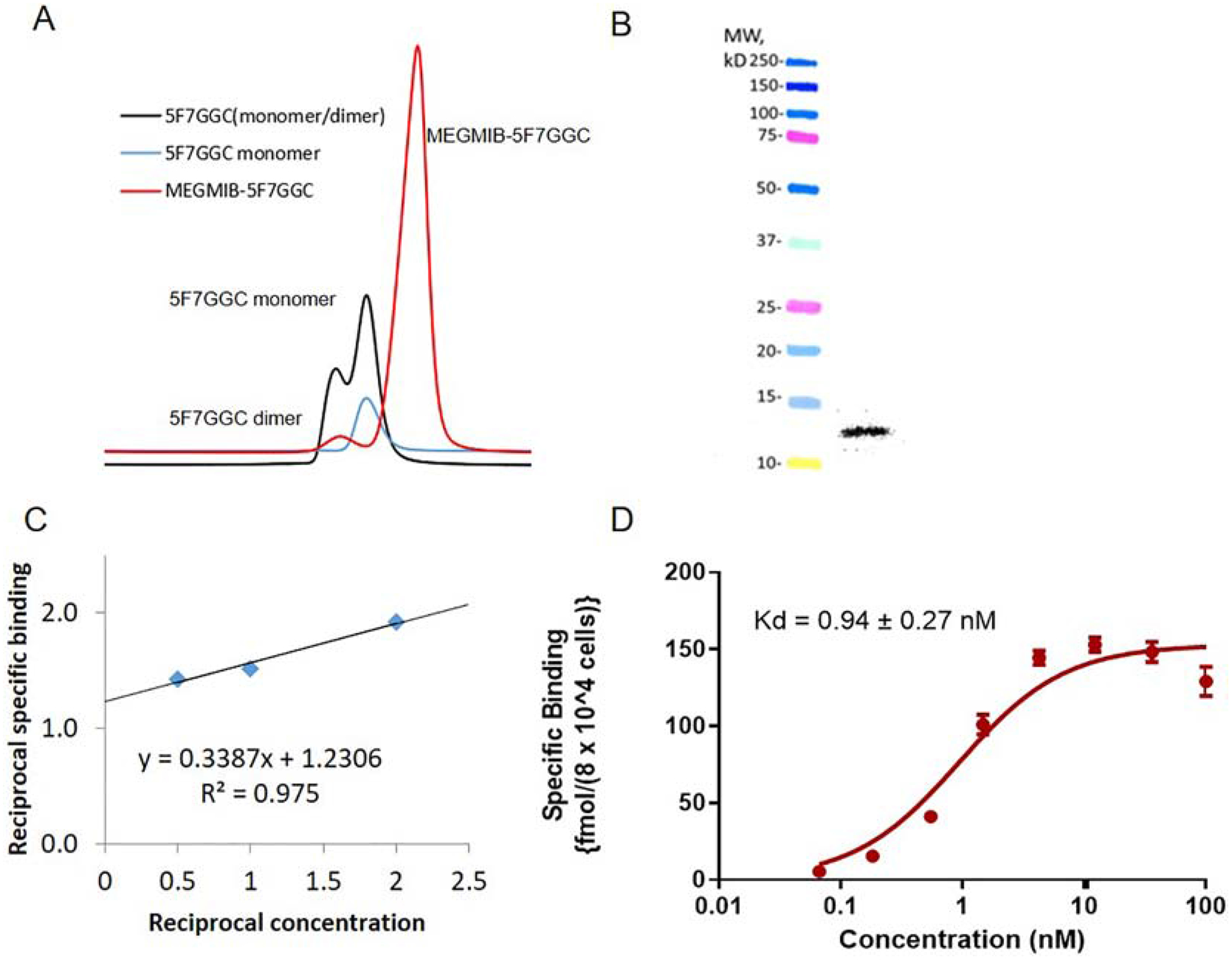
Quality control data for MEGMIB-5FGGC and [131I]MEGMIB-5F7GGC: A) Size-exclusion HPLC of non-reduced 5F7GGC containing both monomer and dimer (black), reduced 5F7GGC (blue) and MEGMIB 5F7GGC conjugate reaction (red) containing MEGMIB-5F7GGC product and 5F7GGC dimer. B) SDS-PAGE/Phosphor Imaging of [131I]MEGMIB-5F7GGC (right lane) and molecular weight markers (left lane). C) Double reciprocal plot of data from the immunoreactivity assay. D) Data from the saturation binding affinity assay performed with SKOV-3 cells.
3.3. Radiochemistry
Boc2-MEGMTB was radioiodinated using conditions previously reported for analogous compounds [17, 34] to yield Boc2-[131I]MEGMIB in 87 ± 6% radiochemical yield (n=11). Because reversed-phase HPLC was used for the separation of Boc2-[131I]MEGMIB, in preliminary experiments, a solid-phase extraction method was employed to isolate Boc2-[131I]MEGMIB from the HPLC fractions but this process was not efficient. Subsequently, the labeled product was isolated by direct ethyl acetate extraction of these HPLC fractions in 89 ± 3% yield (n=3). The labeled prosthetic agent, [131I]MEGMIB, was obtained in almost quantitative yield by the treatment of Boc2-[131I]MEGMIB with TFA. The labeled product had a retention time identical to that for the unlabeled standard and radiochemical purity was >99%. Conjugation of [131I]MEGMIB with freshly reduced 5F7GGC was achieved in 45 ± 7% yield (n=10). The molar activity of [131I]MEGMIB-5F7GGC was 930 ± 25 GBq/mmol (n=6).
3.4. Quality control
SDS-PAGE/phosphor imaging of [131I]MEGMIB-5F7GGC revealed a single radioactive band corresponding to the expected molecular weight of ~13 kDa (Figure 2B). Size-exclusion HPLC analysis of the [131I]MEGMIB-5F7GGC conjugate also indicated a single peak with a retention time corresponding to that of the unlabeled conjugate (Figure 2A). The immunoreactive fraction (IRF) determined by Lindmo assay was 81.3% (Figure 2C). A saturation binding assay using the SKOV-3 ovarian carcinoma cell line as the HER2-expressing target gave a Kd = 0.94 ± 0.27 nM for [131I]MEGMIB-5F7GGC (Figure 2D).
3.5. In vitro stability study
The radiolabeled conjugate iso-[125I]SGMIB-5F7 exhibited excellent in vitro stability, with 100% of activity associated with the intact conjugate after incubation for 48 h at 37°C in PBS, fetal bovine serum (FBS) and human serum (HS). With [131I]MEGMIB-5F7GGC, 78%, 90% and 87% of radioiodine activity was associated with the intact conjugate after a 24 h incubation in PBS, HS and FBS, respectively; at 48 h, these were 71%, 88% and 91%.
3.6. Paired-label internalization
The percentage of input activity initially bound to the cells after a 1 h incubation at 4°C was 10.96 ± 0.64% for [131I]MEGMIB-5F7GGC, which was significantly higher (P<0.05) than that for co-incubated iso-[125I]SGMIB-5F7 (8.84 ± 0.32%). For both conjugates, non-specific binding, determined by co-incubation with excess trastuzumab, was <1% of input activity. When incubated at 37°C, the proportion of this initially cell-bound [131I]MEGMIB-5F7GGC activity that was effluxed into the cell culture supernatant was 34.2 ± 2.5%, 45.1 ± 1.2%, 46.6 ± 2.4% and 66.6 ± 1.6% at 1, 2, 4 and 24 h, respectively (Figure 3). Iso-[125I]SGMIB-5F7 exhibited a similar trend albeit with significantly lower values at all time points: 26.4 ± 1.8%, 30.9 ± 0.5%, 29.2 ± 0.4% and 53.9 ± 2.2% at 1, 2, 4 and 24 h, respectively. The fraction activity present on the cell surface for [131I]MEGMIB-5F7GGC was 15.5 ± 1.0%, 17.6 ± 0.4%, 12.4 ± 0.9% and 12.1 ± 1.0% of the initially cell-bound activity at 1, 2, 4 and 24 h, respectively, values significantly lower than those for iso-[125I]SGMIB-5F7 at each time point (17.6 ± 1.6%, 22.3 ± 0.3%, 17.3 ± 1.1% and 15.0 ± 1.2%). For [131I]MEGMIB-5F7GGC, the fraction of initially cell-bound activity trapped intracellularly was 50.3 ± 3.4%, 37.4 ± 1.5%, 41.0 ± 2.4% and 21.4 ± 2.6% at 1, 2, 4 and 24 h, respectively, compared with 56.0 ± 3.3%, 46.8 ± 0.5%, 53.5 ± 0.9% and 31.1 ± 3.4% for iso-[125I]SGMIB-5F7. Differences in residualized activity for the two conjugates was significant at all time points except 1 h (P=0.0005, 2 h; P=0.0011, 4 h; P=0.017, 24 h).
