

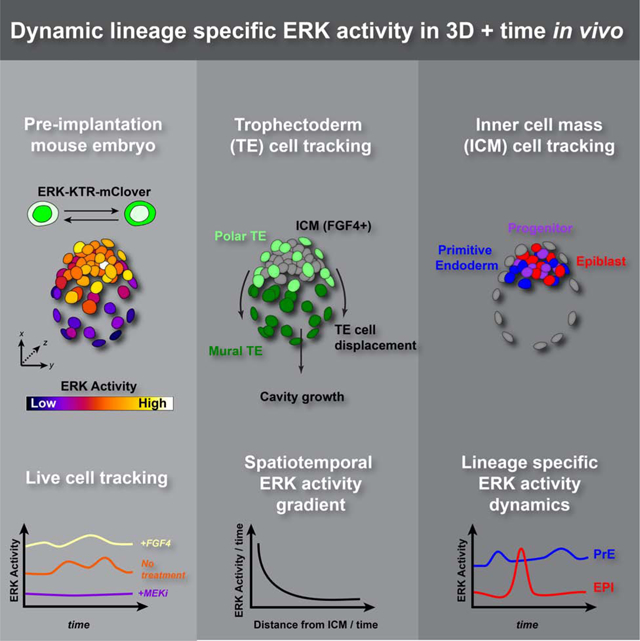
SUMMARY
FGF/ERK signaling is crucial for the patterning and proliferation of cell lineages that comprise of the mouse blastocyst. However, ERK signaling dynamics have never been directly visualized in live embryos. To address whether differential signaling is associated with particular cell fates and states, we generated a targeted mouse line expressing an ERK-kinase translocation reporter (KTR) that enables live quantification of ERK activity at single cell resolution. 3D time-lapse imaging of this biosensor in embryos revealed spatially graded ERK activity in the trophectoderm prior to overt polar-versus-mural differentiation. Within the inner cell mass (ICM), all cells relayed FGF/ERK signals with varying durations and magnitude. Primitive endoderm cells displayed higher overall levels of ERK activity, while pluripotent epiblast cells exhibited lower basal activity with sporadic pulses. These results constitute a direct visualization of signaling events during mammalian pre-implantation development and reveal the existence of spatial and temporal lineage specific dynamics.
Keywords: Blastocyst, FGF, ERK, biosensor, mouse, signaling, kinase-translocation reporter, inner cell mass, trophectoderm, dynamics
Graphical Abstract
eTOC Blurb:
FGF/ERK signaling is crucial for the patterning and proliferation of cell lineages that comprise of the mouse blastocyst. Using an ERK -kinase translocation biosensor, Simon et al. show the trophectoderm is patterned by spatially graded ERK activity, and primitive endoderm and epiblast cells have distinct ERK dynamics.
INTRODUCTION
Cell signaling, modulated by extracellular stimuli and cell intrinsic regulators, is critical for the patterning of tissues during development. In the mouse embryo, the FGF/ERK signaling pathway plays a central role for the coordinated cell fate specification and tissue growth across the embryo before implantation in the maternal uterus (Simon, et al., 2018a; Brewer, et al., 2016). The first cell specification event during pre-implantation development allocates outer and inner cells to extra-embryonic trophectoderm (TE) or inner cell mass (ICM), respectively, at around embryonic day (E)3.0 (Mihajlovic and Bruce, 2017). TE cells express FGF receptors Fgfr1, Fgfr2 and Fgfr4 (Kurowski, et al., 2019; Nowotschin, et al., 2019; Molotkov, et al., 2017; Guo, et al., 2010). Although dispensable for initial TE fate decisions, FGF/ERK signaling is required for maintaining trophoblast stem cell identity and TE proliferation (Christodoulou, et al., 2019; Kurowski, et al., 2019; Nichols, et al., 2009; Yang, et al., 2006; Tanaka, et al., 1998).
Progenitor cells within the ICM are progressively and asynchronously specified to either the pluripotent epiblast (EPI), or extra-embryonic primitive endoderm (PrE) lineages from E3.25 to E3.75 (~32–100cells) (Saiz, et al., 2016b; Schrode, et al., 2014; Grabarek, et al., 2012; Plusa, et al., 2008; Chazaud, et al., 2006). Abrogation of the FGF/FGFR/GRB2/MEK/ERK signaling cascade by genetic or pharmacological perturbation prevents the formation of PrE and impairs pluripotency progression in the EPI (Kang, et al., 2017; Molotkov, et al., 2017; Saiz, et al., 2016b; Ohnishi, et al., 2014; Kang, et al., 2013; Krawchuk, et al., 2013; Yamanaka, et al., 2010; Nichols, et al., 2009; Chazaud, et al., 2006). Conversely, administration of exogenous FGF to embryos induces a PrE fate in uncommitted ICM progenitors (Saiz, et al., 2016b; Kang, et al., 2013; Krawchuk, et al., 2013; Yamanaka, et al., 2010). Therefore, cell fate specification in the ICM is driven by FGF signaling acting predominantly via the ERK pathway. Fgf4 is the only FGF ligand expressed in the blastocyst at the time of ICM cell fate specification, expressed from E3.0-E3.25 (~16–32 cell stage) in uncommitted ICM progenitors, and then later by EPI cells (Nowotschin, et al., 2019; Ohnishi, et al., 2014; Guo, et al., 2010). The FGF receptor Fgfr1 is expressed from E3.25 (~32 cells) by all cells of the ICM, and a second receptor Fgfr2 is expressed in specified PrE cells (Nowotschin, et al., 2019; Kang, et al., 2017; Molotkov, et al., 2017; Brewer, et al., 2015; Ohnishi, et al., 2014).
Given the early pan-ICM expression of Fgfr1, the predominant receptor for PrE specification, heterogeneity in FGF/ERK signaling is considered the main driver of ICM lineage segregation (Saiz, et al., 2020; Simon, et al., 2018a; Kang, et al., 2017; Molotkov, et al., 2017; Tosenberger, et al., 2017; De Mot, et al., 2016; Bessonnard, et al., 2014). How FGF signal transduction differs between cells of the blastocyst is a key open question in the field. However, previous approaches used to measure ERK activity in the blastocyst have been unable to detect dynamic ERK-activities described in other systems (Deathridge, et al., 2019; Zhang, et al., 2018; de la Cova, et al., 2017; Gillies, et al., 2017; Conlon, et al., 2016; Ryu, et al., 2016; Regot, et al., 2014). For example, using a transcriptional reporter for Spry4, a target of FGF/ERK signaling, we could not detect differences between ICM lineages (Morgani, et al., 2018). However, the design of this reporter was not permissive for visualizing short term changes in signaling activity. By contrast, p-ERK immunostaining directly measures the amount of activated ERK in fixed embryos and provided direct evidence that primitive endoderm cells have a higher proportion of p-ERK positive cells compared with epiblast cells (Azami, et al., 2019). However, a direct comparison of p-ERK levels between lineages remains outstanding, and immunostaining only provides a snapshot of this dynamic process, in which heterogenous labelling may be due to transient ERK phosphorylation. Quantification of FGF/ERK signaling transduction using rapidly responsive live reporters is therefore essential for furthering our understanding of signaling dynamics and how they impact cellular behaviors such as tissue patterning in vivo.
To achieve this, we generated a targeted mouse line expressing an ERK-kinase translocation reporter (KTR) in all cells. To perform in toto embryo imaging during pre-implantation development, we co-visualized this KTR reporter with a ubiquitously expressed nuclear reporter which facilitated segmentation, tracking and quantitation in 3D time-lapse data. In total, we manually tracked and analyzed ERK activity over time in approximately 2,000 cells from 60 blastocyst stage embryos across different experiments. Live imaging of this ratiometric sensor in embryos revealed dynamic, lineage specific ERK activities at single-cell resolution. We observed a spatiotemporal ERK activity gradient along the embryonic-abembryonic axis across the TE that presaged its differentiation to polar and mural subtypes. Within the ICM we found heterogenous activities, where all cells relayed FGF/ERK signals to differing extents. Primitive endoderm cells displayed higher overall levels of ERK activity, while pluripotent EPI cells exhibited lower basal activity with infrequent pulses over time. Together, these observations reveal how distinct modes of transduction of an individual signal within a population can mediate symmetry breaking and generate cellular diversity during embryonic development.
RESULTS
An ERK-KTR targeted mouse line for live visualization of ERK activity in vivo
To follow ERK activities in vivo, we used an ERK kinase translocation reporter (ERK-KTR) (Kudo, et al., 2018; Regot, et al., 2014). ERK-KTR consists of a docking site (fragment of human Elk1, an ERK substrate), nuclear localization (NLS) and export (NES) signals, and a mClover fluorescent protein (Lam, et al., 2012) (Figure 1A). When the ERK signaling pathway is active, activated p-ERK phosphorylates the sensor and nuclear export predominates, resulting in cytoplasmic enrichment. Conversely, when the ERK signaling pathway is inactive, the sensor is unphosphorylated and nuclear import predominates, resulting in increased nuclear enrichment of mClover fluorescence. Therefore, the cytoplasmic to nuclear ratio of ERK-KTR serves as proxy for ERK activity.
Figure 1: An ERK-KTR targeted mouse line for live visualization of ERK activity in vivo.
