

Abstract
Genome editing has powerful applications in research, health care and agriculture. However, the range of possible molecular events resulting from genome editing has been underestimated and the technology remains unpredictable on and away from the target locus. This has considerable impact in providing a safe approach for therapeutic gene editing, agriculture and other applications. This Opinion article discusses how to anticipate and detect those editing events by a combination of assays to capture all possible genomic changes. It also discusses strategies for preventing unwanted effects, critical to appraise the benefit or risk associated with the use of the technology. Anticipating and verifying the result of genome editing are essential for the success for all applications.
Keywords: Genome editing, on-target activity, off-target activity, ectopic insertions, validation
Gene editing: a transformative technology
The application of genome editing is transforming agriculture, biomedical research and health care. The many proposed purposes include the generation of more productive or robust crops and farm animals, animal hosts for the production of tissues for graft purposes and therapies that employ ex vivo or somatic tissue engineering [1–3]. The promise of applicability is turning into reality as illustrated by a first non-randomized Phase I clinical triali in which the use of clustered regularly interspaced short palindromic repeats (CRISPR)-engineered T cells was recently found to be safe [4]. To date, over 20 Phase I/II human clinical trials are underway for a broad range of diseases including cancers, β-thalassemia, sickle cell disease and Duchenne muscular dystrophy (summarized and discussed in [1,5]).
Genome editing is generally based on either zinc finger nucleases [6], transcription activator-like effector nucleases [7] or the CRISPR/CRISPR-associated (Cas) system (see Glossary, [8]). These molecules act by inducing a double-stranded cut in a specific DNA sequence, which results in a genetic alteration as the gap is being repaired. In the clinic, the initial applications aim for deletions of genomic DNA intervals and do not yet involve precision at the nucleotide level, so these can be executed through the sole delivery of a genome editing nuclease. But for more precise editing, such as the generation of point mutations or more intricate changes or even accurate deletion of a genomic segment, single- or double-stranded DNA templates are also delivered, together with the nucleases, to direct the repair to result in a given sequence by homology directed repair (HDR) [9–11] or non-homologous end-joining [12]. Base editors [13] and prime editors [14] are alternative strategies for more precise editing. Overall, the range of genome editing tools is ever increasing and their transformative potential across a wide range of fields of application is immense.
Genome editing: a disruptive but still erratic technology
The safety of genome editing technologies is just as critical as their efficiency for their successful application in health or agriculture. Common to all fields of application are the risks associated with undesired genetic changes that can be triggered by genome editing.
The potential for unwanted off-target nuclease activity was recognized early in the application of the CRISPR/Cas9 system as a genome editing tool [15]. The frequency of such events and the attached risks were the subjects of much debate [16–18]. The general consensus is that, with careful molecular design, off-target events are rare and generally can be segregated away from the allele of interest in genome-edited animals, unless the off-target region is in linkage disequilibrium with the target site; however, they are potentially more pernicious in cultured cells or in a somatic delivery system [19–23]. In those cases, it is particularly essential that off-target events are captured [24]. However, on-target effects and ectopic insertions of donor template are less predictable and have often been underestimated.
But on-target effects of genome editing enzymes are also now better documented. These can take many forms: single nucleotide variations, indels, large and/or complex genomic rearrangements, segmental duplications, chromosomal translocation, terminal chromosomal truncation up to several megabases, or loss of one or both arms of a chromosome ([25–31], Figure 1) and some mutagenic events are not compatible with efficiently populating a cell lineage in vivo [30]. These effects are intimately linked to the kinetics of the enzyme’s interaction with the DNA and with the DNA repair pathways [32,33]. This results in multiple cutting at the target site and it can lead to the alteration of a larger than expected segment by ~1–2 kb up to 50 kb at a frequency of ~15 to 20% of the target DNA in somatic cells [26,25,29]. In early embryos, this additionally translates to mosaicism of the mutated alleles [34–36].
