

Abstract
Malignant Hyperthermia(MH) was described 60 years ago and our understanding of the clinical and pathophysiologic features of this syndrome continue to advance. MH is attributed to a genetically determined susceptibility to an abnormal skeletal muscle cell calcium flux triggered by potent inhalation anesthetics (e.g., isoflurane) or succinylcholine. Since dantrolene sodium was approved in 1979 the mortality from malignant hyperthermia dropped substantially, but remains 4-10%. The exact incidence of malignant hyperthermia reactions is unknown but may be as high as one in 25,000 anesthetics. Variants in three genes cause the susceptibility to this disorder. The RYR1 gene is most common followed by CACNA1S and STAC3. While the caffeine-halothane contracture test and the in vitro contracture test are considered the gold standards for diagnosis of susceptibility to malignant hyperthermia, there have been important advances in genetic testing. Due to these advances, it is timely for the field to consider the utility and practicability of screening for malignant hyperthermia susceptibility using genomic testing. Here we pose a simple, but bold question; what would it take to end deaths from malignant hyperthermia? We review recent advances and propose a scientific and clinical pathway toward this audacious goal to provoke discussion in the field.
Malignant hyperthermia is a syndrome of acutely disordered skeletal muscle excitation-contraction coupling leading to fever, acidosis, hypercapnia, tachycardia, hyperkalemia, muscle rigidity, and rhabdomyolysis that can be triggered by potent inhalation anesthetics and depolarizing neuromuscular blocking agents (e.g., succinylcholine).1 A malignant hyperthermia reaction is challenging to manage, requiring rapid interventions to halt the procedure, discontinue the triggering agents, administer dantrolene, correct dysrhythmias, and other crucial supportive measures.2,3 Even though early intervention using these measures is effective in aborting or ameliorating the reaction, the mortality for a malignant hyperthermia reaction is still 4 – 10%.4,5 Morbidity is more common, can be severe, and in some cases long lasting (e.g., renal failure). Malignant hyperthermia susceptibility can be a component of some congenital myopathies but it is most commonly the only manifestation in an affected individual and it is this latter manifestation we are focused on here. Malignant hyperthermia susceptibility is a heritable trait, primarily associated with variants in either the type 1 ryanodine receptor (RYR1) intracellular calcium channel or the alpha 1S subunit (CACNA1S) of the voltage-dependent L-type Ca2+ channel. The disorder is heritable, but it is not always inherited: rare cases have been shown to be due to de novo mutation events. Another gene associated with malignant hyperthermia reactions is STAC3, although all the reported occurrences involve individuals with biallelic variants who have an apparent myopathy: here we are focused on individuals who are asymptomatic until exposed to a triggering agent. A recent report6 suggested that TRPV1 is also associated with malignant hyperthermia, but this has not been confirmed. Estimates of the prevalence of malignant hyperthermia susceptibility vary widely, from 1/200 to 1/3,000,7–9 although the clinical incidence of malignant hyperthermia reactions is much lower, between 1:10,000 and 1:150,000 general anesthetics.10,11 From 50% to >70% of those who have experienced a malignant hyperthermia reaction are found to have at least one of more than 200 variants in either RYR1 or CACNA1S, indicating that there is both locus and allelic heterogeneity.1,12
Research into malignant hyperthermia susceptibility over the past decades has provided important insights into the epidemiology, pathophysiology, clinical management, and genetics of this disorder. At the same time, it is recognized that the mortality associated with malignant hyperthermia has declined little since the widespread adoption of dantrolene. Given the advancement in scientific understanding and medical management that has occurred, we pose to the field a simple and direct question; what would it take to end deaths from malignant hyperthermia?
We are posing this rhetorical question to organize our thinking and direct our clinical and scientific resources toward an ideal objective. The complete elimination of morbidity and mortality from malignant hyperthermia is likely impossible – since a complete understanding of the biology of this trait, identification of all at-risk individuals, and changing their anesthetic management to the degree needed to drive the mortality to zero is complex. We argue that it is conceivable that we can come close to eradicating all deaths from malignant hyperthermia susceptibility or to sufficiently reduce the death rate that the efforts and expenses would be worthwhile. Going forward, malignant hyperthermia susceptibility is an attractive target for a genomic screening effort for a number of reasons.