Figure 3.
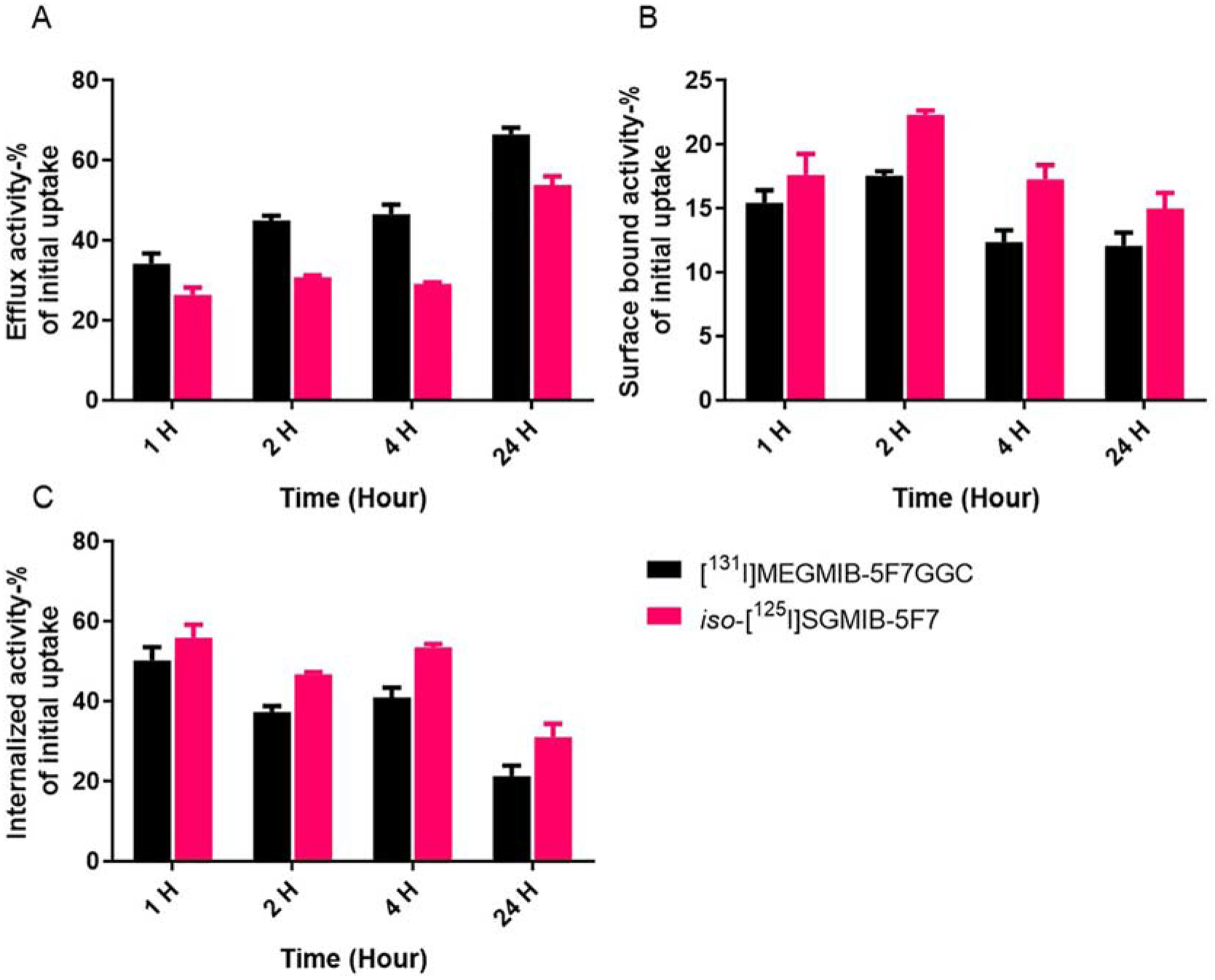
Paired-label internalization of iso-[125I]SGMIB-5F7 (red bars) and [131I]MEGMIB-5F7GGC (black bars) in HER2-expressing SKOV-3 cells in vitro: A) percentage of radioactivity in efflux after 1, 2, 4 and 24 h incubation. B) percentage of radioactivity bound to cell surface after 1, 2, 4 and 24 h incubation. C) percentage of radioactivity internalized in the cells after 1, 2, 4 and 24 h incubation.
3.7. Paired-label biodistribution of [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7
The uptake of activity in tumor and normal tissues after co-injection of iso-[125I]SGMIB-5F7 and [131I]MEGMIB-5F7GGC in athymic mice bearing subcutaneous HER2-expressing SKOV-3 xenografts is given in Table 1. No significant difference (P>0.66 at all three time points) in tumor uptake between the two tracers was observed. Tumor uptake of [131I]MEGMIB-5F7GGC was 8.43 ± 2.84% ID/g, 6.11 ± 1.63% ID/g and 3.60 ± 1.75% ID/g compared with 8.35 ± 2.66% ID/g, 6.11 ± 1.56% ID/g and 3.13 ± 1.52% ID/g for iso-[125I]SGMIB-5F7 at 1, 4, and 24 h, respectively. On the other hand, uptake of [131I]MEGMIB-5F7GGC in most normal tissues was generally lower than that seen for iso-[125I]SGMIB-5F7, especially at 1 h and 4 h. Notably, the kidney activity level for [131I]MEGMIB-5F7GGC at 1 h (20.39 ± 5.34 %ID/g) was more than 2-fold lower (P=0.01) compared with that seen for iso-[125I]SGMIB-5F7 (41.82 ± 13.33 %ID/g). At 4 h, the renal activity for [131I]MEGMIB-5F7GGC also was lower (P=0.02), albeit to a lesser degree. The liver uptake of [131I]MEGMIB-5F7GGC at 1 h (1.74 ± 0.45 %ID/g) also was significantly lower (P=0.003) than that seen for iso-[125I]SGMIB-5F7(4.88 ± 0.71 %ID/g); a similar difference was seen at 4 h. For both conjugates, uptake in other normal tissues generally was low with no significant retention of activity. The thyroid uptake for both conjugates was low at 1 h (0.18 ± 0.08 %ID and 0.08 ± 0.05 %ID for iso-[125I]SGMIB-5F7 and [131I]MEGMIB-5F7GGC, respectively), indicative of minimal deiodination of both conjugates in vivo. At 4 h, the thyroid uptake for [131I]MEGMIB-5F7GGC (0.12 ± 0.03 %ID) was significantly lower (P=0.0012) than that for iso-[125I]SGMIB-5F7 (0.35 ± 0.10 %ID); a similar trend persisted at 24 h but the difference was not quite statistically significant (P=0.06).
Table 1.