Figure1: Possible outcomes of Cas9 double stranded break of the DNA:
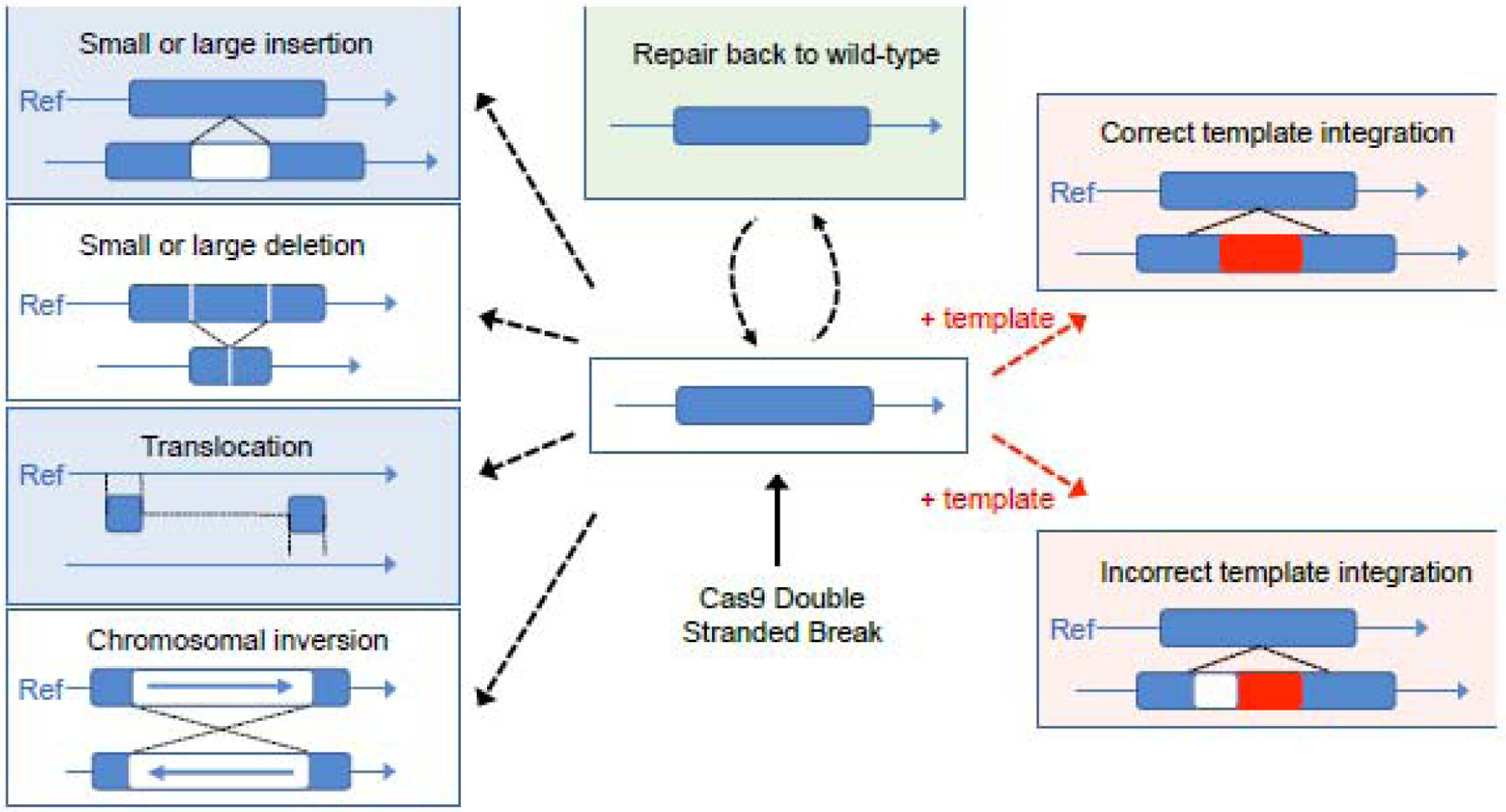
Cas9 induced double stranded break leads to a series of expected and unexpected outcomes. Final editing outcome results in small insertions, deletions or chromosomal translocations or incomplete template integration.
Similarly, repair with template can result in a variety of sequence changes even when the insertion of the repair template is on target (Figure 1). It can yield unpredictable and sometimes complex events at the target site, such as partial insertion of the template, sequence duplications, inversions or rearrangements of the template in combination with endogenous sequences [37–40], as well as ectopic insertions of the repair template. The delivery of template for repair or vectors for nuclease expression can also yield the ectopic insertions that may potentially affect the safety of the approach [41,42]. The recent example of gene editing of the POLLED allele in cattle [43], in which the full outcome had not been identified from the first analysis of the edited cows [44] illustrates the difficulties involved in thoroughly identifying ectopic insertions of the repair template. Additional studies show that pervasive insertion of the donor template across the genome can remain undetected with conventional methods such as PCR and Sanger sequencing [40]. These examples underline questions on the prevalence and type of on-target modifications and ectopic insertions of the donor DNA following co-delivery with nucleases. They also articulate the importance of employing the appropriate assays to evaluate the correctness of the resulting genome editing event(s).
Unknown unintended consequences of genome editing
As genome editing is increasingly used, further unexpected and potentially negative outcomes of its application are still being uncovered:
For example, the perdurance and transmission of DNA DSBs is a phenomenon so far overlooked and that forms a molecular basis for several mixed alleles arising from a single cutting event [31,45].
Also, the occurrence of potentially extensive gene conversion [46] of edited alleles went unnoticed until recently: This consists of the transfer of DNA from one genomic location to another by homologous recombination. Examples of gene conversion are the transfer of the DNA from delta-hemoglobin to beta-hemoglobin in the case of dividing or non-dividing cells, or transfer to the use of the paternal allele as a repair template in the case of embryonic cells [47–49]. Gene conversion could result in a partial or full repair of the allele; it was hypothesized that such a mechanism could be utilised as an internal template repair for precision editing, but this is still disputed [48]. In particular, as conversion tracks may expand well beyond the targeted region, the resulting loss of heterozygocity represents an additional risk for clinical application [31,50].
Finally, and very importantly, it is now becoming increasingly apparent that genomic segments can be inadvertently altered at comparatively large distances from the cutting site [29,51]. The frequency of such outcomes and the genetic range susceptible to alteration following genome editing intervention remain to be fully appraised.