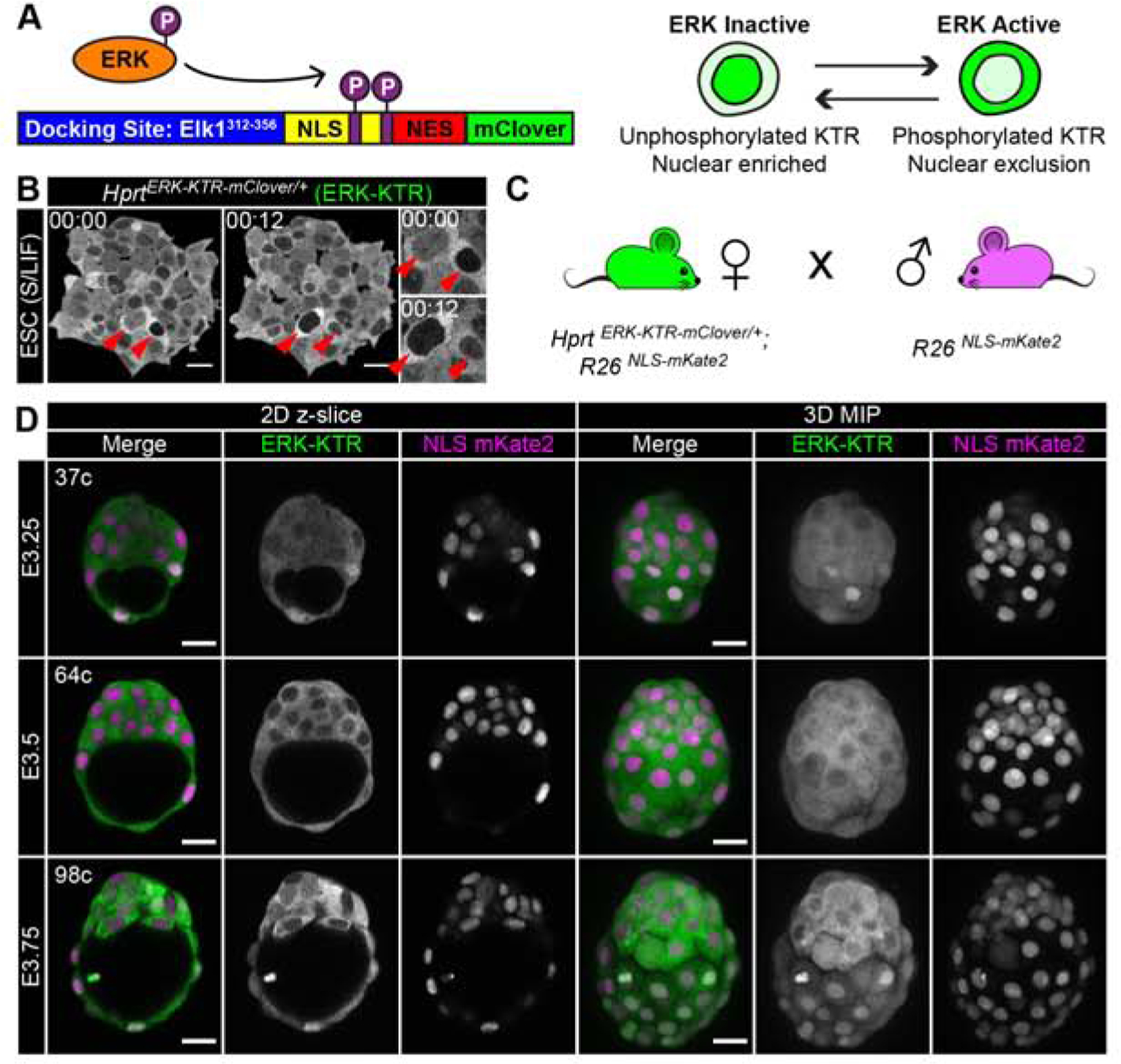
(A) Schematic of ERK kinase translocation reporter (ERK-KTR). ERK-KTR is a biosensor, which shuttles between cytoplasm and nucleus as a read-out of ERK kinase activity. NLS: nuclear localization signal. NES: nuclear export signal. P: Phosphorylation. (B) Time-lapse confocal images of mouse embryonic stem cells (ESC) expressing HprtCAG-ERK-KTR-mClover, abbreviated to HprtERK-KTR. Red arrows indicate two cells at different times. Inset shows higher magnification. Timestamp shows Hour:Min. S/LIF: Serum LIF culture conditions. (C) Summary diagram of mouse cross used to generate hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryos in this study. (D) Confocal images of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryos during pre-implantation development. Embryonic day (E) and cell number are indicated for staging. Scale bars: 20μm.
To generate a bright reporter suitable for 3D time-lapse imaging at single-cell resolution, we coupled the ERK-KTR-mClover construct to a strong CAG promoter (Niwa, et al., 1991). We targeted the construct to the Hprt locus on the X chromosome to generate an HprtCAG-ERK-KTR-mClover allele (abbreviated to HprtERK-KTR), thereby avoiding unpredictable site-specific integration and copy-number effects on expression levels associated with random-integrant transgenes. Mouse embryonic stem cells (ESC) containing the targeted allele robustly expressed the ERK-KTR-mClover (Figure 1B). In 2i conditions (MEK and GSK3 inhibitors) the reporter was homogenously localized throughout the cell (data not shown) (Ying, et al., 2008). By contrast, in serum/LIF conditions, which are permissive for FGF/ERK signaling, the reporter was predominantly localized in the cytoplasm, indicating high ERK activity, consistent with p-ERK levels in these conditions (Figure 1B) (Deathridge, et al., 2019; Chen, et al., 2015; Ying, et al., 2008). However, the localization of the reporter was variable between different cells and within a cell over time, typical of the heterogeneity seen in serum/LIF cultured ESCs (Lanner and Rossant, 2010) (Figure 1B, inset, Supplemental Figure 1A–B, and Supplemental Movie 1).
The HprtERK-KTR ESCs were used to generate HprtERK-KTR mice, which were crossed with mice ubiquitously expressing a nuclear localized red fluorescent protein, mKate2 (Rosa26CAG−3xNLS-mKate2, abbreviated to R26NLS-mKate2) (Susaki, et al., 2014) for segmentation of nuclei, cell counting and tracking (Figure 1C). We collected hemizygous HprtERK-KTR, homozygous R26NLS-mKate2 embryos for our analyses. To account for within-litter variation and unknown fertilization time, we also precisely staged embryos using total cell number (32 cells ≈ embryonic day [E]3.25, 64 cells ≈ E3.5, 100 cells ≈ E3.75). We visualized expression of the ERK-KTR (mClover) and NLS-mKate2 reporters in live embryos in 3D using laser-scanning confocal microscopy. At E3.0-E3.25 ERK-KTR showed a relatively even nuclear-to-cytoplasmic distribution in cells of the inner cell mass (ICM) and some cells with nuclear enriched reporter localization, indicative of low signaling activity. The reporter then progressively became more cytoplasmic from E3.5 to E4.25, and in the early post-implantation EPI at E5.5 (Figure 1D, Supplemental Figure 1C). We were unable to correlate the dynamic range of the ERK-KTR reporter and pERK staining within the same experiment as the staining procedure for pERK in embryos is incompatible with the localization based ratiometric read-out of the reporter. The high percentage (8%) PFA fixation and Triton-X100 permeabilization protocols used for pERK immunodetection (Azami, et al., 2019; Corson, et al., 2003) do not preserve the nuclear and cytoplasmic localization of the KTR reporter, a common artifact encountered when fixing fluorescent proteins (Schnell, et al., 2012; Melan and Sluder, 1992). However, we note these changes in reporter localization indicate an increase in ICM and EPI ERK activity with developmental stage, consistent with recent p-ERK immuno-localization (Azami, et al., 2019).
ERK-KTR is a read out of FGF/ERK signaling in the mouse blastocyst
The ERK-KTR ratiometric reporter system can be used to quantify dynamic ERK activity in individual cells. Although the blastocyst is relatively small, (E3.5 = ~90μm, ~64 cells), segmentation and tracking of nuclei during cellular rearrangements within the densely packed ICM presents a challenge for automated image analysis in 4D. Therefore, we developed a pipeline to segment and calculate the ratio between mean cytoplasmic and nuclear ERK-KTR fluorescence intensity (C:N ratio) per Z-slice (optical section) as a proxy for ERK activity. Determination of a C:N ratio normalizes for KTR expression between cells within any given sample, and loss of fluorescence signal along the Z-axis due to imaging depth (Kudo, et al., 2018). We then reconstructed these images in 3D and over time (3D+t) to manually track and measure the ERK activity of individual cells in time-lapse movies (Figure 2A, Supplemental Figure 2A, Supplemental Movie 2, and see STAR methods).
Figure 2: ERK-KTR is a read out of FGF/ERK signaling in the mouse blastocyst.