Pair-labeled biodistribution of iso-[125I]SGMIB-5F7 and [131I]MEGMIB-5F7GGC in athymic mice bearing Her2-expressing SKOV-3 xenografts.
| Percent injected dose per gramc | ||||||
|---|---|---|---|---|---|---|
| 1 h | 4 h | 24 h | ||||
| tissue | 125Ia | 131Ib | 125I | 131I | 125I | 131I |
| Liver | 4.88 ± 0.71 | 1.74 ± 0.45 | 2.12 ± 0.84 | 0.76 ± 0.36 | 0.46 ± 0.14 | 1.53 ± 0.55 |
| Spleen | 1.05 ± 0.51 | 0.36 ± 0.14 | 0.74 ± 0.23 | 0.20 ± 0.05 | 0.22 ± 0.16 | 0.65 ± 0.48 |
| Lungs | 2.50 ± 0.58 | 2.48 ± 0.51 | 1.08 ± 0.40 | 1.06 ± 0.39 | 0.84 ± 0.85 | 0.54 ± 0.60 |
| Heart | 0.49 ± 0.12 | 0.41 ± 0.11 | 0.11 ± 0.03 | 0.08 ± 0.02 | 0.03 ± 0.03 | 0.03 ± 0.02 |
| Kidneys | 41.82 ± 13.33 | 20.39 ± 5.34 | 2.88 ± 0.46 | 1.77 ± 0.28 | 0.12 ± 0.02 | 0.40 ± 0.14 |
| Stomach | 2.00 ± 0.66 | 0.94 ± 0.30 | 1.20 ± 0.64 | 0.61 ± 0.49 | 0.07 ± 0.05 | 0.09 ± 0.08 |
| Sm. Int. | 1.26 ± 0.27 | 2.09 ± 0.47 | 0.75 ± 0.27 | 0.31 ± 0.11 | 0.05 ± 0.01 | 0.09 ± 0.02 |
| Lg. Int. | 0.38 ± 0.15 | 0.52 ± 0.15 | 2.80 ± 0.99 | 3.20 ± 2.33 | 0.13 ± 0.03 | 0.18 ± 0.04 |
| Muscle | 0.29 ± 0.07 | 0.22 ± 0.08 | 0.08 ± 0.05 | 0.05 ± 0.03 | 0.01 ± 0.01 | 0.01 ± 0.01 |
| Blood | 0.83 ± 0.28 | 0.59 ± 0.18 | 0.22 ± 0.09 | 0.12 ± 0.03 | 0.17 ± 0.33 | 0.13 ± 0.10 |
| Bone | 0.69 ± 0.24 | 0.48 ± 0.23 | 0.18 ± 0.09 | 0.09 ± 0.02 | 0.13 ± 0.26 | 0.13 ± 0.10 |
| Brain | 0.06 ± 0.03 | 0.05 ± 0.02 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Thyroid | 1.25 ± 0.87 | 0.57 ± 0.44 | 1.79 ± 0.59 | 0.60 ± 0.20 | 2.03 ± 1.17 | 0.83 ± 0.48 |
| Tumor | 8.35 ± 2.66 | 8.43 ± 2.84 | 6.11 ± 1.56 | 6.11 ± 1.63 | 3.13 ± 1.52 | 3.60 ± 1.75 |
iso-[125I]SGMIB-5F7.
[131I]MEGMIB-5F7GGC.
%ID/g, mean ± SD, n=5
Tumor-to-normal tissue ratios for the two radioconjugates are presented in Figure 4. In general, tumor-to-normal tissue ratios were (> 3:1 by 1 h for both conjugates except for the kidneys. Compared with those for iso-[125I]SGMIB-5F7, tumor-to-normal tissue ratios for [131I]MEGMIB-5F7GGC were higher especially at 1 h and 4 h. The tumor-to-kidney ratio for [131I]MEGMIB-5F7GGC at 1 h was 0.41 ± 0.09, 2-fold higher (P=0.008) than that for iso-[125I]SGMIB-5F7 (0.21 ± 0.09), with a similar difference seen at 4 h. Tumor-to-liver ratios for [131I]MEGMIB-5F7GGC were substantially higher than those for iso-[125I]SGMIB-5F7 at 1 h and 4 h with an almost 3-fold difference observed at 1 h ([131I]MEGMIB-5F7GGC, 4.85 ± 1.23; iso-[125I]SGMIB-5F7, 1.70 ± 0.45).
Figure 4.
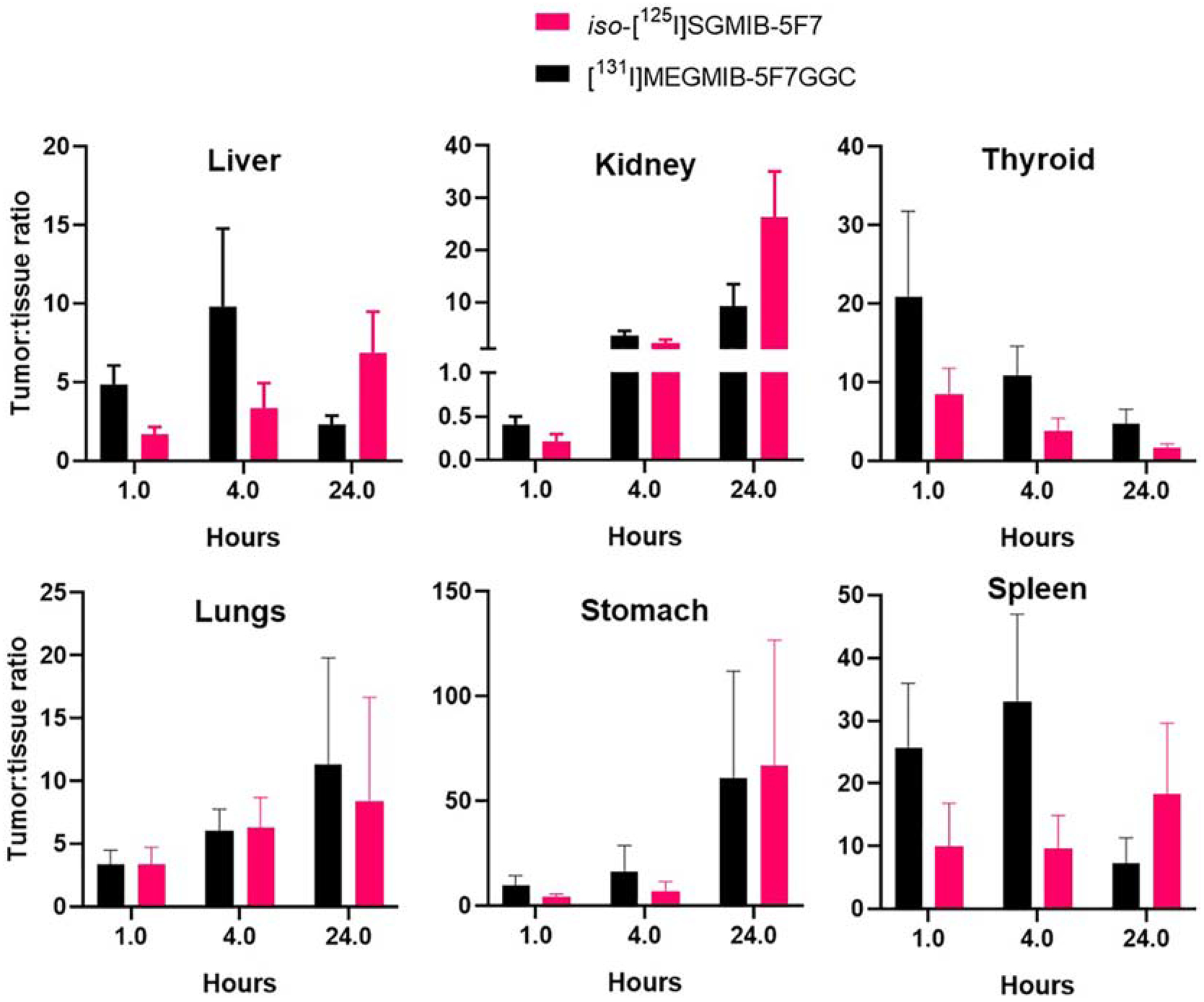
Tumor to healthy tissue ratios after injections of iso-[125I]SGMIB-5F7 and [131I]MEGMIB-5F7GGC in athymic mice bearing Her2-expressing SKOV-3 xenografts.
3.8. Catabolism of labeled conjugates in athymic mice bearing SKOV-3 xenografts
The in vivo fate of [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7 were compared by evaluating the molecular weight profile of the radioactivity present in urine, tumor and kidney. After homogenization, the fraction of radioactivity recovered in the tissue supernatant was calculated for each of 5 mice:
After injection of [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7, the 131I and 125I activity recovered in the kidney homogenate supernatants represented 84.5 ± 10.2% and 89.2 ± 9.7%, respectively, of the total activity in the kidneys at 1 h. With tumor, the 125I and 131I activity recovery in homogenate supernatants was 55–100% and 58–100%, respectively. When the tissue homogenates and the urine samples were passed through 0.22 μm SpinX filter columns, 88–100% (n=15) of both 125I and 131I activity was isolated in the filtrates. To evaluate the nature of the radiolabeled molecules that were present, samples were subjected to filtration through 5-kDa and 30-kDa MWCO Vivaspin® filters. In urine, 81.4 ± 7.1% (n=3) of 131I and 92.3 ± 2.9% (n=3) of 125I activity was associated with molecules of molecular weight >5 kDa with no significant difference between these two values (P=0.067). In tumor homogenates, 93.9 ± 1.6% of 131I and 98.4 ± 1.2% of 125I activity (P=0.019) was associated with >5kDa molecules, with the vast majority associated with molecules of >30 kDa was for both 131I and 125I (131I, 95.8 ± 0.8%; 125I, 94.6 ± 2.0%). In kidney homogenate supernatants, the percentage of activity associated with >5 kDa molecules was 80.9 ± 5.5% for 131I and 83.9 ± 2.7% for 125I with the difference not statistically significant. Compared with tumor, a lower percentage of kidney homogenate activity was associated with >30kDa molecules for both radionuclides (131I, 54.3 ± 6.0%; 125I, 66.3 ± 4.1%) with the difference between the two radionuclides statistically significant (P=0.044).