All of these poorly understood consequences pertain to changes to the DNA sequence. Other potential unknown consequences of on- and off-target effects could have an entirely different molecular basis, such as deregulation of the chromatin environment or the three-dimensional organisation of the nucleus, which could change the genome stability or gene expression. The incidence of such potential consequences is as yet largely unexplored,
The context of genome editing application changes the question
In all instances, the challenge is to fully apprehend the editing outcomes that may have adverse consequences. This is likely to require the application of a suite of molecular techniques to interrogate the different artefactual features that can be encountered in genome editing. These features can be diverse in scale (single base to megabase) and may involve additional template insertions. All of these outcomes can occur at the targeted site or ectopically. Secondly, to add more complexity, the extent and nature of lesions varies considerably depending on whether the editing occurs in non-dividing somatic, dividing somatic or germinal (early embryonic) cells. In the case of euploid clonal cell populations or the progeny of founder animals, there are only two alleles for each autosomal locus, and therefore a maximum of two variants may need to be identified. On the other hand, animals born from genome editing of early embryos are generally mosaic [34,37,38]. Modification of pools of cultured cells yield heterogenous cell populations [18] and tissues modified by somatic modification [52] represent yet a larger degree of genetic complexity. In all these examples multiple and potentially very diverse allelic variants are represented at different frequencies. It is critical to elaborate a clear strategy that takes into account these different degrees of genetic complexity to uncover, fully characterize, or ideally prevent, these unwanted events.
The stakes are high as the impact of incomplete characterisation of editing effects is potentially important in all areas of application: in the laboratory, the risk is of irreproducible or artefactual research. Therefore, information obtained with genetically edited founder animals (likely to be mosaic) must be interpreted on the basis of the intrinsic genetic complexity of these animals and the genetic content of progeny must be extensively re-validated. Equally, interpretation of data obtained with edited culture cell pools (where repair may result in many different alleles with various rates) requires an understanding of the genetic composition of these complex cell populations such as mosaicism [25]. When editing is used for the production of agricultural products (plants or animals), it remains unclear whether uncontrolled outcomes may pose a risk to the users. Such variability may prevent licensing for commercialisation by regulators or negatively affect the confidence in the safety of those products by consumers [44,53]. For use in the clinic, in tissue engineering or by somatic delivery, the degree of variability of genomic outcome that may be acceptable in terms of safety remains to be appreciated and may not represent an insurmountable obstacle [4]. However, the range of edited sequences that can result from a given therapeutic intervention still must be thoroughly understood to evaluate the associated benefit/risk balance [24]. Finally, an inability to fully validate the consequences of CRISPR/Cas activity throughout the embryo represents a practical barrier to germline editing in the clinic [54].
In all applications of genome editing (whether in biomedical research, agricultural production or in the clinic) a thorough evaluation of these outcomes is necessary for a realistic appraisal of the benefit:risk associated with the use of the technology. The required level of investigation depends on the specific application, but all demand the ability to anticipate the whole range of potential (wanted and unwanted) consequences of each genome editing intervention.
Strategy for validation
Capturing the variability of genome editing outcomes requires increasing investment in resources as the attention extends away from the target site:
As a minimum, amplification and sequencing of the target site and of chromosomally linked potential off-target sites should be achieved.
Quantification of the number of copies of deleted segments or donor template to capture on target duplications and ectopic integrations should be included. (This is also essential for all applications.)
The use of more elaborate assays to inform on potential larger-scale chromosomal rearrangements is desirable, as an increasing number of examples have been identified, in which additional sequence changes away from the cutting site have been found.
Where required, analysis should be extended to the whole genome to predict or capture potential off-target sites.
Equally, a pragmatic approach to the interrogation of genome editing takes into account the likely genetic complexity of the edited material (whether all cells have identical genomes or constitute a genetically diverse population) and the context of application. For example, mosaic founder small laboratory animals will be bred, thus allowing for the segregation of most unwanted edits at the next generation. It is therefore only essential to search for the presence of an allele of interest and linked off-target effects. Definitive characterisation of the model can await transmission of the allele of interest to the subsequent generation. On the other hand, the whole gamut of mutations arising from CRISPR/Cas activity is to be considered when this technique is used in large livestock (as associated financial and welfare costs are high and timelines extended by long gestations) or for somatic treatment in the clinic [24,44]. Depending on the genome editing application, the strategy for validation will therefore either aim to identify the presence of a specific variant, seek to capture complexity or definitively ascertain an entire genetic make-up. In summary, the genotyping strategy will take into account the ability of each molecular assay to cope with the genetic complexity of the material and will customize effort for the context of utilization.
Capturing the variability of genome editing outcome on target
Appraising the on-target outcome of CRISPR/Cas activity was initially perceived as a straightforward exercise and was therefore performed by a simple set of standard molecular biology protocols: surveyor assays or PCR amplification and Sanger sequencing, with in some instances prior cloning of the PCR product [9]. Although such approaches are generally sufficient to detect the presence of the desired mutation [36], they do not support capturing the entire range of sequences that arises from the CRISPR/Cas mutagenesis effect on target sites in materials of complex genetic make-up. For example, larger deletions that include the sequences annealed by at least one of the PCR primers employed for genotyping are not detected [26]. Equally, low frequency events may be overlooked or relevant cell lineages may be inaccessible for sampling, or underrepresented in samples; for example, a variant may not be detected within the somatic cells of a founder animal (ear biopsy) but may be identified in their progeny [38].