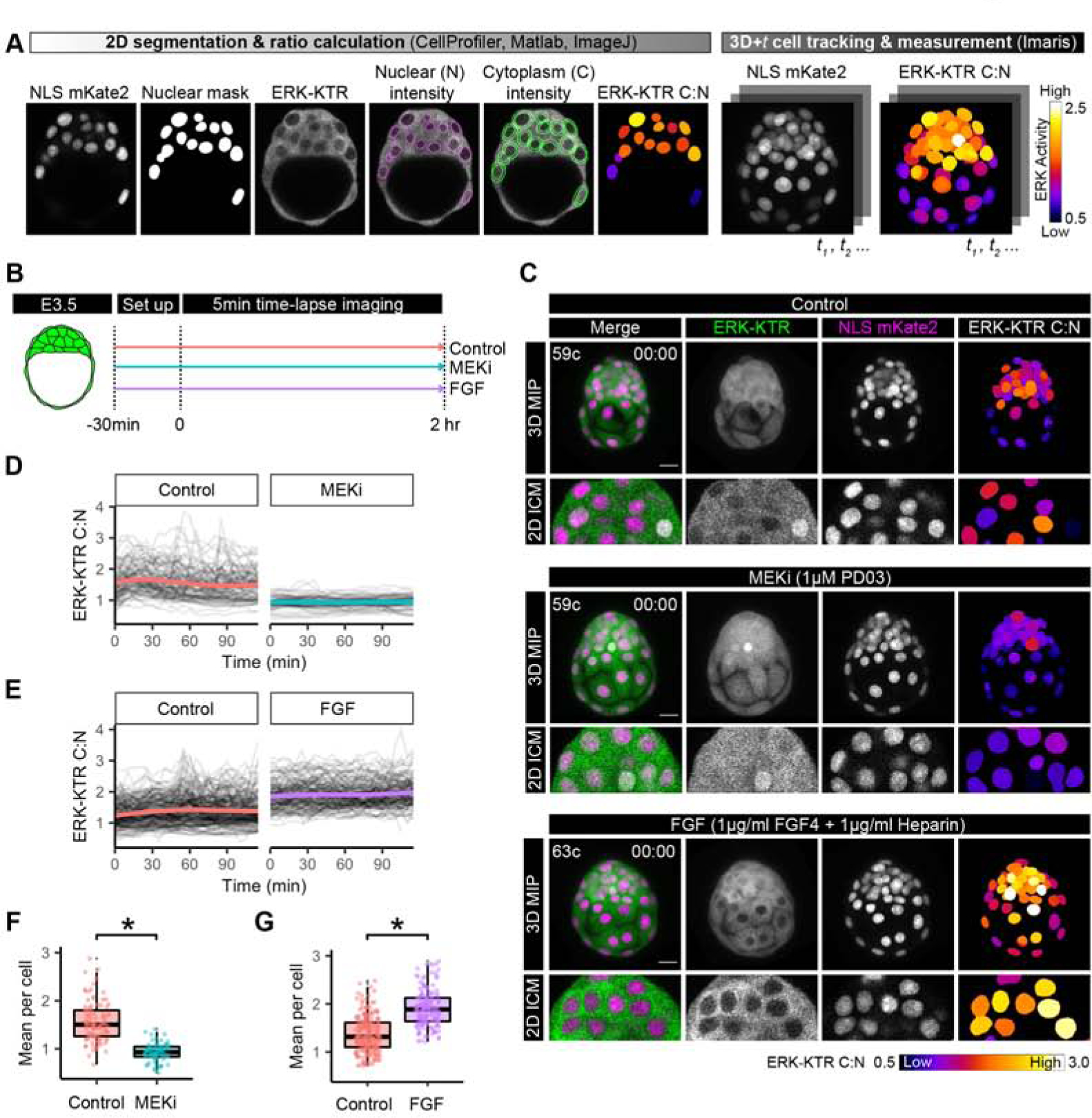
(A) Pipeline for quantification of ERK-KTR for 3D+t embryo time-lapse imaging. ERK-KTR cytoplasmic to nuclear (C:N) ratiois a proxy for ERK activity and can be visualized with a heatmap (see STAR Methods).. (B) Schematic of embryo culture treatments and time-lapse imaging. (C) First frame of time-lapse confocal images of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryos in control, MEKi and FGF culture conditions. Embryonic day (E) and cell number are indicated for staging. Max intensity projections (MIP) and magnification of a single z-slice at mid-point through the ICM are shown. Scale bar: 20μm. ERK-KTR C:N values corresponding to ERK activity is shown as a heatmap as indicated. (D-E) Traces of ICM cell ERK-KTR C:N values over the course of the 2 hour time-lapse imaging in littermate control versus MEKi conditions (D) and control versus FGF conditions (E). Single-cell traces are shown in black, and population mean in color; control: orange, MEKi: turquoise, FGF: purple. (F-G) Boxplot showing integrated ERK activities in individual ICM cells over the 2 hour time-lapse imaging. Mean ERK-KTR C:N values per cell are shown for every cell as individual points. Color coding for treatments as indicated before. Asterisk indicates statistical significance. (F) Control versus MEKi: p = 0.016 (Wilcoxon test) Control: n=5, MEKi n=4 embryos. (G) Control versus FGF: p=0.003 (Wilcoxon test). Control: n=10, FGF: n=8 embryos.
To test the responsiveness of ERK-KTR as a read-out of FGF/ERK signaling during preimplantation development, we performed 3D time-lapse imaging of mid-stage blastocysts (E3.5, ~64 cells) in different treatment regimes. The sensitivity of embryos to pharmacological perturbation of FGF signaling is maximal in a narrow time window between E3.5 and E3.75 (Bessonnard, et al., 2017). Therefore, we collected E3.5 embryos, and cultured them in control (untreated) conditions, in the presence of a MEK inhibitor (1μM PD0325901, MEKi) or FGF ligand (1μg/ml FGF4 + 1μg/ml Heparin). We imaged the embryos for a total of 2 hours, with a 5 min time-lapse interval (Figure 2B–C, Supplemental Figure 2B, and Supplemental Movie 3).
We tracked individual ICM cells in control, MEKi and FGF cultures and measured the ERK C:N ratio (Supplemental Figure 2D–E, Supplemental Movie 3). In control embryos, ERK-KTR localization was heterogeneous between individual cells within the ICM (Supplemental Figure 2E–F), but it was predominantly cytoplasmic, corresponding to higher C:N values and high ERK activity (Figure 2C, top). When we treated embryos with MEKi to block ERK kinase activity, ERK-KTR fluorescence intensity became equivalent between the cytoplasm and the nuclei of ICM cells or nuclear enriched, corresponding to lower C:N values and low ERK activity (Figure 2C, middle). By contrast, when we treated embryos with FGF, ERK-KTR localized almost exclusively to the cytoplasm, hence high ERK activity (Figure 2C, bottom, Supplemental Figure 2B). When analyzing ERK-KTR C:N traces of single cells followed throughout the time-lapse movie, we found that MEKi abolished fluctuations that were seen in control cells and cells treated with FGF (Figure 2D–E, Supplemental Figure 2E). Due to this dynamic behavior, we used the mean ERK-KTR C:N for each cell over the entire time-course as an integrated measure of ERK activity. In MEKi treated embryos, the mean ERK-KTR C:N per cell was significantly reduced when compared to control (untreated) embryos (Figure2D, colored lines, Figure 2F), whereas FGF treated embryos had significantly higher values of mean ERK-KTR C:N per cell than control embryos (Figure 2E,G).
We used the ERK-KTR C:N values obtained from the FGF and MEKi treatments, to set a threshold C:N value of 1.21 to delineate “high” and “low” signaling activity (Supplemental Figure 2G, and see STAR Methods). Control embryos had a mix of cells with high and low signaling activity following thresholding for C:N values (Supplemental Figure 2H–I). Upon embryo culture with FGF or MEKi, most treated ICM cells instead have low or high ERK activity, respectively (Supplemental Figure 2H–I). In conclusion, these data demonstrate that ERK-KTR provides a quantifiable dynamic readout of FGF/ERK activity in live blastocysts in 3D at single-cell resolution.
The trophectoderm is patterned by a spatial and temporal ERK activity gradient
FGF/ERK signaling is required for trophoblast stem cell identity, acting to inhibit differentiation, promote proliferation, and maintain Cdx2 expression (a trophoblast stem-cell marker) (Christodoulou, et al., 2019; Nichols, et al., 2009; Yang, et al., 2006; Tanaka, et al., 1998). In the embryo, FGF4 produced by EPI cells acts as a positional cue to drive the lineage divergence at E4.5 (~150–200 cell stage) of polar TE cells (CDX2high) overlying the ICM, and mural TE cells (CDX2low) overlying the blastocyst cavity (Christodoulou, et al., 2019; Goldin and Papaioannou, 2003; Gardner, 2000; Chai, et al., 1998). We applied our reporter system to understand how FGF4 signaling patterns the TE. In static images, we noticed differential ERK activity at E3.75 between both TE regions (Figure 1D, Figure 3A) with high activity in polar TE cells and low activity in mural TE cells, consistent with p-ERK staining of the late-stage blastocyst (Azami, et al., 2019),
Figure 3: The trophectoderm is patterned by a spatial and temporal ERK activity gradient.