4. Discussion
Site-specific labeling strategies have been evaluated extensively for antibody drug conjugate (ADC) development [35–37] because: 1) they can provide a more homogenous ADC due to the fewer conjugation sites compared to random methods, which can improve the overall in vivo stability of the ADC [35]; 2) they allow more precise control of payload conjugation to avoid conjugation on mAb binding sites, thereby preserving binding affinity; and 3) for clinical translation, site-specific conjugation provides a reproducible and homogeneous product that should be viewed favorably by regulatory agencies.
The maleimido-thiol Michael addition chemistry exploited herein targets the free thiol groups on protein cysteine residues to allow site-specific conjugation. Generally, this method requires a reduction of constituent disulfide bonds to afford free thiols for conjugation; however, many disulfide bonds are critical for mAb structural integrity and hence this reduction procedure may compromise binding affinity and other critical properties [38]. On the other hand, insufficient reduction may decrease conjugation yields [39]. Engineering a cysteine tail on either the C- or N-terminus can be advantageous in this case [15, 35], although the location of the engineered cysteine tail can influence conjugate’s performance [40].
The in vivo behavior of proteins modified via thiosuccinimide conjugation using the maleimido-thiol Michael addition reaction is expected to be different from the same protein modified with a similar prosthetic moiety via an amide bond formed using an active ester. Arano et al. reported faster clearance of radioiodine activity from kidney, liver and spleen when an intact mAb was labeled using a thiosuccinimide linkage [39]. Similar results have been reported for an intact mAb labeled with 211At via a thiolmaleimide conjugation [35]. With smaller mAb fragments such as diabodies, the pattern of normal tissue distribution also was dependent on whether the labeled prosthetic agent was attached via an amide or a thiosuccinimide bond [41, 42]. For sdAbs, when conjugated with fluorophores, maleimido-thiol site-specific labeling out-performed the NHS conjugation method [11]. On the other hand, a head-to-head comparison of sdAb 2Rs15d labeled with 211At using the two linkages showed an advantage for NHS ester conjugation [26]. However, these results may not be indicative of those obtainable with radioiodine because of differences between the two halogens with regard to affinity for sulfur atoms and susceptibility to dehalogenation.
To the best of our knowledge, this is the first report of site-specific labeling of an sdAb via a radioiodinated residualizing prosthetic group, in this case placed on a C-terminus cysteine tail. Based on previous studies with anti-HER2 5F7 sdAb showing excellent trapping in tumor cells in vitro and in xenografts in vivo with low accumulation in normal tissues [18, 26], a guanidine-containing residualizing prosthetic agent, iso-[131I]SGMIB served as the template for design of the thiol reactive prosthetic agent for this site specific labeling strategy. With 5F7, iso-SGMIB could react with any of its 5 lysine amino side chains or the N-terminal amine of the sdAb potentially resulting in a heterogeneous mixture of randomly labeled conjugates. Although heterogeneity from random labeling of an sdAb like 5F7 should be less of an issue compared with an intact mAb, site-specific conjugation may offer advantages and thus a head-to-head comparison is worth evaluating [43–45]. Moreover, with one of the 5F7 lysines in its CDR regions, a decline in affinity after random lysine conjugation is a distinct possibility.
A maleimide moiety-bearing analogue of SGMIB, MEGMIB, was successfully developed for the site-specific radioiodination of a 5F7 variant with a C-terminus GGC tail. First, an improved method was developed for the synthesis of both the Boc2-iso-SGMTB tin precursor and the unlabeled Boc2-iso-SGMIB standard. This new approach, using 3-bromo-5-methylbenzoic acid as the starting material, decreased the number of steps from 6 to 5 and increased overall yield from 4 to 15% [30]. Unlike the previously reported method [24], unlabeled Boc2-iso-SGMIB was synthesized by iodination of Boc2-iso-SGMTB using KI in the presence of an oxidant by modifications of the reaction conditions reported for the iododestannylation reaction [24, 46]. Initially chloramine-T was used [39] but resulted in poor yields but switching to NCS as the oxidant [38] gave excellent yields (90%). The tin precursor for the synthesis of radioiodinated MEGMIB, Boc2-MEGMTB, was synthesized in good yields by conjugating Boc2-iso-SGMTB with an excess of 2-maleimidoethylamine. Because it does not contain a hydrolytically unstable NHS ester, it could be purified by reversed-phase HPLC. Synthesis of unlabeled Boc2-MEGMIB was performed as done for Boc2-iso-SGMIB, except that after deprotection, reversed-phase HPLC was used to purify the final product.
For synthesis of the cold conjugate, the ratio of iso-SGMIB to 5F7 was optimized at 3:1 to maximize the yield of the 1:1 iso-SGMIB-5F7 conjugate. For radiolabeled iso-[125/131I]SGMIB-5F7 multiple conjugation is unlikely due to the low molar concentration of iso-[125/131I]SGMIB; however, using iso-[131I]SGMIB-5F7 for therapeutic applications might involve multiple conjugation sites per sdAb molecule. As noted above, there are five lysine amino groups in 5F7 sdAb one of which is located in the CDR2 [15], and conjugation of iso-SGMIB to that lysine could potentially affect the binding affinity of 5F7 to HER2. In order to best mimic characteristics of a radiolabeled iso-SGMIB-5F7 conjugate yet allow SPR binding analysis, a 1:1 cold conjugate (MW=13118.5 Da) was isolated in >95% purity. As with the radioactive version, this 1:1 conjugate can potentially be a mixture of molecules modified at any of the six potential conjugation sites. SPR analysis gave a Kd of 0.21 nM for the 1:1 iso-SGMIB-5F7 conjugate(s) compared with 0.22 nM for unmodified 5F7. This suggests that at a 1:1 substitution level, the iso-SGMIB-5F7 conjugate(s) either didn’t include a significant CDR2 lysine-modified component and/or CDR2 conjugation did not compromise HER2 binding.
In addition to avoiding conjugation at biologically crucial sites on a protein, other potential advantages of site-specific conjugation have been documented in a recent review [47]. As it is expected to yield a more homogeneous product, we explored this possibility for labeling the anti-HER2 sdAb with a residualizing prosthetic agent. Because of the excellent results obtained with site-specific conjugation by modification of cysteine residues with maleimide moiety-bearing molecules [23–26, 48, 49], we developed the MEGMIB agent, an analogue of the reported prosthetic agent iso-SGMIB [17], that contained a maleimide function. This was then used for site-specific labeling of 5F7GGC, containing a cysteine at its C-terminus, a location distant from the CDR loops on an sdAb [11].
Because of the susceptibility of its free thiol to oxidation, 5F7GGC can exist as a mixture of monomer and dimer, making it essential to reduce it to predominantly monomeric form before conjugation. Several reducing agents including tris(2-carboxyethyl) phosphine hydrochloride (TCEP) and dithiothreitol (DTT) have been used for this purpose, and removal of these reducing agents is often necessary for optimal conjugation [48]. We developed an effective reduction protocol using an immobilized form of TCEP (gel) and a 0.2 M NH4OAc, 5 mM EDTA reduction buffer. The advantages of immobilized TCEP gel are that it provides a mild reducing condition so that the native sdAb disulfide bond can be preserved, and that the reduced sdAb can be easily separated from excess reducing agent, which can compromise maleimide conjugation yields. Using our reducing conditions, >95% monomer (determined using size-exclusion LC-MS) could be obtained in 1 h.