Southern blot analysis appraises a wider genetic interval and can identify genomic changes away from the immediate vicinity of the targeted sequence [55,42]. Cytogenetic methods and fiber fluorescence in situ hybridization (fiber-FISH) ([56], [28] and see Glossary) support the survey of an even broader region and can identify unwanted insertion or deletion of genetic material as well as large-scale sequence rearrangements. Because of the variability of the outcome and the length of the modified segments, a fuller examination requires more elaborate and expensive assays such as targeted locus amplification (TLA) [57] (see Glossary) or, in the case of material of complex genetic make-up, high throughput short-read [58] or long-read [26,59] sequencing. For the last two, targeted sequencing can rely on the isolation of the loci of interest by simple PCR [26,58,59], but this limits the size of the interval that can be interrogated. Other approaches for larger template isolation are emerging to lift this constraint (for example, biotin-labelled primers [60] and Cas9-aided capture [61,62], see Glossary).
Scanning the genome for wider consequences of CRISPR/Cas activity
Gene editing nucleases are powerful tools to introduce sequence changes at a target locus but they can also lead to changes in other similar sequences genome-wide. Copy counting of a deleted segment will inform on the possibility that an unexpected rearrangement has occurred instead of the simple removal of an interval of interest [27,36]. Equally, copy counting of the DNA template (single- or double-stranded) will identify additional integrations. Digital PCR (dPCR) generally is a straightforward assay for this but standard quantitative PCR (qPCR) can also be employed [63]. At a larger genomic scale, array-comparative genomic hybridization (CGH, see Glossary) and FISH enable a whole genome to be surveyed and can identify large sequence alterations away from the nuclease cutting site [29].
Whole-genome sequencing allows for a broad and unbiased capture of genome editing outcome [64] but such an approach is expensive and inadequate for complex genetic materials such as heterogeneous cultured cell populations, or when CRISPR/Cas9 is used with somatic delivery. The complexity of the question demanded the development of bespoke analysis approaches for more effective identification of off-target effects. Many solutions have evolved, based on sequencing of captured susceptible sites (for example, GUIDE–seq [65], CIRCLE–seq [66], LAM–HTGTS [67], UDiTaS™ [68], Digenome-Seq [69] and CHANGE–seq [70], see Glossary). An alternative method captures off-target CRISPR/Cas activity “red-handed” by detecting the DSB repair complex MRN, binding to genomic DNA using ChIP–Seq, a method called DISCOVER–seq ([71], see Glossary). Methods are continuously being evolved in particular to address the remaining challenges of capturing the rarer events in samples of high genetic complexity and of eliminating bias towards particular types of sequence modifications.
No single assay captures all the potential outcomes of genome editing
Crucially, no single technology is able to capture all of the unexpected sequence changes that can result from genome editing. Targeted sequencing using Sanger, or next-generation methods [26,59,72], affords validation of the targeted locus to the single-base level, but only reports on sequence variation at loci that are chosen as relevant and on the integrity of an interval of a limited size, and these techniques do not identify additional sequence changes elsewhere. Droplet digital PCR [27,40] or even Southern blot analysis [42] help to identify unexpected copy numbers of given sequences but do not report on the exact sequences. Neither of these techniques unravels the complexity of non-clonal materials that contain many genetic identities. Technologies based on the visualisation of chromosome segments with fluorescent probes permit the survey of large regions but generate data of low resolution.
All strategies to identify distal or off-target activity also have sensitivity limitations and biases. Sanger sequencing can be applied to many off-target sites, but the loci for analysis must be predicted. Protocols based on Sanger or short-read sequencing do not readily identify structural variations [73]. On the other hand, FISH cytogenetic analysis and the elegant variation of DNA combing (see Glossary) allow for the documentation of large structural variation at the expense of the granularity of sequencing information. Methods for capturing potential off-target sites [65–70] may not reveal all events, whereas DISCOVER–seq technology [71] captures events contemporary to the assay, but not those that occurred earlier in the time course of the intervention. Finally, even whole-genome sequencing technologies, when they can be afforded, will have their own biases that leave remaining ambiguities in terms of CRISPR/Cas activity consequences: short reads-based sequencing is likely to miss structural rearrangements whereas long-read sequencing can lack accuracy if ample coverage is not obtained. With either modality, achieving close to full genome sequence requires heavy investment, even with genetically homogeneous material.
Nevertheless, characterisation of potentially complex genome-editing events is essential to establish the reliability of genome-edited materials and their suitability for their intended use. Defining the appropriate validation strategy will determine the best possible combination of assays in terms of their scope and available resources, and requires the anticipation of potential outcome, genetic complexity of the edited material and essential quality criteria for a given application. Understanding the frequency of the different variants that result from each genome-editing application will also underpin the development of refined strategies for a more exact outcome in future attempts [59].
Preventing the damage
Whilst it is important to identify unwanted events, their prevention seems even more desirable. To alleviate the risk of unwanted outcomes following CRISPR/Cas9 editing, many strategies have been proposed. Early in the development of the technology, predicting the mutagenesis pattern of the guide RNA through its computational design was a major focus for precision editing. Guide efficiency, as well as the prediction of mutagenesis effects and their potential off-target effects is now better understood. In addition, non-canonical sgRNAs, Cas9 variants selected for enhanced specificity [74] and nickases [75], were used in initial strategies to achieve accurate interventions, in many cases to the detriment of efficiency. An alternative approach aims to focus activity on the desired target by increasing protospacer adjacent motif (PAM) selectivity, thereby diminishing activity at some other non-target sites that harbour alternative PAM variants [76]. In addition, temporally controlling its activity by employing ribonucleoprotein or using a ubiquitin-proteasome degradation signal could help to restrict extensive DNA cutting [77]. The introduction of a spatial control by expressing Cas9 in a specific cell type or targeting its delivery could also reduce the risk of DNA damage [24,78]. Finally, competition with inactive RNPs targeting off-target sites has been proposed as a mean to focus genome editing onto the target site [79].