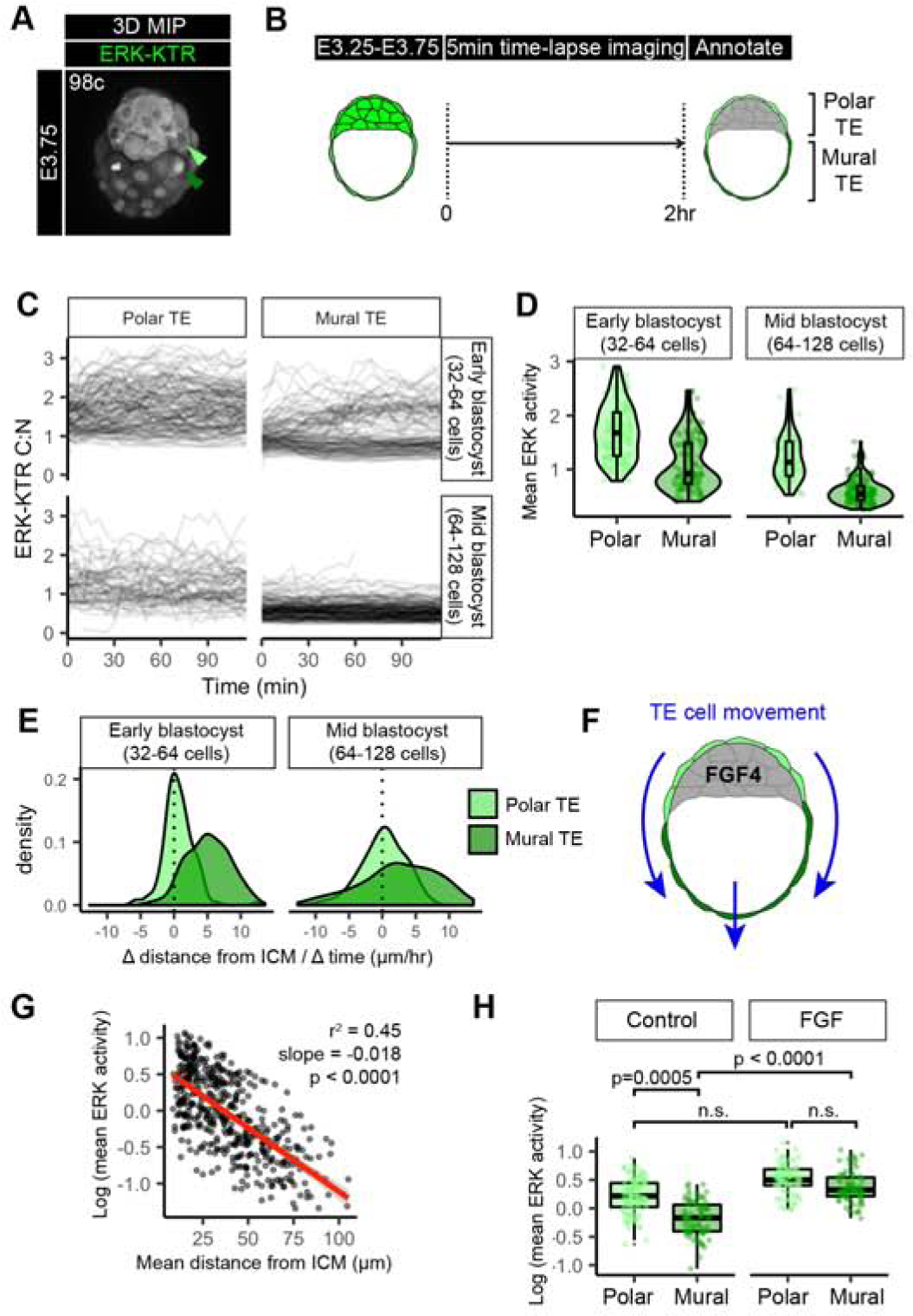
(A) Confocal image of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryos during preimplantation development. Embryonic day (E) and cell number are indicated for staging. Reproduced from panel within Figure 1D. (B) Schematic of time-lapse confocal imaging where polar versus mural TE identity was annotated by position at the end of the movie. (C) Traces of single cell ERK-KTR C:N values over the course of the 2 hour time-lapse imaging. Single-cell traces are shown in black, for polar TE, and mural TE. Stage of embryo at the beginning of the movie is indicated, early blastocyst (32–64 cell) stage and mid-blastocyst (64–128 cell) stage. (D) Violin and boxplots showing mean ERK activities in individual TE cells over the 2 hour time-lapse imaging. Mean ERK-KTR C:N values per cell are shown for every cell as individual points. Polar TE: light green, Mural TE: dark green. Cell stages at beginning of time-lapse are indicated. early-blastocyst stage: n=14, mid-blastocyst stage: n=10 embryos. (E) Density plots showing the change (Δ) in distance from the nearest ICM cell over the duration of the time-lapse for individual Polar TE and Mural TE cells. (F) Schematic of TE movement along the embryonic – abembryonic axis away from the inner cell mass (ICM, grey) derived FGF4 signals. (G) Scatterplot comparing the mean distance over the lifetime of that cell to its nearest ICM neighbor in each frame, with the mean ERK activity of that cell. Linear regression line of best fit shown in red. Slope, adjusted r2 goodness of fit, and p value are indicated. (H) Boxplot showing mean ERK activities in individual ICM cells over the 2 hour time-lapse imaging. Mean ERK-KTR C:N values per cell are shown for every cell as individual points. Color coding for TE sub-types as indicated before. Control: n=10, FGF: n=8 embryos (as in Figure 2). P values for statistical significance of pairwise comparisons are shown (Wilcoxon test), horizontal lines indicate groups compared. Not significant (n.s.) pairwise comparisons indicated where p > 0.05.
To establish when these signaling patterns emerge, we performed time-lapse imaging of E3.25-E3.75 embryos for 2 hours with a 5 min interval (Figure 3B and Supplemental Movie 4). To precisely stage each embryo, we counted the total cell number using the NLS-mKate2 nuclear marker and analyzed embryos between the early blastocyst (32–64 cells) and the mid-blastocyst (64–128 cells). Polar versus mural TE identities were manually assigned based on the final position of cells relative to the ICM. Individual ERK-KTR C:N traces showed differences within the TE (Figure 3C). By the mid-blastocyst stage, mural TE cells had a distinct signaling profile from polar TE cells, showing much lower activity (Figure 3C). At the early blastocyst stage differences were already apparent between polar and mural TE cells (Figure 3D, Supplemental Figure 3A). This observed ERK activity in the polar TE from early blastocyst (32–64 cells) stage was earlier than reported p-ERK staining from E3.75 or E4.5 (Azami, et al., 2019; Christodoulou, et al., 2019), demonstrating the high sensitivity of the ERK-KTR reporter (Regot, et al., 2014).
During embryo growth and expansion of the blastocyst cavity mural TE cells progressively move farther away from the ICM, the source of the FGF4 signal, concomitant with a flow of polar TE cells to a mural TE position (Figure 3E–F and Supplemental Figure 3B) (Christodoulou, et al., 2019; Gardner, 2000). Therefore, we hypothesized that mean ERK activities within the TE may depend on changes in their signaling environment over time. To test this, we measured distance of each TE cell to its nearest ICM cell at every time-point, then calculated the mean distance over the time-course for that cell. We noticed an exponential decline in a cell’s mean ERK activity with increasing mean distance from the ICM across early and mid-blastocyst stages (Figure 3G). A similar relationship was observed in longer time-lapse imaging experiments, where early blastocyst stage embryos were imaged for a total of 12 hours with a 15 min time interval (Supplemental Figure 3C–F). Together, this suggests that ERK activity emerges in a spatiotemporal gradient along the embryonic – abembryonic axis of the TE, such that mean ERK activity is related to the spatial position of a cell within the TE of the blastocyst over time. This spatial-temporal gradient in ERK activity in the TE is in contrast to, and precedes, the binary difference, high (Polar TE) versus low (Mural TE) p-ERK immunostaining reported previously in pre- and peri-implantation embryos (Christodoulou, et al., 2019).
Differential ERK signaling between TE subtypes could be caused by cell autonomous transcriptional changes (Frias-Aldeguer, et al., 2020) and/or changes in mechanical properties (i.e. higher cortical tension from overlying blastocyst cavity vs ICM) (Chan, et al., 2019; De Belly, et al., 2019). Alternatively, non-cell autonomous effects, namely, the availability and distance from source of FGF4 ligand may drive the difference in ERK activity. To differentiate between these possibilities, we assessed the response of polar and mural TE cells by tracking TE cells in control (untreated) and FGF treated embryo experiments (Figure 3H). In control embryos, there was a significant difference in ERK activity between mural and polar TE cells. Upon FGF treatment, however, mural TE cells responded to FGF at comparable levels to polar TE cells, and the association of ERK activity with distance was diminished (Figure 3H, Supplemental Figure 3E). These data argue that differences in ERK activities across the TE at this stage are not cell-intrinsic, and that mural TE are able to respond to exogenous ligand. Since FGF ligands are expressed exclusively in the ICM and not the TE (Nowotschin, et al., 2019; Guo, et al., 2010), these experiments suggest that the ICM spatially and temporally patterns ERK activities in TE cells. This patterning begins in the early blastocyst stage, preceding the expression of markers of polar and mural TE differentiation.
As FGF/ERK signaling is required for maintain Cdx2 expression in trophoblast stem cells and peri-implantation TE (Christodoulou, et al., 2019; Nichols, et al., 2009; Yang, et al., 2006; Tanaka, et al., 1998), we suspected the early TE ERK gradient may also affect levels of CDX2 in preimplantation embryos. Therefore, we analyzed 3D spatial information of published CDX2 immunostained wild-type embryos (Saiz, et al., 2016b). We discovered a spatial CDX2 protein gradient in the TE along the embryonic-abembryonic axis from 120 cell late blastocyst stage embryos (Supplemental Figure 3H). Intriguingly, these data reveal that a CDX2 gradient earlier than the distribution of high-Cdx2 and low-Cdx2 corresponding to polar and mural trophectoderm at peri-implantation (Christodoulou, et al., 2019). This suggests a link between the gradient of ERK activity and the spatial gradient of Cdx2 activity along the embryonic-abembryonic axis in pre-implantation embryos.