It should be noted that a significant fraction of reduced 5F7GGC reoxidizes back to dimeric form within 2 h of separation from TCEP at room temperature. Also, it is important to note that sdAbs have an intradomain disulfide bond that provides conformational stability. Initially, harsher reducing conditions (10 mM TCEP at 37°C for 2 h) were evaluated for 5F7GGC reduction; however, conjugates with 3 prosthetic moieties per protein were observed, reflecting reduction of the intradomain disulfide. This was not observed with the optimized immobilized TCEP gel reduction protocol even when higher molar ratios (>5:1 of MEGMIB:5F7GGC), higher temperatures (45°C) and longer reaction times were evaluated. SPR analysis indicated that the purified 1:1 MEGMIB-5F7GGC conjugate had a Kd of 0.20 nM, compared with 0.22 nM for unmodified 5F7. The high affinity for this conjugate under the optimized reduction and conjugation conditions, is consistent with the maleimide conjugation occurring at the C-terminal cysteine with preservation of the interdomain disulfide bond. For this reason, the same reducing conditions were utilized for synthesis of the radiolabeled version of the conjugate [131I]MEGMIB-5F7GGC.
Synthesis of Boc2-[131I]MEGMIB from its tin precursor was achieved essentially following the method used for the synthesis of Boc2-iso-[131I]SGMIB [19] in similar radiochemical yields. Initially, solid-phase extraction with a C-18 cartridge was used to isolate Boc2-[131I]MEGMIB from the HPLC fractions; however, better recovery (~90%) of Boc2-[131I]MEGMIB was achieved when liquid-liquid extraction using ethyl acetate was employed. Deprotection of Boc2 -[131I]MEGMIB with TFA delivered the final prosthetic agent in 99% radiochemical purity (determined by HPLC). When stored in PBS at 4°C, [131I]MEGMIB remained >95% intact for up to 48 h. The conjugation of [131I]MEGMIB to 5F7GGC was performed immediately after its reduction to minimize re-oxidation. A 1-h conjugation reaction was found to be optimal; longer incubation times were counterproductive due to re-oxidization of 5F7GGC to its dimer, as indicated by LCMS. [131I]MEGMIB-5F7GGC had excellent immunoreactivity and binding affinity, indicating that [131I]MEGMIB conjugation did not compromise HER2 binding. Indeed, the Kd of 0.94 ± 0.27 nM measured for [131I]MEGMIB-5F7GGC binding on SKOV-3 cells is perhaps the highest affinity determined for a radiolabeled sdAb monomer in a cell based assay.
Unlike the results noted above at 4°C, [131I]MEGMIB-5F7GGC exhibited relatively low stability, especially in PBS. In contrast, excellent in vitro stability was observed for iso-[125I]SGMIB-5F7 with all radioactivity associated with the intact molecule even after incubation at 37°C for 48 h. These results are consistent with the previously reported limited stability of thiosuccinimide at pH>7 solution [41, 50], however, they do not explain the significantly higher stability observed for this radioconjugate in serum, which is more relevant to in vivo use. We note that the stability of thiosuccinimide does increase under moderately acidic conditions (5.8–7.0) [50], which are often present in the tumor extracellular microenvironment [51].
It is encouraging that the uptake of [131I]MEGMIB-5F7GGC in SKOV-3 cells after a 1-h incubation at 4°C was slightly higher (P<0.05) than that for co-incubated iso-[125I]SGMIB-5F7, which might reflect its somewhat, albeit not statistically significantly higher, affinity (0.94 ± 0.27 nM vs. 1.3 ± 0.2 nM; P=0.1) after site-specific radiolabeling. Given that both prosthetic agents contain the same residualizing guanidine moiety, similar intracellular trapping for the two conjugates would be expected. Indeed, at 1 h, the percentage of initially bound activity that was internalized was similar (50–65%) for [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7 (~50%) and in agreement with previous results with this cell line for 5F7 radiolabeled with other residualizing prosthetic agents [18, 52]. On the other hand, intracellular trapping was higher than that reported for another anti-HER2 sdAb, 2Rs15d, for SKOV-3 cells [26, 28], presumably reflecting the lower degree of internalization as well as lower on-rate of 2Rs15d [26].
At longer incubation times, internalized and membrane-bound activity for [131I]MEGMIB-5F7GGC was significantly lower than that for co-incubated iso-[125I]SGMIB-5F7. These trends were consistent with a previous study involving direct comparison of an anti-HER2 mAb labeled via both an amide (via NHS ester) and thiosuccinimide linkage [9]. We speculate that this may reflect a lower rate of hydrolysis of the NHS-derived amide linkage. Additionally, if the instability was due to transthiolation, it can be minimized by having an adjacent positively charged group that will facilitate opening the thiosuccinimide ring, thereby preventing the thiol exchange reaction [23]. The lower degree of intracellular trapping seen with [131I]MEGMIB-5F7GGC at later time points might also be due to catabolites that are less polar - and hence more permeable to lysosomal and cellular membranes - than those generated from iso-[125I]SGMIB-5F7. Future studies are planned to directly address these possibilities.
Despite the lower stability and cellular retention of activity noted above for [131I]MEGMIB-5F7GGC compared with iso-[125I]SGMIB-5F7, its tumor targeting in vivo was excellent. Moreover, no significant differences were observed in tumor uptake between the two labeled sdAbs at any time point. Unfortunately, there are no published biodistribution results with other 5F7 radioconjugates in this tumor model to serve as benchmarks for comparison. On the other hand, there are several studies evaluating the biodistribution of radioiodinated anti-HER2 2Rs15d sdAb in the same SKOV-3 xenografts model [28, 53, 54], and in all cases, tumor uptake was lower than that observed in the current study for [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7. A recent study evaluated different methods for labeling 2Rs15d including [211At]SAGMB (similar to iso-[125I]SGMIB but different radionuclide and substitution position) and [211At]MSB (a nonresidualizing malemido agent attached via a C-terminal cysteine tail on 2Rs15d) [26]. The uptake for these 211At-labeled 2Rs15d conjugates in SKOV-3 xenografts at 1 h was similar to that observed herein for both [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7; however, at 24 h, 211At tumor uptake was about three times lower than seen in the current study. Contributory factors to the different tumor retention at later time points between the two studies include differences in prosthetic agent residualization, sdAb internalization and halogen stability. With regard to effects related to linkage between the prosthetic agent and the sdAb, stomach uptake for [211At]MSB-2Rs15d was about 10 times higher than that for [211At]SAGMB-2Rs15d, suggesting considerably higher dehalogenation of the 211At-labeled maleimido-derived conjugate had occurred. In contrast, thyroid and stomach uptake of radioiodine activity for [131I]MEGMIB-5F7GGC were quite low and generally about half those for co-administered iso-[125I]SGMIB-5F7.
Consistent with the in vitro data in SKOV-3 cells, retention of radioiodine activity in xenografts from this cell line for both [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7 was not significantly different for the two prosthetic agents. With both labeling methods, about 73% and 40% of the activity levels observed at 1 h were retained at 4 and 24 h, respectively, which compares favorably with results obtained with direct iodination albeit in a different xenograft model [15]. In the catabolism study performed 1 h after labeled sdAb administration, extraction of radioactivity into the tumor homogenate supernatant was variable, and lower than observed from kidney homogenate, suggesting some intracellular trapping had occurred. In addition, ~95% of the radioactivity for both 131I and 125I in the tumor homogenate supernatant was associated with molecules with a molecular weight > 30 kDa, which likely reflects a complex between the labeled sdAbs and HER2 or its extracellular domain.
Activity levels in kidneys and liver 1 h after injection of [131I]MEGMIB-5F7GGC were significantly lower than those for co-administered iso-[125I]SGMIB-5F7 (P=0.01 for kidney uptake, P=0.0001 for liver uptake). The lower activity levels in kidneys observed for [131I]MEGMIB-5F7GGC is particularly encouraging because renal radiation toxicity is the most likely dose-limiting factor for radiolabeled sdAbs. In most cases, tumor-to-normal tissue ratios were considerably higher for [131I]MEGMIB-5F7GGC than those for iso-[125I]SGMIB-5F7 with an almost a two-fold difference seen for kidneys at 1 h. In order to evaluate the cause for this difference in behavior, the nature of the labeled catabolites present in kidneys and urine 1 h after labeled sdAb injection were evaluated. With both [131I]MEGMIB-5F7GGC and iso-[125I]SGMIB-5F7, the fraction of activity associated with >5 kDa molecules was >80% for both in kidneys and urine, which would suggest similar in vivo stability for the two conjugates.