The primary disease manifestation is typically dramatic, severe, and quantifiable
Most people have almost zero risk of malignant hyperthermia, a few people have a high risk, and most of the latter group can be identified
There is relatively little stigma associated with a diagnosis of malignant hyperthermia susceptibility so pre-symptomatic diagnosis is not highly aversive
An operating room malignant hyperthermia reaction is completely avoidable in known susceptible individuals by avoiding exposure to the triggering agents, which involves decontamination of the anesthetic workstation and use of alternative anesthetics
Genetic tools with the potential to identify individuals with malignant hyperthermia susceptibility are increasingly powerful and costs are falling rapidly
Here we outline some ideas about what an organized program to substantially reduce deaths from malignant hyperthermia ought to comprise:
Develop a robust and practical physiologic diagnostic test
Research to identify all genetic loci that cause or contribute to malignant hyperthermia susceptibility
Establish the pathogenicity of all variants in genes that cause or contribute to malignant hyperthermia susceptibility
Develop and pilot genomic screening techniques
Consultation services to confirm malignant hyperthermia susceptibility diagnoses and educate individuals with malignant hyperthermia susceptibility
Health care information systems for real-time support and resources for the management of a malignant hyperthermia reaction and management of at-risk individuals
One can readily envision that accomplishing these objectives is feasible and if accomplished, we could reduce the risks of malignant hyperthermia at each step of the process from operative planning to discharge. For example, if we can reduce the number of susceptible individuals with who are exposed to a triggering agent by 75% and reduce the mortality rate of a malignant hyperthermia reaction by 75%, then deaths from malignant hyperthermia would be reduced by more than 90%. This is an exciting and worthy aim and we outline some important considerations for the unmet objectives below.
Develop a robust and practical physiologic diagnostic test
Accurate phenotyping is essential in the genetic investigation of any trait. Singly, none of the clinical signs of a malignant hyperthermia reaction is specific, but a nascent reaction can be recognized by an astute clinician and the management imperative is to abort a reaction as soon as it is suspected. It is now rare for a reaction to reach such a fulminant stage that the diagnosis is unequivocal. Even when the proband’s diagnosis could be made on the basis of their clinical reaction, clinical phenotyping for other family members is challenging. Scientific advances in the genetics of malignant hyperthermia have substantially been founded on the use of the malignant hyperthermia susceptibility phenotype determined using contracture testing. Indeed, the original identification of the RYR1 and CACNA1S susceptibility loci, and many other large genetic studies of malignant hyperthermia have come from countries where contracture testing of affected families is quality-controlled and practicable.
While it might be ideal that contracture testing was universally available, there are numerous barriers to this goal, which are beyond the scope of this commentary. Therefore, the development of a physiologic diagnostic confirmation test that is analytically robust, but which can use tissue that can be sampled locally (ideally less invasively) and transported to the testing center, would greatly improve accessibility to malignant hyperthermia testing. The challenge here is daunting – although we would be eager to work toward an alternative clinical phenotyping test, there are no existing data to our knowledge that point to a ready path to such an assay.
Research to identify all genetic loci that cause or contribute to malignant hyperthermia susceptibility
Genomic technologies are rapidly advancing, primarily due to chip-based DNA testing platforms 13 and next generation sequencing.14 Whereas Sanger sequencing of RYR1 and CACNA1S has been and remains expensive, next generation sequencing panel tests that include these genes are now available at costs well below that of Sanger sequencing. Next generation exome and genome sequencing are increasingly available in many countries and becoming an affordable part of research and health care. These rapid advances and falling costs enable both research and clinical genomic testing that were inconceivable just a few years ago. They enable rapid identification of sequence variants in individuals with putative inherited diseases. However, these variants may number several thousand in each sample and predicting which variant(s) is(are) implicated in the disease can be challenging. In malignant hyperthermia susceptibility where a single missense variant may be all that is required, once variants in RYR1, CACNA1S, and STAC3 have been excluded, this approach has proved fruitless to date. So far, relatively few samples from malignant hyperthermia susceptible individuals have undergone exome or genome sequencing. If a larger number can be sequenced we will more likely be able to identify rare recurrent variants or genes that have an increased burden of rare variants. We propose that there should be a coordinated program of clinical and research testing such that every individual with a malignant hyperthermia reaction or positive contracture test is evaluated by next generation sequencing to increase the chances of identifying the causative variant(s). This should be a mix of both clinical testing and clinical research testing. Clinical sequencing of known malignant hyperthermia susceptibility-associated genes is available from a number of laboratories (See “Online Resources”, below). De-identified data from all who are sequenced and found to harbor a pathogenic or likely pathogenic variant (determined as per Refs 15,16) should be deposited in a public repository, such as ClinVar or a dedicated malignant hyperthermia database so that all can benefit from this knowledge. Individuals who are not found to have an unambiguously pathogenic variant should be referred to a clinical research program to be further evaluated to better understand the genetic basis of this disease. The pooling and organization of these cases and data will add immeasurably to efforts to fully catalog genetic variation associated with malignant hyperthermia susceptibility.