Signaling heterogeneity between ICM cells corresponds to lineage identity
Endogenous FGF signaling is required for the emergence and distribution of PrE and EPI progenitors within the ICM population (Kang, et al., 2013; Krawchuk, et al., 2013). To address whether there is differential transduction of an FGF signal in ICM lineages, we focused on ICM cells in our time-lapse movies of early (32–64 cell) and mid (64–128 cell) blastocyst stage embryos (Figure 4A). We tracked ICM cells at 5 min intervals throughout 2 hour time-lapse periods (Figure 4A, Supplemental Figure 4A). We immediately fixed and immunostained embryos to assign lineage identities after the time-lapse, and were able to identify terminal ICM cell fates. Embryos were processed individually to keep track of their origin, and ICM lineage identities were assigned based on NANOG and GATA6 staining as described previously (Saiz, et al., 2016a; Saiz, et al., 2016b; Schrode, et al., 2014; Plusa, et al., 2008) (Figure 4A,B). Fixed embryos were then manually registered to the final frame of the time-lapse movie to retrospectively assign ICM lineage identities to tracked cells (Figure 4A,C, Supplemental Movie 4 and Supplemental Movie 5, see STAR Methods).
Figure 4: Signaling heterogeneity between ICM cells corresponds to lineage identity.
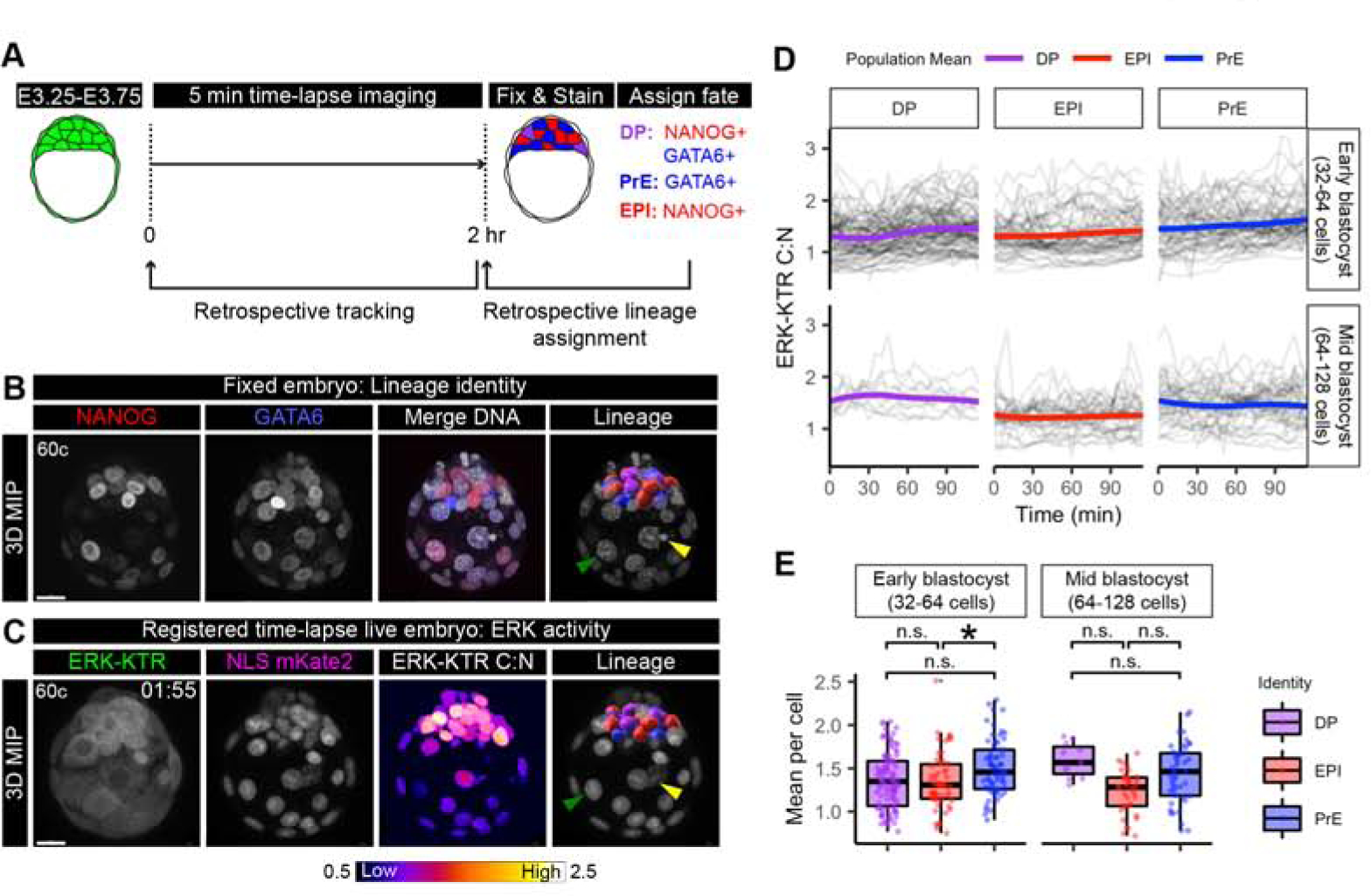
(A) Schematic of time-lapse confocal imaging as in (Figure 3C). Embryos were fixed immediately after the end of the time lapse and stained to retrospectively identify inner cell mass (ICM) lineages. ICM progenitors are double positive (DP) for NANOG and GATA6. DP cells are specified to primitive endoderm (PrE) GATA6+ cells, and Epiblast (EPI) NANOG+ cells. (B) Max intensity projection (MIP) confocal image of an embryo stained for NANOG (red) and GATA6 (blue). Merge with Hoechst (grey) to label nuclei. Lineage includes 3D rendering in Imaris (Bitplane) corresponding to a cell’s lineage identity after k-means clustering of relative NANOG and GATA6 levels (Saiz, et al., 2016b). DP: purple, PrE: blue, EPI: red. Merged with Hoechst (grey) to show nuclei. Arrows indicate landmarks within trophectoderm (TE) used for manual image registration with final time frame of time-lapse. Yellow: apoptotic cell, Green: Mural TE. Scale bars: 20μm. (C) Max intensity projection of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryos in final time frame of 2 hour time-lapse movie. ERK-KTR C:N heatmap showing level of ERK activity. Retrospective ICM lineage identification when manual registered to fixed and stained (B) arrows indicate the same landmarks. Lineage is a 3D rendering in IMARIS colored corresponding to retrospective lineage identification (DP: purple, PrE: blue, EPI: red) merged with NLS mKate2 (grey) to show nuclei. (D) Traces of single-cell ERK-KTR C:N values over the course of the 2 hour time-lapse imaging. Single-cell traces are shown in black. Population means are overlaid in color corresponding to lineage identity. Stage of embryo at the beginning of the movie is indicated, early blastocyst (32–64 cell) stage and mid-blastocyst (64–128 cell) stage. (E) Boxplot showing integrated ERK activities in individual ICM cells over the 2 hour time-lapse imaging period. Mean ERK-KTR C:N values per cell are shown for every cell as individual points. Color coding for lineage as indicated before. Asterisk indicates statistical significance (linear mixed effects model, post-hoc Tukey pairwise test, see STAR Methods). Early blastocyst stage EPI-PrE p=0.0271 n=14 embryos. Mid-blastocyst stage: n=8 embryos. Not significant (n.s.) pairwise comparisons indicated where p > 0.05.
Individual cell traces revealed a range of baseline ERK activities and temporal variation within cells of the same identity (Figure 4D). In early blastocyst stage embryos ERK activity in individual cells gradually increased (+18%), which was not seen in mid-blastocyst stage embryos (+0.6%)(Figure 4D), consistent with increasing levels of Fgf4 mRNA between E3.25 (~32 cells) and E3.5 (~64 cells) (Ohnishi, et al., 2014). Thresholding to discriminate intervals of high and low ERK activity revealed that all ICM lineages contained differing proportions of cells with periods of high-signaling activity (Supplemental Figure 4B). This heterogeneous pan-ICM activation of FGF/ERK is in line with observations made using p-ERK staining, as well as a Spry4H2B-GFP transcriptional reporter (Azami, et al., 2019; Morgani, et al., 2018). Together, these data indicate that all ICM cells, irrespective of lineage identity, transduce FGF/ERK signals at early- and - mid blastocyst stages.
Recent work on ERK signaling dynamics in vitro suggests that in single cells mean ERK activity is the strongest predictor of lineage-specific gene induction (Gillies, et al., 2017). We therefore assessed whether the mean ERK activity per cell is associated with lineage outcomes within the ICM. From early blastocyst stage, those cells that had adopted a PrE identity on average had the highest mean ERK activity (Figure 4E, left). At the mid-blastocyst stage although there were no statistically significant differences between lineages when performing pairwise comparisons, PrE cells showed a higher mean ERK activity than EPI cells (Figure 4E, right). These lineage differences were also apparent between in individual embryos. There was marked embryo-to-embryo variability, and in many, but not all embryos, PrE cells had higher mean ERK activity than EPI cells (Supplemental Figure 4C–D).