It is likely that the differences in the in vivo behavior of the two sdAb conjugates is due to differential metabolism of thiosuccinimide and amide linkages. Several mechanisms have been proposed to account for the in vivo fate of thiosuccinimides. First, thiosuccinimide linkages can be prone to hydrolysis under moderately basic conditions (7.4–8.8) [23, 41, 50] but have higher stability in a slightly acidic environment (pH 6). The extracellular environment in tumors is often acidic [55] whereas the pH in normal tissues are near neutral. Thus, the pH dependent stability of thiosuccinimide could function as selectively cleavable linker, facilitating clearance of activity from normal tissues. Second, a maleimide can be regenerated from a thiosuccinimide via the retro-Michael reaction and the released maleimide can react with other free thiols on endogenous compounds [56]. In this case, the stability of the thiosuccinimide depends on thiol pKa; those with higher pKa will form a more stable thiosuccinimide [57]. Finally, thiosuccinimide linkages can undergo oxidation to sulfoxide; subsequent elimination of sulfoxide would result in loss of the conjugated species [58]. We speculate that the lability of the thiosuccinimide linkage contributes to the generally faster clearance of activity from normal tissues for [131I]MEGMIB-5F7GGC.
Because serum has a high concentration of molecules with free thiols ~0.6 mM total thiol concentration [59] in human serum, trans-thiolation of [131I]MEGMIB-5F7GGC in blood would seem probable. Consistent with this, the in vitro stability of [131I]MEGMIB-5F7GGC in serum was lower than that for iso-[125I]SGMIB-5F7; however, perplexedly, its stability in PBS was lower than in serum. In addition, the low uptake of [131I]MEGMIB-5F7GGC in blood rich organs such as blood, liver, and spleen does not seem compatible with a high degree of thiol-transfer of [131I]MEGMIB to free thiols. Given the small size of sdAbs, it is likely that tumor targeting/internalization and normal tissue clearance happened much faster than thiol-exchange. In the catabolism studies, a significantly lower percentage of the kidney homogenate supernatant activity from [131I]MEGMIB-5F7GGC (58.5%) was associated with molecules >30 kDa compared with iso-[125I]SGMIB-5F7 (76.1%), an observation counter indicative for thiol transfer to proteins like serum albumin. We speculate that the >30 kDa species observed for both radio-conjugates may represent complexes with HER2 extracellular domain, which is known to be shed from SKOV-3 cells in vitro [60].
Of potential relevance to the current study is a head-to-head comparison of the biodistribution of conjugates formed via amide and thiosuccinimide linkages that reported lower kidney activity levels with the amide linkage [9]. Although this finding is in contradiction with our own, the maleimide moiety evaluated by Pruszyinski et al. was conjugated to thiols generated via treatment of amino groups on the protein with Traut’s reagent, which was different from the engineered cysteine tail 5F7GGC. Moreover, an intact antibody (trastuzumab) with a size greater than the cutoff for renal filtration was evaluated unlike the sdAb evaluated herein. Nonetheless, the earlier work demonstrates that different conjugation strategies (NHS active ester or maleimide) can have significant effects on the biodistribution of radiolabeled protein conjugates. This is also supported by the work of Li et al. [41] evaluating different thiol-based site-specific conjugation methods demonstrating that differences in conjugation chemistry had dramatic effects on the biodistribution of the radio-conjugates [41, 42].
5. Conclusion
In summary, we developed a site-specific and residualizing labeling agent for the radioiodination of the anti-HER2 sdAb 5F7GGC in radiochemical yields comparable to those obtained for random labeling with the analogous lysine-specific prosthetic agent.
Compared with our previous lead agent iso-[125I]SGMIB-5F7, the site-specific conjugate [131I]MEGMIB-5F7GGC exhibited nearly identical binding affinity, immunoreactivity and intracellular trapping capacity, but with the prospect of offering a product with greater homogeneity. In mice with HER2-expressing SKOV-3 xenografts, [131I]MEGMIB-5F7GGC has tumor targeting and retention similar to that for iso-[125I]SGMIB-5F7, but had significantly lower uptake in kidney and other normal tissues. Although the exact mechanism responsible for the faster clearance of [131I]MEGMIB-5F7GGC from normal tissues is unclear, given its excellent tumor targeting and fast renal clearance, further evaluation of [*I]MEGMIB-5F7GGC as an imaging agent and targeted radiotherapeutic is warranted.
Scheme 2.
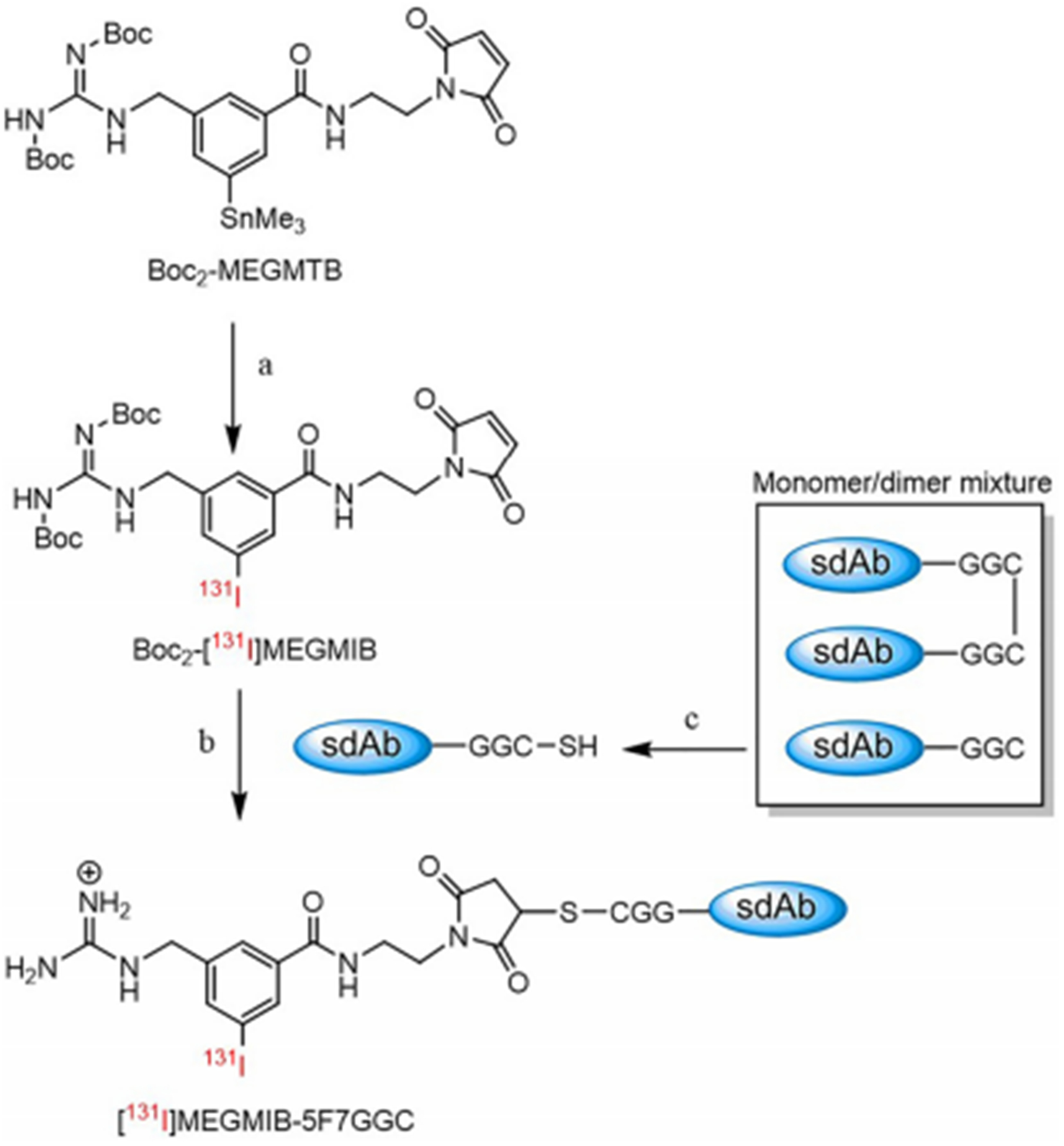
Radiolabeling of the Boc2-MEGMTB tin precursor and the radioconjugation of [131I]MEGMIB to the anti-Her2 sdAb 5F7GGC. a) i: [131I]NaI, NCS, acetic acid, MeOH, ii: RP-HPLC; b) i: TFA, ii: 0.2 M NH4OAc, 5 mM EDTA buffer, 37 °C, 45 min; c) immobilized TCEP gel, 0.2 M NH4OAc, 5 mM EDTA buffer, 37 °C, 1 h.