Escalation to next generation sequencing may also prove useful in cases where malignant hyperthermia susceptibility is apparently more genetically complex,12,17 especially if combinations of rare variants are involved. However, if the genetic model involves combinations of several more common variants, sample sizes will need to be even larger and SNP-chip genotyping is likely to be more cost-effective.
Establish the pathogenicity of all variants in genes that cause or contribute to malignant hyperthermia susceptibility
Efforts are underway to comprehensively characterize the pathogenicity of reported variants in RYR1 and CACNA1S through the ClinGen Variant Curation Expert Panel process (https://www.clinicalgenome.org/affiliation/50038/). This effort is initially focused on the variants proposed by the European Malignant Hyperthermia Group (https://www.emhg.org/) with an adaptation of the American College of Medical Genetics and Genomics variant pathogenicity standards.15 These standards, which must be adapted to take into account knowledge of the biology of RYR1 and MHS, comprise 27 criteria including observations of inheritance, case-control studies, functional studies, in silico predictors, and conforms to the current international standard for variant pathogenicity assertions. A major deficit in being able to assign pathogenic status is the small number of variants that have been robustly functionally characterized in relevant model systems. Current testing is robust, but low throughput. A recent revolution in functional genomics heralds a realistic prospect of overcoming this bottleneck. Prime editing, an adaptation of CRISPR technology18, coupled with strategies for high-throughput functional assays19 have the potential to support highly robust functional assessments of all variants, even before they are detected in a human. If these technologies can be adapted to RYR1 and other genes implicated in malignant hyperthermia susceptibility, they have the potential to provide for high-throughput, low cost functional in vitro assays for every potential variant. This task is not trivial, but it should be feasible. Even achieving a goal of assessing the pathogenicity of variants that can account for 80% of known cases of malignant hyperthermia susceptibility would create a set of pathogenic variants that could be employed for clinical research to test the practicality of pre-operative screening.
Develop and pilot genomic screening techniques
A future is coming where large numbers of individuals undergo genome-wide screening that encompasses many disease and pharmacogenetic susceptibilities: it is essential to develop evidence to support this approach on a disease-by-disease basis. We propose that a trial of preoperative screening for malignant hyperthermia susceptibility would serve as a proof of principle to test the applicability and utility of extracting malignant hyperthermia-associated variants from genomic or exomic data. Once a suitable set of pathogenic variants is identified, genomic screening for malignant hyperthermia susceptibility could be piloted on a population of individuals scheduled for elective surgery. The size and power analysis of such a study will require more accurate estimates of prevalence and the diagnostic yield of a given set of pathogenic variants. We propose that this could be fruitful, even without a clear understanding of penetrance of the variants, because one could eliminate MH reactions if every person with an at-risk genotype was administered a non-triggering agent. While general population screening for malignant hyperthermia susceptibility will likely not be practical for some time, an ever-increasing number of individuals with variants in RYR1 and CACNA1S are being identified through secondary findings from exome and genome sequencing.20,21 These individuals provide opportunities to study and pilot approaches to presymptomatic diagnosis. When malignant hyperthermia-susceptible individuals are identified through preoperative screening or secondary findings and there is no personal or family history of suspected malignant hyperthermia, the presence of the variant represents the only known risk factor for malignant hyperthermia susceptibility in the family. Identifying an individual with malignant hyperthermia susceptibility is an opportunity to classify all members within a family, where the risk of having malignant hyperthermia susceptibility (50% for first degree relatives) is orders of magnitude higher than the general population. Prospective determination of risk of relatives can therefore be made by testing for the single variant, which is simpler to perform and interpret than is exome, panel, or even full gene testing as the laboratory does not need to interpret other variants.