Where a DNA template is employed, tipping the balance in favour of homology directed repair HDR against non-homologous end joining and other repair events is beneficial to ensuring quality. This may be achieved by co-delivery of HDR effectors [80], by pharmacological intervention using small molecule compounds ([81], (although this may reduce cell viability), or by directing Cas protein expression to specific cell-cycle phases [82,83]. The choice of the repair template is also of primary importance to reduce or eliminate the prevalence of ectopic insertions. For example, circular DNA repair templates [11] or templates tethered to the CRISPR complex [11,84] could result in a higher proportion of on-target integrations compared to double-stranded and single-stranded linear DNA donors. Equally, delivery of the template at a lower concentration would result in a lower copy number being ectopically inserted across the genome [85], although this may affect overall efficiency of the genome editing attempt. All of these techniques have been shown to enhance the frequency, or proportion, of desired outcomes but none of them guarantees it. Thorough monitoring of outcomes remains essential in all cases and for all applications. New generations of genome-editing tools such as base editing [13] and prime editing [14] represent further progress towards controlling genome-editing outcome, but are not yet capable of absolute precision editing.
Concluding Remarks
Although genome editing is being developed for application across many fields (biomedical research, agricultural production or in the clinic) much work remains to be done towards understanding the frequency, extent and mechanisms of the sequence changes that the system produces (See Outstanding Questions). Existing genome editing systems all rely on the uncontrolled participation of endogenous DNA repair factors and consequently are error-prone and not entirely predictable. An error-free system will await much more sophisticated approaches that will both carry out the desired DNA repair and shield the modified locus from the intervention of endogenous DNA repair machineries. An additional possible avenue would be to further develop and engineer additional DSB-free -and ideally DNA nick-free- editing systems, such as transcriptome engineering using Class VI CRISPR enzymes or others classes of effectors [86]. Finally, an important area of development in the gene editing field consists of uncovering and harnessing the tremendous diversity and versatility of anti-phage defence systems in bacteria discovered through metagenomic sequencing [87,88]. In the near future, this will lead to improvements in the specificity and efficiency of the current gene editing tools, and to alternatives to the existing ones.
Outstanding questions box:
What would be the acceptable balance of specificity versus efficiency in editing for health or agriculture application?
Will double stranded break free gene editing tools offer higher specificity and efficiency in targeting than classical CRISPR editing tools?
What would be the place of RNA editing enzymes or others classes of CRISPR effectors in the expansion of the gene editing toolbox?
Meanwhile existing genome editing tools are still extraordinarily powerful: while the technology cannot yet aim for one single ideal product, a pragmatic version of precision editing would ensure an outcome of acceptable results when benefit/risk evaluation is performed. However, in all instances of application, the bottom line is that the potential collateral genetic damage must be anticipated and identified.
Table 1:
Methods for the analysis of the targeted locus.
| Technique | Advantages | Disadvantages | Reference |
|---|---|---|---|
| PCR and Sanger sequencing | Easy to implement | Can be difficult to interpret and may overlook some alleles | [9] |
| PCR, sub-cloning and sequencing | Easy to implement, appropriate to disentangle mosaic cell population | Work intensive and may overlook some alleles | [9] |
| Surveyor assay (ie T7 nuclease assay) | Easy to implement | Does not provide sequence granularity | [9] |
| Southern blot | Interrogates a large genomic segment | Does not provide sequence granularity | [42,55] |
| FISH | Interrogates a large genomic segment | Does not provide sequence granularity | [56] |
| Fiber-FISH | Interrogates a large genomic segment | Does not provide sequence granularity | [28] |
| dPCR or qPCR | Detects duplications | Does not provide sequence granularity | [27,40,63] |
| TLA | Interrogates a large genomic segment | Expensive to implement | [57] |
| PCR and short read based-NGS | Appropriate to analyse many genome editing samples | Expensive to implement unless large numbers of samples analysed | [58] |
| PCR and long read based-NGS | Appropriate to disentangle mosaic cell population and interrogates a large genomic segment | Expensive to implement unless large numbers of samples analysed | [26,59] |
Box 1: Factors that should be assessed when validating genome editing outcome:
What is/are the new genetic modification(s) on target? What is the length of the interval potentially altered by the intervention?
Are there sequence changes in potential off-target sites? Are these sites physically linked to the target locus?
What is the number of copies of the mobilized segments (deleted or introduced as template)?
Can the purpose of the application cope with potential unwanted changes in the genome?
Highlights.
Genome editing has a transformative potential in healthcare or to improve crops or livestock. However, the use of Cas9 or other nucleases can yield to unpredictable events at the target site or off target.
To overcome the challenge, it is critical to understand and accurately predict the whole range of possible editing outcomes.
The key to success is to combine molecular assays to evaluate the sequence changes at the target site and to quantify the number of copies of segments deleted/inserted across the genome.
For all application, a thorough evaluation of these outcomes is essential to identifying all collateral damages from nuclease activity and to a real appraisal of the benefit/risk associated to applying the technology.