Given that EPI cells express high levels of Fgf4, we wondered if their proportion within the ICM might influence ERK activity across the entire ICM population. We calculated the percentage of EPI cells within the ICM for each embryo using the fixed immunostained time-lapse end-points (Figure 4B), and compared this to the mean ERK activity within lineages for each embryo (Supplemental Figure 4E). There was no correlation between the proportion of EPI cells on the mean ERK activity in DP progenitors and EPI cells. By contrast, there was a positive association between the percentage of EPI cells and mean ERK activity in PrE cells. Therefore, ERK activity in cells that express Fgf4, DP (moderate levels) and EPI (high levels) (Nowotschin, et al., 2019), that can signal in an autocrine (from the same cell) or paracrine (from neighboring cells) manner, seemingly are not correlated with global changes in FGF4. Whereas, ERK activity in cells specified as PrE by the end of the time-lapse are greatly influenced by population based changes in FGF4, via paracrine signaling. This agrees with models of FGF4 as a dynamic readout of lineage size to determine cell fate specification as a means of tissue size control (Saiz, et al., 2020).
Given that ERK signaling dynamics can span different timescales (Conlon, et al., 2016; Ryu, et al., 2016), we also analyzed long-term signaling trends in the ICM. We imaged ERK-KTR in early blastocysts for a total of 12 hours with 15-min time-lapse intervals (Supplemental Figure 4F). Cells specified to PrE by the 12 hour end-point had significantly higher activity than EPI cells, and DP progenitor cells had intermediate activity (Supplemental Figure 4G). These long-term trends were consistent with our results from short-term time-lapse imaging (Figure 4D–E). In the subset of cells that divided over the time-course, ERK activity following cell division was initially low, then gradually increased over the remainder of the cell cycle for all ICM lineages (Supplemental Figure 4H). Within ICM lineages, PrE cells exhibited elevated activity relative to EPI cells across the cell cycle (Supplemental Figure 4I).
Lineage-specific dynamics of ERK activity in ICM cells
Integrated signaling activity is dependent on the strength and duration of the response. We noticed temporal variation in ERK activity within ICM cells in our short-term time-lapse imaging. Over the 2 hour time-lapse ICM cells spent a varying percentage of time in a high versus low signaling state (Figure 4D, Supplemental Figure 4B). To characterize this temporal ERK activity we classified peaks to extract dynamic information from single-cell traces. The peak classification parameters were optimized to identify local maxima of ERK activity for visually identifiable peaks in different cell types tracked across the ICM in 3D+t every 5 min for 2 hours (Figure 5A–D). To further validate this approach, we assessed peaks in our different treatment regimes of embryos cultured in MEKi or FGF (Figure 2). Using these settings only 3% of ICM cells were called with one or more peaks in MEKi treated embryos (Supplemental Figure 5A). By contrast, treatment with FGF increased the peak number and fraction of ICM cells with peaks as compared to untreated embryos (Supplemental Figure 5B).
Figure 5: Lineage-specific dynamics of ERK activity in ICM cells.
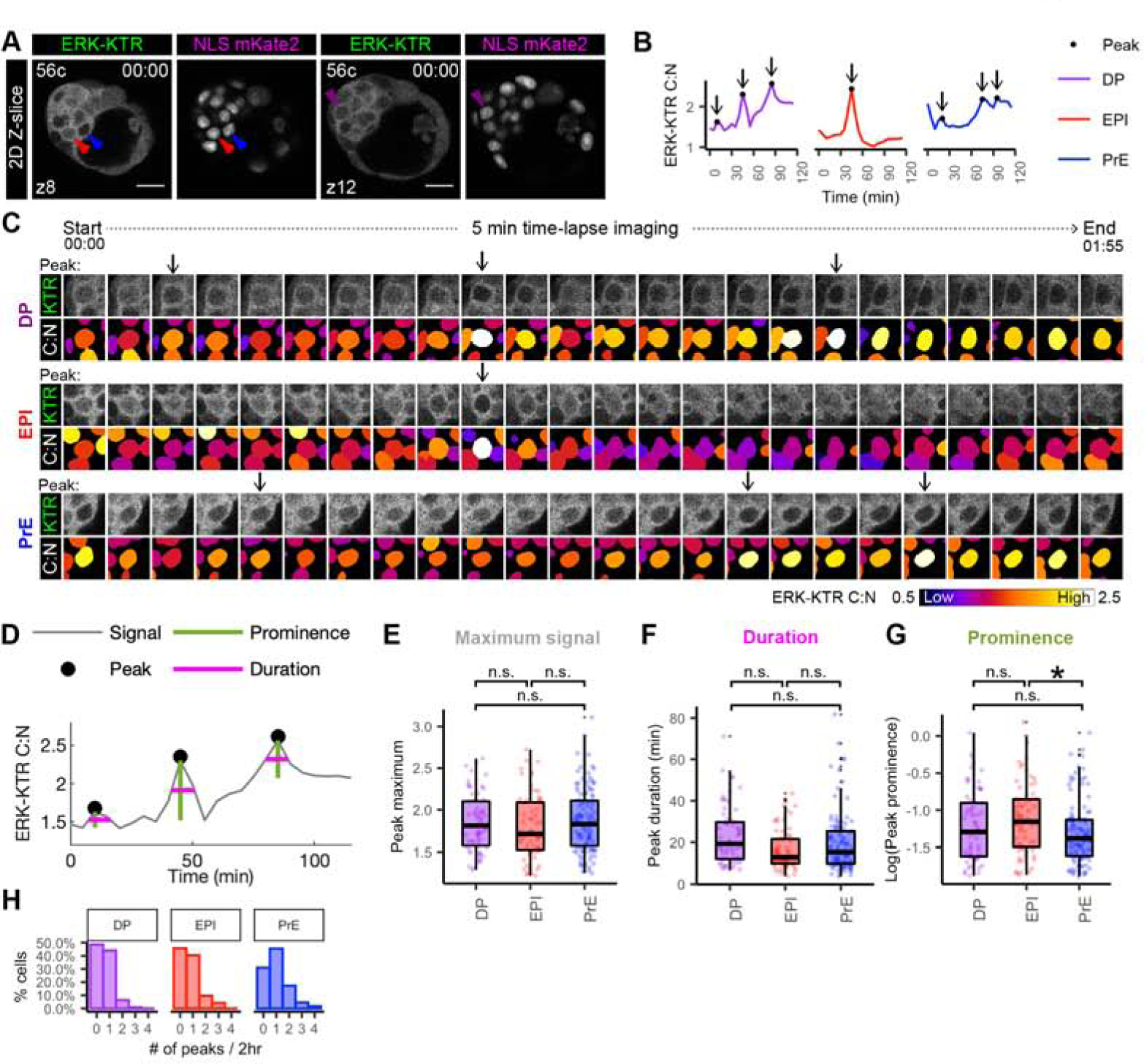
(A) Single Z-slice of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryos at beginning of 2 hour 5 min time-lapse movie (Figure 4A). Cell number and position of z-slice is shown. Arrowheads indicate cells shown in traces (B) and sequential frames (C). (B) Single-cell traces of ERK-KTR C:N over 2 hour time-lapse. Color coding indicate lineage as shown. Arrows indicate peaks. (C) Sequential frames of example DP, EPI and PrE cells from the same embryo shown in A. ERK activity illustrated by C:N heatmap. Arrows indicate peaks. (D) Peak calling (of the example DP trace from the embryo shown in A) to extract dynamic information from individual traces. Peaks identified as local maxima. Prominence is peak height relative to nearest minima. Duration is width at half prominence. (E-G) Boxplot showing dynamic information extracted from peak-calling (D) Each peak is shown as individual points. Color coding for lineage as indicated before. Asterisk indicates statistical significance. Prominence EPI versus PrE p=0.016 (Wilcoxon test, Bonferroni adjustment). n=22 embryos. Not significant (n.s.) pairwise comparisons indicated where p > 0.05. (E) Histogram showing the number of peaks detected within each cell as a percentage of cell in that lineage.
To assess differences between ICM lineages at early and mid-blastocyst stages, we compared the maximum signal, prominence (height relative to nearest minimum), and duration (width at half prominence) of each peak (Figure 5D). The maximum signal and duration of peaks was comparable between ICM lineages (Figure 5E, F). The peak prominence, however, was significantly higher in EPI cells compared to PrE cells (Figure 5G). Given that the maximum signal was similar between EPI and PrE cells, we hypothesized that the difference in prominence was due to a higher baseline ERK activity in PrE cells caused by sequential or sustained ERK activation. By comparing the nearest minimum (maximum signal – prominence) as an approximation of baseline levels, PrE cells indeed were elevated compared to EPI cells (Supplemental Figure 5G). Furthermore, PrE cells had a higher proportion of cells with one or more peaks, relative to both EPI cells and DP progenitor cells (Figure 5H).
The peak duration were not significantly different between control (untreated) and FGF treated embryos (Supplemental Figure 5E,F). However, the maximum signal and nearest minimum (approximation of baseline) were elevated, while the peak prominence was reduced in cells of FGF treated embryos compared to cells in controls (Supplemental Figure 5D,G), indicating that the similar maximum signal of peaks in ICM lineages in untreated conditions (Figure 5E) was not due to saturation of the ERK-KTR biosensor, but limited by the concentration of endogenous FGF ligand.