Consultation services to confirm diagnoses and educate individuals with malignant hyperthermia susceptibility
A genetic test, even with physiologic confirmation is not enough – these individuals also need access to a knowledgeable provider (most likely anesthesiologist, neurologist specializing in myopathy, or a clinical geneticist) to engage with the affected individual to analyze the test results, make the clinical-molecular diagnosis, and educate the patient, their family, and care provider about their disorder. The affected individual is a key part of the puzzle – it will be critical that they accept and understand their diagnosis and its implications to maximize the likelihood that the information is used to their benefit. Support groups such as MHAUS (in North America) can be helpful to identify experts and provide information (see links, below).
There must be support also for care providers unfamiliar with incorporating genomic test information into anesthetic management decisions. Taking the data from the advances we propose into account, professional bodies (such as ASA) will need to develop policies and practice standards that are based on the risk stratification of genomic predictive testing. An analogous approach has been adopted in obstetrics, where the highly complex non-invasive prenatal genomic screening test has been rapidly taken up, with clear guidelines and risk determinations. Such guidelines are no guarantee of good care nor are they a perfect shield from liability, but they give providers clear guidance and substantially lessen risks.
Health care information systems for real-time support resources for the management of a malignant hyperthermia reaction and management of at-risk individuals
In North America, the Malignant Hyperthermia Association of the United States (MHAUS) provides 24/7 hotline support for clinicians managing patients with a known or suspected malignant hyperthermia reaction (similar resources are available in other countries). These valuable resources should be universally recognized and readily used, but additional resources for the identification and management of malignant hyperthermia should be developed. Artificial intelligence-driven22 patient monitoring and clinical decision support tools23 within the electronic health record could be developed to support preoperative decision-making regarding test results, facilitate real-time early recognition of a malignant hyperthermia event, and other decisions. Finally, information on malignant hyperthermia susceptibility should be readily portable so that patients can benefit from it no matter where they receive their care.
Conclusion
None of these approaches alone will accomplish our objective. Instead, we recognize that it will be essential that research, screening, education, and management are integrated into a functional whole systems-based approach to end deaths from malignant hyperthermia. This proposal is centered on a genomics-centered approach to malignant hyperthermia susceptibility. This is not to say that a major, disruptive advance in muscle physiologic testing could not occur that would change this assessment entirely – disruptive advances are by their nature unpredictable. We propose a model for organizing the necessary genetic and anesthetic activities needed to build an integrated system that will capture all events, maximize knowledge, and reduce death and disability from this disease. We propose a flow diagram that incorporates all of these activities and builds data and expertise into the future. (Figure)
Figure:
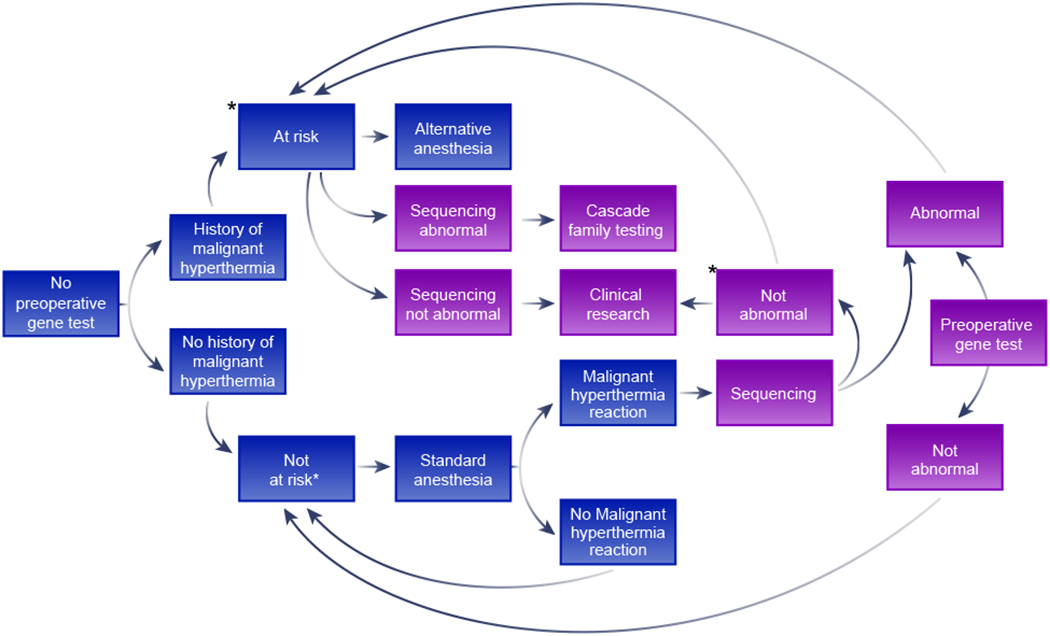