Acknowledgements
The authors thank Dr Louise Tinsley for expert assistance with the preparation of the manuscript, three anonymous reviewers and the Editor for their comments and suggestions. L.T. was supported by Medical Research Council grant A410 and the National Institute for Health (Grant U42OD011174). G.B. was supported by the National collaborative Research Infrastructure (NCRIS) via the Australian Phenomics Network, the National Health and Medical Research Council of Australia (Grant APP1143008), the Australian Research Council (Grant DP180101494) and the National Natural Science Foundation of China (Grant 81772214).
Glossary box
- Biotin-labeled probe
Enzymatic incorporation of biotin at the 5’ end of a DNA oligonucleotide to label a PCR product in conjunction with fluorophores and enzymes.
- Cas9-aided capture
Use of Cas9 enzyme to enrich a genomic target for high throughput sequencing without utilising PCR amplification methods.
- Cas9 variants for genome editing
Cas9 nickases cut a single strand of the DNA. Dead Cas9 (dCas9) is a catalytically inactive Cas9. HFCas9 and eSpCas9 are highly specific Cas9 variants. Base editing is the fusion of a cytidine or adenosine deaminase with dCas9 or a Cas9 nickase. Prime editing is the fusion between a reverse transcriptase enzyme and dCas9.
- CRISPR-Cas systems
Clustered Regularly Interspaced Short Palindromic Repeats–Cas associated proteins. Defence systems in bacteria against phage infection consisting of an acquisition system to memorize phage attack and an effector complex that recognizes a protospacer adjacent motif (PAM) of the intruder genome resulting in a single- or double-stranded break of the DNA or single-stranded break of the RNA. Type II, V or VI CRISPR systems with Cas9, Cas12 and Cas13 as effector protein signature are harnessed as gene editing tools.
- Cytogenetic methods
Fluorescence in situ hybridization (FISH), fiber fluorescence in situ hybridization (fiber-FISH) are fluorescent probe-based methods to visualise a specific region or an entire chromosome within a particular genome. Fiber fluorescence in situ hybridization (fiber-FISH or DNA Combing) uses FISH on mechanically stretched DNA to increase its resolution. Comparative genomic hybridisation (CGH) array: competitive in situ hybridisation between two different DNA genomic samples in conjunction with a DNA microarray to evaluate copy number variation.
- CIRCLE-seq,and Digenome-seq
Unbiased high throughput genome-wide profiling techniques to detect detection of randomly sheared genomic DNA and cleaved by Cas9 ribonucleoprotein that are circularised (CIRCLE-seq) or not (Digenome-seq).
- DISCOVER-seq
Unbiased high throughput genome-wide profiling techniques to detect DSB repair protein MRE11 binding sites.
- Double stranded break of the DNA (DSB)
Results in the cellular machinery repairing DNA breakage via the non-homologous end-joining (NHEJ) pathway, the micro-homology end-joining (MMEJ) repair pathway based on 3–5 bp micro-homology sequences as a repair template, or the homology directed repair (HDR) system, which uses a longer template for repair such as a single-stranded DNA.
- GUIDE-seq
Unbiased high throughput genome-wide profiling technique to detect integration of a double-stranded oligonucleotide cassette in a double stranded break.
- LAM-HTGS
Unbiased high throughput genome-wide profiling technique to detect chromosomal translocations using a “bait” and “prey” DSB.
- UDiTaS™ and CHANGE-seq
harnessing of Tn5 transposon tagmentation followed by a series of PCR amplifications (UDiTaS™) or genomic DNA circularisation (CHANGE-seq).
- Targeted locus amplification (TLA)
Amplification of a targeted gene of interest using the physical proximity of the nucleotides within this locus of interest by DNA cross-linking, fragmentation, ligation and high-throughput sequencing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The study NCT03399448 is registered at ClinicalTrial.gov (https://clinicaltrials.gov/ct2/show/NCT03399448?cond=crispr&draw=2&rank=7)
References
- 1.Pickar-Oliver A and Gersbach CA (2019) The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol 20, 490–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajizadeh Dastjerdi A et al. (2019) The Expanding Class 2 CRISPR Toolbox: Diversity, Applicability, and Targeting Drawbacks. BioDrugs Clin. Immunother. Biopharm. Gene Ther 33, 503–513 [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y et al. (2018) Applications and potential of genome editing in crop improvement. Genome Biol. 19, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stadtmauer EA et al. (2020) CRISPR-engineered T cells in patients with refractory cancer. Science DOI: 10.1126/science.aba7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamilton JR and Doudna JA (2020) Knocking out barriers to engineered cell activity. Science DOI: 10.1126/science.aba9844 [DOI] [PubMed] [Google Scholar]
- 6.Urnov FD et al. (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651 [DOI] [PubMed] [Google Scholar]
- 7.Miller JC et al. (2011) A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol 29, 143–148 [DOI] [PubMed] [Google Scholar]
- 8.Jinek M et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H et al. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quadros RM et al. (2017) Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 18, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu B et al. (2018) Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol 36, 632–637 [DOI] [PubMed] [Google Scholar]