In conclusion, these data demonstrate that the ERK-KTR reporter enables the study of temporal changes in ERK activity during ICM lineage specification. ERK activity is dynamic with activation profiles that differ between cells of distinct lineage identities: EPI have lower baseline signaling with sporadic pulses of activity, while PrE cells have elevated baseline activity.
DISCUSSION
Genetic and pharmacological perturbation of FGF/ERK signaling has demonstrated its critical role for the proper patterning and proliferation of cells of the blastocyst stage embryo prior to implantation into the maternal uterus. FGF/ERK signaling impacts all three blastocyst lineages; it drives PrE specification, regulates EPI maturation, and has been suggested as the mitogen which promotes TE proliferation (Christodoulou, et al., 2019; Kurowski, et al., 2019; Kang, et al., 2017; Molotkov, et al., 2017; Saiz, et al., 2016b; Kang, et al., 2013; Krawchuk, et al., 2013; Yamanaka, et al., 2010; Nichols, et al., 2009; Chazaud, et al., 2006; Yang, et al., 2006; Goldin and Papaioannou, 2003; Chai, et al., 1998; Tanaka, et al., 1998; Feldman, et al., 1995). To directly and quantitively visualize the ERK signaling activities of individual cells within the entire blastocyst population we developed an ERK-KTR targeted mouse reporter line. Modulation of the FGF/ERK pathway in embryos revealed that our reporter was acutely responsive to these perturbations, thus constituting a faithful readout of ERK activity. Although several ligands could potentially feed into the Raf/MEK/ERK cascade during pre-implantation stages, the absence of p-ERK staining and the complete loss of PrE specification in Fgf4−/− embryos suggests that in the context of the blastocyst, ERK is the main effector of FGF signaling, and therefore ERK-KTR primarily detects FGF4 driven ERK activity at this stage (Azami, et al., 2019; Kang, et al., 2013; Krawchuk, et al., 2013; Chazaud, et al., 2006).
By visualizing ERK signaling activities live in the TE, we quantified a spatiotemporal ERK activity gradient within the TE beginning from the early blastocyst stage, which was not possible with a previous transcriptional reporter of FGF signaling (Morgani, et al., 2018). We observed decreasing ERK activity along the growing embryonic to abembryonic axis. Although FGF ligands may be freely diffusible (Yu, et al., 2009), these data suggest there is no substantial luminal accumulation of FGF4 in the blastocyst cavity as has been observed in other systems (Ryan, et al., 2019; Durdu, et al., 2014). Alternatively, the presence of extra-cellular matrix components, such as heparin sulphate (HS), might act to concentrate ICM progenitor and EPI-derived ligands in the immediate vicinity of the ICM (Matsuo and Kimura-Yoshida, 2014; Ornitz, 2000). At early post-implantation stages, HS-deficient Ext2 embryos show defective extra-embryonic ectoderm (of polar TE origin) development, due to a requirement for HS-chains in retaining locally high concentrations of FGF ligands (Shimokawa, et al., 2011). In support of the short-ranging FGF action, a recent study in mouse ESCs revealed that paracrine FGF4 signals act over distances of ~1 cell diameter (Raina, et al., 2020). We found that signaling within the mural TE increased upon FGF treatment, indicating that ligand availability can shape the spatiotemporal ERK activity gradient within the TE. In vivo and in vitro studies have revealed that FGF4 maintains trophoblast stem-cell identity (Nichols, et al., 2009; Goldin and Papaioannou, 2003; Chai, et al., 1998; Tanaka, et al., 1998). Competition within the TE for locally concentrated FGF4 mitogens emanating from the ICM niche could therefore act as a mechanism to balance the ratio of stem cell and differentiated progenitors, as seen in other systems (Kitadate, et al., 2019)
FGF signaling via ERK is required within the ICM for PrE specification, and EPI maturation, which is abrogated upon MEK inhibition (Nichols, et al., 2009). We observed some ERK activity in all ICM cells, in agreement with observations made using p-ERK staining, Spry4H2B-GFP transcriptional reporter and pan-ICM expression of Fgfr1 and its functional requirement in both PrE and EPI lineages (Azami, et al., 2019; Morgani, et al., 2018; Kang, et al., 2017; Molotkov, et al., 2017). ERK signaling was highest in those cells that had acquired a PrE identity by the end of the time-lapse movies. This differential ERK signaling could be explained by the local signaling environment where higher concentrations of the FGF4 ligand leads to signaling via FGFR1/ERK on progenitor cells to induce a PrE fate (Kang, et al., 2017; Molotkov, et al., 2017). Or additionally, by upregulation of RTK receptors FGFR2 and PDGFRa or modifiers of the pathway upon PrE specification (Azami, et al., 2019; Kang, et al., 2017; Molotkov, et al., 2017). Although, as both FGFR2 and PDGFRA act via the PI3K axis for PrE-survival (Bessonnard, et al., 2019; Molotkov and Soriano, 2018; Artus, et al., 2013), ERK activity is likely relayed by FGFR1, which is the predominant receptor for PrE specification (Kang, et al., 2017; Molotkov, et al., 2017). Therefore, it remains possible that, even though they are subtle, the higher ERK activities seen here in the PrE might be instructive for lineage acquisition. However, since we relied on end-point analyses for assignment of lineage identities, we could not assign the exact point at which individual cells commit to a particular fate. Lineage-specific expression changes to FGF pathway components may also lead to differential ERK activity between lineages. Therefore, a limitation of this analysis is our inability to attribute causality between ERK activities and cell fate specification. Further study involving multiple spectrally-distinct lineage-specific (PrE and EPI) reporters (Xenopoulos, et al., 2015; Plusa, et al., 2008) alongside the ERT-KTR would be needed to delineate the causality and timing of ERK activities and eventual fates.
Transient and pulsatile modes of ERK activity have been described in diverse systems including cultured mammalian cells, mouse epidermis, Drosophila, C. elegans embryos and yeast through single-cell time-lapse imaging using ERK FRET, phase-separation, or KTR-based biosensors (Deathridge, et al., 2019; Moreno, et al., 2019; Zhang, et al., 2018; de la Cova, et al., 2017; Gillies, et al., 2017; Conlon, et al., 2016; Ryu, et al., 2016; Hiratsuka, et al., 2015; Regot, et al., 2014). Here, we quantified short-term temporal dynamics operating within ICM cells of the mouse blastocyst stage embryo. PrE cells exhibit sustained ERK activity, whereas EPI cells exhibit infrequent pulses while maintaining lower baseline ERK activity, reminiscent of the steady-state in ESCs (Supplemental Figure 1A, B). We were limited in the total length of time we could image short-term ERK dynamics, as we sought to reduce the overall light exposure time to reduce phototoxicity in embryos imaged by confocal microscopy. Moving forward, the use of alternative imaging modalities, for example selective plane illumination microscopy (SPIM), which require lower levels of light exposure should enable in toto embryo imaging at high temporal resolution throughout the entire ICM cell fate specification process (~48hrs) (Strnad, et al., 2016).
How cells decode signaling dynamics has been studied in other systems (Patel and Shvartsman, 2018; Levine, et al., 2013; Purvis and Lahav, 2013), but been a challenge to investigate within mammalian embryos. Reflecting on our observations with PrE fate acquisition, integrated ERK activity over time is associated with cell fate determination in multiple cell types (Gillies, et al., 2017; Albeck, et al., 2013; Aoki, et al., 2013; Santos, et al., 2007; Murphy, et al., 2002). We hypothesize that ICM lineage identity is encoded by pulsatile versus sustained signaling dynamics, as has been shown for ERK pathway in neurogenesis (PC-12 cells) (Santos, et al., 2007), and the Notch pathway during myogenesis (C2C12 cells) (Nandagopal, et al., 2018). In these cultured mammalian cell lines, differential dynamics are elicited by distinct ligands, but it is an open question how differential dynamics are established in response to a single ligand within the ICM of the mouse blastocyst. Treatment of blastocysts with exogenous FGF4 lead to a sustained signaling response, due to increased baseline ERK activity, as well as a moderate increase in the frequency of pulses. Thus, differential activity of PrE and EPI cells may be indicative of heterogenous FGF4 concentrations across the ICM. In addition, differential activity could also be due to the expression of distinct negative feedback regulators (EPI: ETV5, DUSP1/6, PrE: DUSP4) or receptors (EPI: FGFR1, PrE: FGFR1, FGFR2, PDGFRa) upon specification to EPI versus PrE identities (Azami, et al., 2019; Nowotschin, et al., 2019; Kang, et al., 2017; Ohnishi, et al., 2014; Guo, et al., 2010). Alternatively, ICM cell fate decisions may operate through amplitude modulation (Li and Elowitz, 2019), as has been shown for p53 regulation of cell-cycle transitions (Reyes, et al., 2018). Taken together, our results constitute direct visualization of dynamic signaling events underlying mammalian pre-implantation development, leading us to uncover spatial and temporal lineage-specific dynamics. The approach and tools we have developed will be broadly applicable to study signaling dynamics in other in vivo contexts using the mouse model.