A model for the future management of malignant hyperthermia susceptibility risk through genomic screening. The blue boxes represent the current, phenotypic ascertainment approach to malignant hyperthermia susceptibility, where neither contracture tests nor DNA testing is practical and the purple boxes represent the proposed future approach, supplementing the present approach. ‘Abnormal testing’ means the presence of a variant that is likely to cause malignant hyperthermia susceptibility. ‘Not abnormal testing’ is the converse of that result. Boxes with an asterisk indicate steps that contracture testing should be considered to assess malignant hyperthermia risk. Note that contracture testing may be done prior to DNA testing or reserved for those who show no abnormality on sequencing. The phrase ‘history of malignant hyperthermia’ should be considered as at least a reasonably strong history and ‘malignant hyperthermia reaction’ should be considered as at least reasonably strong evidence of a malignant hyperthermia reaction.
Population screening for genetic disease is not risk-free. There will be false-positive and false-negative results. The risk of false positives would be that individuals would be offered non-triggering agents unnecessarily. False negatives could lead to very rare occurrences of malignant hyperthermia. There is also a risk that by reducing the incidence of malignant hyperthermia, anesthesiologists would be become less familiar with the recognition and management of a reaction. While in the US the Genetic Information Non-discrimination Act should be protective in most cases, it is possible that some individuals who are diagnosed by screening (true or false-positive) could be, for example, denied entry to the Armed Forces.
We recognize that these goals are grand and challenging. We also recognize, and indeed hope, that others will debate and help us to refine our proposals and weigh in with different approaches that could help us work toward the goal of ending deaths from malignant hyperthermia.
Acknowledgments
Funding Sources:
This work was supported by: NIH grant AR053349 to Robert T. Dirksen, Ph.D.
NIH grants HG200359, HG200387 to Leslie G. Biesecker, M.D.
Competing Interest Disclosure:
James Weber, Ph.D., is the President of Prevention Genetics, Marshfield, Wisconsin. Leslie G. Biesecker has received research support from ArQule (now wholly owned by Merck, Inc.), Pfizer, Inc., and honoraria from Cold Spring Harbor Laboratory.
Sheila Riazi has received research support from Norgine Pharmaceuticals
Footnotes
Online Resources:
Genetic Test Registry for RYR1 testing: https://www.ncbi.nlm.nih.gov/gtr/all/tests/?term=6261%5Bgeneid%5D
Genetic Test Registry for CACNA1S testing: https://www.ncbi.nlm.nih.gov/gtr/all/tests/?term=779%5Bgeneid%5D
Malignant Hyperthermia Association of the United States (MHAUS):
The North American Malignant Hyperthermia Registry of MHAUS (https://anest.ufl.edu/namhr)
European Malignant Hyperthermia Group (EMHG):
REFERENCES
- 1.Rosenberg H, Sambuughin N, Riazi S, Dirksen R: Malignant Hyperthermia Susceptibility, GeneReviews((R)). Edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A. Seattle (WA), 1993 [Google Scholar]
- 2.Glahn KP, Ellis FR, Halsall PJ, Muller CR, Snoeck MM, Urwyler A, Wappler F, European Malignant Hyperthermia G: Recognizing and managing a malignant hyperthermia crisis: guidelines from the European Malignant Hyperthermia Group. Br J Anaesth 2010; 105: 417–20 [DOI] [PubMed] [Google Scholar]
- 3.Larach MG, Dirksen SJ, Belani KG, Brandom BW, Metz KM, Policastro MA, Rosenberg H, Valedon A, Watson CB, Society for Ambulatory A, Malignant Hyperthermia Association of the United S, Ambulatory Surgery F, Society for Academic Emergency M, National Association of Emergency Medical T: Special article: Creation of a guide for the transfer of care of the malignant hyperthermia patient from ambulatory surgery centers to receiving hospital facilities. Anesth Analg 2012; 114: 94–100 [DOI] [PubMed] [Google Scholar]