Acknowledgements
The authors acknowledge the National Cancer Institute for providing support for this research under Grant CA42324. We thank Xiao-Guang Zhao for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
G.V. and M.Z are consultants of Cereius, Inc and hold ownership interest, including patents/patent applications in single domain antibody fragment radiolabeling methods.
References
- [1].Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, and McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987;235:177–82. [DOI] [PubMed] [Google Scholar]
- [2].Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989;244:707–12. [DOI] [PubMed] [Google Scholar]
- [3].Holbro T and Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol 2004;44:195–217. [DOI] [PubMed] [Google Scholar]
- [4].Rasaneh S, Rajabi H, Babaei MH, Daha FJ, and Salouti M. Radiolabeling of trastuzumab with 177Lu via DOTA, a new radiopharmaceutical for radioimmunotherapy of breast cancer. Nucl Med Biol 2009;36:363–9. [DOI] [PubMed] [Google Scholar]
- [5].Price E, Zeglis B, Cawthray J, Ramos N, Ramogida C, Lewis J, et al. H4octapatrastuzumab: An acyclic chelator-immunoconjugate with superior properties to DOTA for In-111/Lu-177 imaging and therapy. J Nucl Med 2013;54:500–. [Google Scholar]
- [6].Palm S, Enmon RM, Matei C, Kolbert KS, Xu S, Zanzonico PB, et al. Pharmacokinetics and biodistribution of 86Y-trastuzumab for 90Y dosimetry in an ovarian carcinoma model: correlative MicroPET and MRI. J Nucl Med 2003;44:1148–55. [PubMed] [Google Scholar]
- [7].Kotts C, Su F, Leddy C, Dodd T, Scates S, Shalaby M, et al. 186Re-labeled antibodies to p185HER2 as HER2-targeted radioimmunopharmaceutical agents: comparison of physical and biological characteristics with 125I and 131I-labeled counterparts. Cancer Biother Radio 1996;11:133–44. [DOI] [PubMed] [Google Scholar]
- [8].Chen K-T, Lee T-W, and Lo J-M. In vivo examination of 188Re (I)-tricarbonyl-labeled trastuzumab to target HER2-overexpressing breast cancer. Nucl Med Biol 2009;36:355–61. [DOI] [PubMed] [Google Scholar]
- [9].Pruszynski M, Koumarianou E, Vaidyanathan G, Chitneni S, and Zalutsky MR. D-amino acid peptide residualizing agents bearing N-hydroxysuccinimido-and maleimido-functional groups and their application for trastuzumab radioiodination. Nucl Med Biol 2015;42:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Debie P, Lafont C, Defrise M, Hansen I, van Willigen DM, van Leeuwen FW, et al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J Control Release 2020;317:34–42. [DOI] [PubMed] [Google Scholar]
- [11].Kijanka M, Warnders F-J, El Khattabi M, Lub-de Hooge M, van Dam GM, Ntziachristos V, et al. Rapid optical imaging of human breast tumour xenografts using anti-HER2 VHHs site-directly conjugated to IRDye 800CW for image-guided surgery. Eur J Nucl Med Mol I 2013;40:1718–29. [DOI] [PubMed] [Google Scholar]
- [12].Pruszynski M, Kang CM, Koumarianou E, Vaidyanathan G, and Zalutsky MR. D-amino acid peptide residualizing agents for protein radioiodination: Effect of aspartate for glutamate substitution. Molecules 2018;23:1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gonzalez-Sapienza G, Rossotti MA, and Tabares-da Rosa S. Single-domain antibodies as versatile affinity reagents for analytical and diagnostic applications. Front Immunol 2017;8:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vaneycken I, Devoogdt N, Van Gassen N, Vincke C, Xavier C, Wernery U, et al. Preclinical screening of anti-HER2 nanobodies for molecular imaging of breast cancer. FASEB J 2011;25:2433–46. [DOI] [PubMed] [Google Scholar]
- [15].Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, et al. Targeting breast carcinoma with radioiodinated anti-HER2 Nanobody. Nucl Med Biol 2013;40:52–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].D’huyvetter M, Aerts A, Xavier C, Vaneycken I, Devoogdt N, Gijs M, et al. Development of 177Lu-nanobodies for radioimmunotherapy of HER2-positive breast cancer: evaluation of different bifunctional chelators. Contrast Media Mol I 2012;7:254–64. [DOI] [PubMed] [Google Scholar]
- [17].Vaidyanathan G and Zalutsky MR. Synthesis of N-succinimidyl 4-guanidinomethyl-3-[*I] iodobenzoate: a radio-iodination agent for labeling internalizing proteins and peptides. Nat Protoc 2007;2:282. [DOI] [PubMed] [Google Scholar]
- [18].Choi J, Vaidyanathan G, Koumarianou E, Kang CM, and Zalutsky MR. Astatine-211 labeled anti-HER2 5F7 single domain antibody fragment conjugates: Radiolabeling and preliminary evaluation. Nucl Med Biol 2018;56:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Spicer CD and Davis BG. Selective chemical protein modification. Nat Commun 2014;5:4740. [DOI] [PubMed] [Google Scholar]
- [20].Cal PM, Bernardes GJ, and Gois PM. Cysteine-Selective Reactions for Antibody Conjugation. Angew 2014;53:10585–7. [DOI] [PubMed] [Google Scholar]
- [21].Gunnoo SB and Madder A. Chemical protein modification through cysteine. ChemBioChem 2016;17:529–53. [DOI] [PubMed] [Google Scholar]
- [22].Chalker JM, Bernardes GJ, Lin YA, and Davis BG. Chemical modification of proteins at cysteine: opportunities in chemistry and biology. Chem Asian J 2009;4:630–40. [DOI] [PubMed] [Google Scholar]
- [23].Ravasco JM, Faustino H, Trindade A, and Gois PM. Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chem Eur 2019;25:43–59. [DOI] [PubMed] [Google Scholar]
- [24].Khawli LA, Van Den Abbeele AD, and Kassis AI. N-(m-[125I] iodophenyl) maleimide: an agent for high yield radiolabeling of antibodies. Int J Rad Appl Instrum [B] 1992;19:289–95. [DOI] [PubMed] [Google Scholar]
- [25].Vaidyanathan G, Alston KL, Bigner DD, and Zalutsky MR. N ε-(3-[*I] iodobenzoyl)-Lys5-N α-maleimido-Gly1-GEEEK ([* I] IB-Mal-D-GEEEK): a radioiodinated prosthetic group containing negatively charged D-glutamates for labeling internalizing monoclonal antibodies. Bioconjugate Chem 2006;17:1085–92. [DOI] [PubMed] [Google Scholar]
- [26].Dekempeneer Y, Bäck T, Aneheim E, Jensen H, Puttemans J, Xavier C, et al. Labeling of anti-HER2 nanobodies with astatine-211: Optimization and the effect of different coupling reagents on their in vivo behaviour. Mol Pharm 2019. [DOI] [PubMed] [Google Scholar]
- [27].Chatalic KL, Veldhoven-Zweistra J, Bolkestein M, Hoeben S, Koning GA, Boerman OC, et al. A novel 111In-labeled anti–prostate-specific membrane antigen nanobody for targeted SPECT/CT imaging of prostate cancer. J Nucl Med 2015;56:1094–9. [DOI] [PubMed] [Google Scholar]
- [28].Zhou Z, Chitneni SK, Devoogdt N, Zalutsky MR, and Vaidyanathan G. Fluorine-18 labeling of an anti-HER2 VHH using a residualizing prosthetic group via a strain-promoted click reaction: Chemistry and preliminary evaluation. Bioorg Med Chem 2018;26:1939–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vaidyanathan G, McDougald D, Choi J, Pruszynski M, Koumarianou E, Zhou Z, et al. N-Succinimidyl 3-((4-(4-[18F] fluorobutyl)-1 H-1, 2, 3-triazol-1-yl) methyl)-5-(guanidinomethyl) benzoate ([18F] SFBTMGMB): a residualizing label for 18F-labeling of internalizing biomolecules. Org Biomol 2016;14:1261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Choi J, Vaidyanathan G, Koumarianou E, McDougald D, Pruszynski M, Osada T, et al. N-Succinimidyl guanidinomethyl iodobenzoate protein radiohalogenation agents: Influence of isomeric substitution on radiolabeling and target cell residualization. Nucl Med Biol 2014;41:802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lindmo T, Boven E, Cuttitta F, Fedorko J, and Bunn P Jr. Determination of the immunoreactive function of radiolabeled monoclonal antibodies by linear extrapolation to binding at infinite antigen excess. J Immunol Methods 1984;72:77–89. [DOI] [PubMed] [Google Scholar]