- 12.Danner E et al. (2020) A Homology Independent Sequence Replacement Strategy in Human Cells Using a CRISPR Nuclease. bioRxiv DOI: 10.1101/2020.05.11.088252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komor AC et al. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anzalone AV et al. (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature DOI: 10.1038/s41586-019-1711-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu Y et al. (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol 31, 822–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iyer V et al. (2018) No unexpected CRISPR-Cas9 off-target activity revealed by trio sequencing of gene-edited mice. PLoS Genet. 14, e1007503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng Z et al. (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A 111, 4632–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith C et al. (2014) Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell 15, 12–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim S-T et al. (2018) Response to “Unexpected mutations after CRISPR–Cas9 editing in vivo.” Nat. Methods 15, 239–240 [DOI] [PubMed] [Google Scholar]
- 20.Lareau CA et al. (2018) Response to “Unexpected mutations after CRISPR–Cas9 editing in vivo.” Nat. Methods 15, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lescarbeau RM et al. (2018) Response to “Unexpected mutations after CRISPR–Cas9 editing in vivo.” Nat. Methods 15, 237–237 [DOI] [PubMed] [Google Scholar]
- 22.Nutter LMJ et al. (2018) Response to “Unexpected mutations after CRISPR–Cas9 editing in vivo.” Nat. Methods 15, 235–236 [DOI] [PubMed] [Google Scholar]
- 23.Wilson CJ et al. (2018) Response to “Unexpected mutations after CRISPR–Cas9 editing in vivo.” Nat. Methods 15, 236–237 [DOI] [PubMed] [Google Scholar]
- 24.Teboul L et al. (2020) Variability in Genome Editing Outcomes: Challenges for Research Reproducibility and Clinical Safety. Mol. Ther DOI: 10.1016/j.ymthe.2020.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Owens DDG et al. (2019) Microhomologies are prevalent at Cas9-induced larger deletions. Nucleic Acids Res. DOI: 10.1093/nar/gkz459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosicki M et al. (2018) Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol DOI: 10.1038/nbt.4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birling M-C et al. (2017) Efficient and rapid generation of large genomic variants in rats and mice using CRISMERE. Sci. Rep 7, 43331–43331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boroviak K et al. (2017) Revealing hidden complexities of genomic rearrangements generated with Cas9. Sci. Rep 7, 12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cullot G et al. (2019) CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun 10, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leibowitz ML et al. (2020) Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. bioRxiv DOI: 10.1101/2020.07.13.200998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuccaro MV et al. (2020) Reading frame restoration at the EYS locus, and allele-specific chromosome removal after Cas9 cleavage in human embryos. bioRxiv DOI: 10.1101/2020.06.17.149237 [DOI] [PubMed] [Google Scholar]
- 32.Sternberg SH et al. (2015) Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke R et al. (2018) Enhanced Bacterial Immunity and Mammalian Genome Editing via RNA-Polymerase-Mediated Dislodging of Cas9 from Double-Strand DNA Breaks. Mol. Cell 71, 42–55.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizuno S et al. (2014) Simple generation of albino C57BL/6J mice with G291T mutation in the tyrosinase gene by the CRISPR/Cas9 system. Mamm. Genome 25, 327–334 [DOI] [PubMed] [Google Scholar]
- 35.Renaud J-B et al. (2016) Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 14, 2263–2272 [DOI] [PubMed] [Google Scholar]
- 36.Mianné J et al. (2017) Analysing the outcome of CRISPR-aided genome editing in embryos: Screening, genotyping and quality control. Methods San Diego Calif 121–122, 68–76 [DOI] [PubMed] [Google Scholar]
- 37.Renaud J-B et al. (2016) Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 14, 2263–2272 [DOI] [PubMed] [Google Scholar]
- 38.Mianné J et al. (2016) Correction of the auditory phenotype in C57BL/6N mice via CRISPR/Cas9-mediated homology directed repair. Genome Med. 8, 16–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanza DG et al. (2018) Comparative analysis of single-stranded DNA donors to generate conditional null mouse alleles. BMC Biol. 16, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Codner GF et al. (2018) Application of long single-stranded DNA donors in genome editing: generation and validation of mouse mutants. BMC Biol. 16, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen K et al. (2019) CRISPR/Cas Genome Editing and Precision Plant Breeding in Agriculture. Annu. Rev. Plant Biol 70, 667–697 [DOI] [PubMed] [Google Scholar]
- 42.Skryabin BV et al. (2020) Pervasive head-to-tail insertions of DNA templates mask desired CRISPR-Cas9–mediated genome editing events. Sci. Adv 6, eaax2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlson DF et al. (2016) Production of hornless dairy cattle from genome-edited cell lines. Nat. Biotechnol 34, 479–481 [DOI] [PubMed] [Google Scholar]
- 44.Young AE et al. (2020) Genomic and phenotypic analyses of six offspring of a genome-edited hornless bull. Nat. Biotechnol 38, 225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aitken SJ et al. (2020) Pervasive lesion segregation shapes cancer genome evolution. Nature 583, 265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J-M et al. (2007) Gene conversion: mechanisms, evolution and human disease. Nat. Rev. Genet 8, 762–775 [DOI] [PubMed] [Google Scholar]
- 47.Slightom JL et al. (1980) Human fetal G gamma- and A gamma-globin genes: complete nucleotide sequences suggest that DNA can be exchanged between these duplicated genes. Cell 21, 627–638 [DOI] [PubMed] [Google Scholar]
- 48.Javidi-Parsijani P et al. (2020) CRISPR/Cas9 increases mitotic gene conversion in human cells. Gene Ther. 27, 281–296 [DOI] [PubMed] [Google Scholar]
- 49.Chandrasegaran S et al. (2017) Genome editing of human embryos: to edit or not to edit, that is the question. J. Clin. Invest 127, 3588–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang D et al. (2020) FREQUENT GENE CONVERSION IN HUMAN EMBRYOS INDUCED BY DOUBLE STRAND BREAKS. bioRxiv DOI: 10.1101/2020.06.19.162214 [DOI] [Google Scholar]