- 4.Larach MG, Brandom BW, Allen GC, Gronert GA, Lehman EB: Cardiac arrests and deaths associated with malignant hyperthermia in north america from 1987 to 2006: a report from the north american malignant hyperthermia registry of the malignant hyperthermia association of the United States. Anesthesiology 2008; 108: 603–11 [DOI] [PubMed] [Google Scholar]
- 5.Rosero EB, Adesanya AO, Timaran CH, Joshi GP: Trends and outcomes of malignant hyperthermia in the United States, 2000 to 2005. Anesthesiology 2009; 110: 89–94 [DOI] [PubMed] [Google Scholar]
- 6.Abeele FV, Lotteau S, Ducreux S, Dubois C, Monnier N, Hanna A, Gkika D, Romestaing C, Noyer L, Flourakis M, Tessier N, Al-Mawla R,Chouabe C, Lefai E, Lunardi J, Hamilton S, Faure J, Van Coppenolle F, Prevarskaya N. TRPV1 variants impair intracellular Ca2+ signaling and may confer susceptibility to malignant hyperthermia. Genetics in Medicine 2019;21:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ibarra MC, Wu S, Murayama K, Minami N, Ichihara Y, Kikuchi H, Noguchi S, Hayashi YK, Ochiai R, Nishino I: Malignant hyperthermia in Japan: mutation screening of the entire ryanodine receptor type 1 gene coding region by direct sequencing. Anesthesiology 2006; 104: 1146–54 [DOI] [PubMed] [Google Scholar]
- 8.Monnier N, Krivosic-Horber R, Payen JF, Kozak-Ribbens G, Nivoche Y, Adnet P, Reyford H, Lunardi J: Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology 2002; 97: 1067–74 [DOI] [PubMed] [Google Scholar]
- 9.Mungunsukh O, Deuster P, Muldoon S, O’Connor F, Sambuughin N: Estimating prevalence of malignant hyperthermia susceptibility through population genomics data. Br J Anaesth 2019; 123: e461–e463 [DOI] [PubMed] [Google Scholar]
- 10.Ording H: Incidence of malignant hyperthermia in Denmark. Anesth Analg 1985; 64: 700–4 [PubMed] [Google Scholar]
- 11.Rosenberg H, Davis M, James D, Pollock N, Stowell K: Malignant hyperthermia. Orphanet J Rare Dis 2007; 2: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller DM, Daly C, Aboelsaod EM, Gardner L, Hobson SJ, Riasat K, Shepherd S, Robinson RL, Bilmen JG, Gupta PK, Shaw MA, Hopkins PM: Genetic epidemiology of malignant hyperthermia in the UK. British Journal of Anaesthesia 2018; 121: 944–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dracopoli NC, Haines JL: Genotyping, Current Protocols in Human Genetics. New Yord, Wiley-Blackwell, 2010, pp 2.0.1–2.0.3 [Google Scholar]
- 14.Biesecker LG, Green RC: Diagnostic clinical genome and exome sequencing. N Engl J Med 2014; 370: 2418–25 [DOI] [PubMed] [Google Scholar]
- 15.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison SM, Biesecker LG, Rehm HL: Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr Protoc Hum Genet 2019; 103: e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw MA, Hopkins PM: Mission Impossible or Mission Futile?: Estimating Penetrance for Malignant Hyperthermia. Anesthesiology 2019; 131: 957–959 [DOI] [PubMed] [Google Scholar]
- 18.Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR: Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019; 576: 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, Shendure J: Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018; 562: 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG: ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in Medicine 2013; 15: 565–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, McKelvey KD, Ormond KE, Richards CS, Vlangos CN, Watson M, Martin CL, Miller DT: Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017; 19: 249–255 [DOI] [PubMed] [Google Scholar]
- 22.Topol EJ: High-performance medicine: the convergence of human and artificial intelligence. Nat Med 2019; 25: 44–56 [DOI] [PubMed] [Google Scholar]
- 23.Tolley CL, Slight SP, Husband AK, Watson N, Bates DW: Improving medication-related clinical decision support. Am J Health Syst Pharm 2018; 75: 239–246 [DOI] [PubMed] [Google Scholar]