- [32].Foulon CF, Reist CJ, Bigner DD, and Zalutsky MR. Radioiodination via D-amino acid peptide enhances cellular retention and tumor xenograft targeting of an internalizing anti-epidermal growth factor receptor variant III monoclonal antibody. Cancer Res 2000;60:4453–60. [PubMed] [Google Scholar]
- [33].Zhou Z, Vaidyanathan G, McDougald D, Kang CM, Balyasnikova I, Devoogdt N, et al. Fluorine-18 Labeling of the HER2-Targeting Single-Domain Antibody 2Rs15d Using a Residualizing Label and Preclinical Evaluation. Mol Imaging Biol 2017;19:867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, et al. Improved tumor targeting of anti-HER2 nanobody through N-succinimidyl 4-guanidinomethyl-3-iodobenzoate radiolabeling. J Nucl Med 2014;55:650–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 2008;26:925. [DOI] [PubMed] [Google Scholar]
- [36].Pillow TH, Tien J, Parsons-Reponte KL, Bhakta S, Li H, Staben LR, et al. Site-specific trastuzumab maytansinoid antibody–drug conjugates with improved therapeutic activity through linker and antibody engineering. J Med Chem 2014;57:7890–9. [DOI] [PubMed] [Google Scholar]
- [37].Strop P, Liu S-H, Dorywalska M, Delaria K, Dushin RG, Tran T-T, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Bio 2013;20:161–7. [DOI] [PubMed] [Google Scholar]
- [38].Wilbur DS, Chyan M-K, Nakamae H, Chen Y, Hamlin DK, Santos EB, et al. Reagents for astatination of biomolecules. 6. An intact antibody conjugated with a maleimido-closo-decaborate (2-) reagent via sulfhydryl groups had considerably higher kidney concentrations than the same antibody conjugated with an isothiocyanato-closodecaborate (2-) reagent via lysine amines. Bioconjugate Chem 2012;23:409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Arano Y, Wakisaka K, Ohmomo Y, Uezono T, Mukai T, Motonari H, et al. Maleimidoethyl 3-(tri-n-butylstannyl) hippurate: A useful radioiodination reagent for protein radiopharmaceuticals to enhance target selective radioactivity localization. J Med Chem 1994;37:2609–18. [DOI] [PubMed] [Google Scholar]
- [40].Lindbo S, Garousi J, Mitran B, Altai M, Buijs J, Orlova A, et al. Radionuclide tumor targeting using ADAPT scaffold proteins: aspects of label positioning and residualizing properties of the label. J Nucl Med 2018;59:93–9. [DOI] [PubMed] [Google Scholar]
- [41].Li L, Olafsen T, Anderson A-L, Wu A, Raubitschek AA, and Shively JE. Reduction of kidney uptake in radiometal labeled peptide linkers conjugated to recombinant antibody fragments. Site-specific conjugation of DOTA-peptides to a Cys-diabody. Bioconjugate Chem 2002;13:985–95. [DOI] [PubMed] [Google Scholar]
- [42].Tavaré R, Wu WH, Zettlitz KA, Salazar FB, McCabe KE, Marks JD, et al. Enhanced immunoPET of ALCAM-positive colorectal carcinoma using site-specific 64Cu-DOTA conjugation. Protein Eng Des Sel 2014;27:317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Morais M and Ma MT. Site-specific chelator-antibody conjugation for PET and SPECT imaging with radiometals. Drug Discov Today Technol 2018;30:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fay R and Holland JP. The impact of emerging bioconjugation chemistries on radiopharmaceuticals. J Nucl Med 2019;60:587–91. [DOI] [PubMed] [Google Scholar]
- [45].Zhang Y, Park K-Y, Suazo KF, and Distefano MD. Recent progress in enzymatic protein labelling techniques and their applications. Chem Soc Rev 2018;47:9106–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hylarides MD, Wilbur DS, Reed MW, Hadley SW, Schroeder JR, and Grant LM. Preparation and in vivo evaluation of an N-(p-[125I] iodophenethyl) maleimide-antibody conjugate. Bioconjugate Chem 1991;2:435–40. [DOI] [PubMed] [Google Scholar]
- [47].Adumeau P, Sharma SK, Brent C, and Zeglis BM. Site-specifically labeled immunoconjugates for molecular imaging—part 1: cysteine residues and glycans. Mol Imaging Biol 2016;18:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Grant GA. Modification of cysteine. Curr Protoc Protein Sci 2017;87:15.1. 1–.1. 23. [DOI] [PubMed] [Google Scholar]
- [49].Strand J, Nordeman P, Honarvar H, Altai M, Orlova A, Larhed M, et al. Site-Specific Radioiodination of HER2-Targeting Affibody Molecules using 4-Iodophenethylmaleimide Decreases Renal Uptake of Radioactivity. ChemistryOpen 2015;4:174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lewis MR and Shively JE. Maleimidocysteineamido-DOTA derivatives: new reagents for radiometal chelate conjugation to antibody sulfhydryl groups undergo pH-dependent cleavage reactions. Bioconjugate Chem 1998;9:72–86. [DOI] [PubMed] [Google Scholar]
- [51].Wike-Hooley J, Haveman J, and Reinhold H. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol 1984;2:343–66. [DOI] [PubMed] [Google Scholar]
- [52].Zhou Z, McDougald D, Devoogdt N, Zalutsky MR, and Vaidyanathan G. Labeling Single Domain Antibody Fragments with Fluorine-18 Using 2, 3, 5, 6-Tetrafluorophenyl 6-[18F] Fluoronicotinate Resulting in High Tumor-to-Kidney Ratios. Mol Pharm 2018;16:214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].D’Huyvetter M, De Vos J, Xavier C, Pruszynski M, Sterckx YG, Massa S, et al. 131I-labeled anti-HER2 camelid sdAb as a theranostic tool in cancer treatment. Clin Cancer Res 2017;23:6616–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhou Z, Devoogdt N, Zalutsky MR, and Vaidyanathan G. An efficient method for labeling single domain antibody fragments with 18F using tetrazine-trans-cyclooctene ligation and a renal brush border enzyme-cleavable linker. Bioconjugate Chem 2018;29:4090–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].McCoy C, Parkins C, Chaplin D, Griffiths J, Rodrigues L, and Stubbs M. The effect of blood flow modification on intra-and extracellular pH measured by 31P magnetic resonance spectroscopy in murine tumours. Br J Cancer 1995;72:905–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wu H, LeValley PJ, Luo T, Kloxin AM, and Kiick KL. Manipulation of Glutathione-Mediated Degradation of Thiol–Maleimide Conjugates. Bioconjugate Chem 2018;29:3595–605. [DOI] [PubMed] [Google Scholar]
- [57].Baldwin AD and Kiick KL. Tunable degradation of maleimide–thiol adducts in reducing environments. Bioconjugate Chem 2011;22:1946–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Fishkin N, Maloney EK, Chari RV, and Singh R. A novel pathway for maytansinoid release from thioether linked antibody–drug conjugates (ADCs) under oxidative conditions. ChemComm 2011;47:10752–4. [DOI] [PubMed] [Google Scholar]
- [59].Turell L, Radi R, and Alvarez B. The thiol pool in human plasma: the central contribution of albumin to redox processes. Free Radic Biol Med 2013;65:244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gall VA, Philips AV, Qiao N, Clise-Dwyer K, Perakis AA, Zhang M, et al. Trastuzumab increases HER2 uptake and cross-presentation by dendritic cells. Cancer Res 2017;77:5374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]