- 51.Simeonov DR et al. (2019) A large CRISPR-induced bystander mutation causes immune dysregulation. Commun. Biol 2, 70–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laoharawee K et al. (2018) Dose-Dependent Prevention of Metabolic and Neurologic Disease in Murine MPS II by ZFN-Mediated In Vivo Genome Editing. Mol. Ther 26, 1127–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grohmann L et al. (2019) Detection and Identification of Genome Editing in Plants: Challenges and Opportunities. Front. Plant Sci 10, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fogarty NME et al. (2017) Genome editing reveals a role for OCT4 in human embryogenesis. Nature 550, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rezza A et al. (2019) Unexpected genomic rearrangements at targeted loci associated with CRISPR/Cas9-mediated knock-in. Sci. Rep 9, 3486–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rayner E et al. (2019) CRISPR-Cas9 Causes Chromosomal Instability and Rearrangements in Cancer Cell Lines, Detectable by Cytogenetic Methods. CRISPR J. 2, 406–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldstein JM et al. (2019) Variation in zygotic CRISPR/Cas9 gene editing outcomes generates novel reporter and deletion alleles at the Gdf11 locus. Sci. Rep 9, 18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernández A et al. (2020) Simple Protocol for Generating and Genotyping Genome-Edited Mice With CRISPR-Cas9 Reagents. Curr. Protoc. Mouse Biol 10, e69. [DOI] [PubMed] [Google Scholar]
- 59.Canaj H et al. (2019) Deep profiling reveals substantial heterogeneity of integration outcomes in CRISPR knock-in experiments. bioRxiv DOI: 10.1101/841098 [DOI] [Google Scholar]
- 60.St. John J and Quinn TW (2008) Rapid capture of DNA targets. BioTechniques 44, 259–264 [DOI] [PubMed] [Google Scholar]
- 61.Gabrieli T et al. (2018) Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Res. 46, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ebbert MTW et al. (2018) Long-read sequencing across the C9orf72 “GGGGCC” repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol. Neurodegener 13, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weisheit I et al. (2020) Detection of Deleterious On-Target Effects after HDR-Mediated CRISPR Editing. Cell Rep. 31, [DOI] [PubMed] [Google Scholar]
- 64.Iyer V et al. (2015) Off-target mutations are rare in Cas9-modified mice. Nat. Methods 12, 479. [DOI] [PubMed] [Google Scholar]
- 65.Tsai SQ et al. (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol 33, 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsai SQ et al. (2017) CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 14, 607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu J et al. (2016) Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc 11, 853–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giannoukos G et al. (2018) UDiTaS™, a genome editing detection method for indels and genome rearrangements. BMC Genomics 19, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim D et al. (2015) Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 12, 237–243 [DOI] [PubMed] [Google Scholar]
- 70.Lazzarotto CR et al. (2020) CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat. Biotechnol DOI: 10.1038/s41587-020-0555-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wienert B et al. (2019) Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 364, 286–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McCabe CV et al. (2019) Application of long-read sequencing for robust identification of correct alleles in genome edited animals. bioRxiv DOI: 10.1101/838193 [DOI] [Google Scholar]
- 73.Nelson CE et al. (2019) Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med 25, 427–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Okafor IC et al. (2019) Single molecule analysis of effects of non-canonical guide RNAs and specificity-enhancing mutations on Cas9-induced DNA unwinding. Nucleic Acids Res. 47, 11880–11888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ran FA et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moreb EA et al. (2020) CRISPR/Cas “non-target” sites inhibit on-target cutting rates. bioRxiv DOI: 10.1101/2020.06.12.147827 [DOI] [PubMed] [Google Scholar]
- 77.Tu Z et al. (2017) Promoting Cas9 degradation reduces mosaic mutations in non-human primate embryos. Sci. Rep 7, 42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Port F et al. (2020) A large-scale resource for tissue-specific CRISPR mutagenesis in Drosophila. eLife 9, e53865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coelho MA et al. (2020) CRISPR GUARD protects off-target sites from Cas9 nuclease activity using short guide RNAs. Nat. Commun 11, 4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Charpentier M et al. (2018) CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun 9, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Riesenberg S et al. (2019) Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 47, e116–e116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gutschner T et al. (2016) Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep. 14, 1555–1566 [DOI] [PubMed] [Google Scholar]
- 83.Mizuno-Iijima S et al. (2020) Efficient production of large deletion and gene fragment knock-in mice mediated by genome editing with Cas9-mouse Cdt1 in mouse zygotes. Methods DOI: 10.1016/j.ymeth.2020.04.007 [DOI] [PubMed] [Google Scholar]
- 84.Aird EJ et al. (2018) Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun. Biol 1, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Al Abdallah Q et al. (2017) A Simple and Universal System for Gene Manipulation in Aspergillus fumigatus-Assembled Cas9-Guide RNA Ribonucleoproteins Coupled with Microhomology Repair Templates. mSphere 2, e00446–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Konermann S et al. (2018) Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Doron S et al. (2018) Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao L et al. (2020) Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 369, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]