STAR METHODS
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Anna-Katerina Hadjantonakis (hadj@mskcc.org)
Materials Availability
Mouse lines generated in this study have been deposited to Jackson Labs, JAX#035333
Data and Code Availability
The dataset and code generated and used in this study are available at Github, https://github.com/therealkatlab/Simon2020. Time-lapse microscopy images generated in this study are available at Figshare, DOI: 10.6084/m9.figshare.12794810.
Experimental model details
Cell lines
Mouse embryonic stem cell (ESC) line HprtCAG:ERK-KTR-mClover (abbreviated as ERK-KTR) was targeted as follows. The CAGGS enhancer/promoter sequence and a rabbit beta-globin poly-A sequence were PCR amplified from pCX-H2B-EGFP (Hadjantonakis and Papaioannou, 2004). The ERK-KTR-Clover coding sequence was amplified from pENTR-ERKKTRClover (Addgene plasmid #59138) (Regot, et al., 2014). A targeting vector containing homology arms to the Hprt locus (Doetschman, et al., 1987) as well as a DTA cassette was linearized with AcsI (NEB), and the PCR fragments were inserted via Gibson assembly. A sequence-verified targeting vector was linearized with SalI (NEB) and electroporated into E14tg2a ESCs (Hooper, et al., 1987). ESCs were plated onto HPRT+ mouse embryonic feeder cells in DMEM (Life Technologies) supplemented with 15% FBS (Sigma), 10 ng/ml recombinant leukemia inhibitory factor (LIF) (Department of Biochemistry, University of Cambridge, and protein expression facility, MPI Dortmund), 0.1mM 2-mercaptoethanol, glutamax and non-essential amino acids (NEAA) (all Life Technologies). One day after electroporation, correctly targeted clones were selected by addition of 100 μM hypoxanthine, 0.4 μM aminopterin and 16 μM thymidine (HAT medium, Life Technologies) (Doetschman, et al., 1987). All surviving clones evenly expressed the fluorescent sensor. We selected three independent clones for karyotyping (Nagy, et al., 2008), all of which had a modal chromosome count of n = 40. After targeting, ESCs were maintained on 0.1% gelatin (Millipore) coated tissue-culture grade plates in a humidified 37°C incubator with 5% CO2. Cells were grown in DMEM, supplemented with 2mM L-glutamine, 0.1mM MEM NEAA, 1mM sodium pyruvate, 100U/ml penicillin and 100μg/ml streptomycin, 0.1mM 2-mercaptoethanol (all from Life Technologies), 15% FBS (VWR), and 1000U/ml LIF. ESCs were maintained on a layer of Mitomycin C inactivated mouse embryonic fibroblast (MEF) for the generation of chimeric mice and maintained without feeders for live imaging analysis.
Mouse strains and husbandry
All animal work was approved by MSKCC Institutional Animal Care and Use Committee (IACUC). Animals were housed in a pathogen free-facility under a 12 hour light cycle. Mouse strains used in this study were HprtERK-KTR (this study), R26CAG:3xNLS-mKate2 (abbreviated as NLS-mKate2) (Susaki, et al., 2014) and wild-type CD1 (Charles River). HprtERK-KTR mice were generated by the Mouse Genetics Core at MSKCC. Briefly, HprtERK-KTR ESCs derived from E14Tg2a 129Ola mouse strain were injected into C57Bl6 host blastocyst. Resulting high contribution male germline chimeras were mated to wild-type CD1 females. Animals were maintained on a mixed (CD1/Bl6/129) strain background. Mice were genotyped by PCR and fluorescence of ubiquitously expressed florescent protein reporters. The HprtCAG-ERK-KTR-mClover mouse line will be available from The Jackson Laboratory as JAX stock no. 035333
Method details
Embryo culture
Male mice homozygous for the nuclear reporter R26NLS-mKate2 were mated with female mice homozygous for the nuclear reporter R26NLS-mKate2 and heterozygous for the ERK reporter HprtERK-KTR/+. Embryos for this study were obtained from natural matings of females 5 – 12 weeks of age. The sex of embryos was not determined. E3.0 – E4.5 blastocysts were flushed from uterine horns with flushing and holding medium (FHM, Millipore) as described (Behringer, et al., 2014). Zona pellucidae were removed by brief incubation in acid Tyrode’s solution (Sigma). Embryos were washed three times, and then cultured in 5μl in KSOM-AA medium without phenol red (Millipore) overlaid with mineral oil (Sigma), with approximately 5 embryos per droplet. Embryos expressing the ERK-KTR were identified by epifluorescence microscopy, cultured in 35mm glass-bottomed dishes (Maktek) and imaged on a Zeiss LSM880 confocal microscope in a humidified incubation chamber with 5% CO2 at 37°C for 2 or 12 hours. Negative littermate embryos were cultured in 35mm dishes, in a humidified 37°C incubator with 5% CO2 as a control for the same duration as their live-imaged littermates. For ERK signaling perturbation experiments embryos were cultured in the same way, and littermate embryos were randomly assigned in even sized groups between Control: KSOM and either FGF stimulation: 1μg/ml FGF4 (R&D Systems) and 1μg/ml Heparin (Sigma) in KSOM or ERK inhibition: 1mM PD0325901(MEK inhibitor, Reprocell) in KSOM.
Live image data acquisition
Time-lapse imaging of embryos was performed using Zeiss LSM880 laser-scanning confocal microscope. Images were acquired using an oil-immersion Zeiss EC Plan-Neofluar 40x/NA1.3 with a 0.17mm working distance. Laser power was measured at the beginning of each session; mClover: 488nm Argon laser at 30μW and mKate2: 561nm DPSS 561–10 laser at 80μW. Both colors were acquired simultaneously using 0.85μs pixel dwell, bi-directional line scanning, 2-line averaging, 1.5x zoom and 1 airy unit pinhole. To limit light exposure, the inner cell mass was centered in a 60μm stack, with a 2μm step, generating 12-bit range 512×512px images of 0.277μm/px resolution acquired in ~30s per embryo and time point. Embryos were imaged with a time-interval of 5 min for a total of 2 hours, or 15 min for a total of 12 hours, as indicated. Before each time-lapse movie an image stack encompassing the entire embryo was acquired in only mKate2 to allow for an initial cell-count for staging, and time (~30 min) for the embryos to settle after placement on the scope to minimize movement during time-lapse acquisition. After time-lapse imaging a second complete overview stack was taken to allow for manual image registration to the corresponding fixed immunofluorescence image. Time-lapse images of cells were acquired using the same set-up in mClover only and a single z-slice, with a time-interval of 3 min for a total of 90 min,
Immunofluorescence
Immunofluorescence of embryos was carried out as previously described (Simon, et al., 2018b). Briefly, live-imaged embryos (processed individually) and cultured littermate controls were collected immediately after time-lapse movies were complete and fixed in 4% PFA (Electron Microscopy Sciences) for 10 min at room temperature. Fixation quenched endogenous mKate2 fluorescence. Fixed embryos were stored in PBS and prior to staining washed in PBX; 0.1% TritonX-100 (Sigma) in PBS, permeabilized for 5 min in 0.5% TritonX-100, 100mM glycine (Sigma) in PBS, and washed in PBX. Embryos were blocked in blocking buffer, 2% horse serum (Sigma) in PBS, for 40 min at room temperature and incubated overnight at 4°C with goat anti-GATA6 (R&D, 1:100) and rabbit anti-NANOG (Reprocell, 1:500) in blocking buffer (see Key Resources Table). Then, embryos were washed 3x in PBX, blocked again in blocking buffer for 40 min at room temperature, and incubated with secondary antibodies donkey anti-rabbit A647 and donkey anti-goat A568 (both Invitrogen, 1:500) in blocking buffer for 1 hour at 4°C. Embryos were washed 2x in PBX, then incubated for at least 1 hour in 5μg/ml Hoechst 33342 (Invitrogen) in PBS prior to imaging.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-NANOG | Reprocell | Cat# RCAB001P; RRID: AB_1962694 |
| Goat anti-GATA6 | R&D Systems | Cat# AF1700; RRID: AB_2108901 |
| Donkey anti-goat A568 | Invitrogen | Cat# A-11057; RRID:AB_2534104 |
| Donkey anti-rabbit A647 | Invitrogen | Cat# A-10042; RRID: AB_2534017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hoechst 33342 | Invitrogen | Cat# H3570 |
| FGF4 | R&D Systems | Cat# 235-f4 |
| Heparin | Sigma | Cat# H3149 |
| PD0325901 | Reprocell | Cat# 04-0006-10 |
| EmbryoMax KSOM (+AA w/o PhenolRed) | Sigma | Cat# MR-106-D |
| Mineral oil | Sigma | Cat# M5310 |
| FHM | Millipore | Cat# MR-025-D |
| Proteinase K | Roche | Cat# 03115801001 |
| PFA | Electron microscopy sciences | Cat# 15710 |
| Acid Tyrode’s | Millipore | Cat# MR-004-D |
| Glycine | Sigma | Cat# G7403 |
| Triton X-100 | Sigma | Cat# X100 |
| Horse Serum | Sigma | Cat# H0146 |
| DMEM | Life technologies | Cat# 11995073 |
| NEAA | Life technologies | Cat# 11140-050 |
| Glutamine | Life technologies | Cat# 25030164 |
| Glutamax | Life Technologies | Cat # 35050038 |
| Sodium Pyruvate | Life technologies | Cat# 11360070 |
| 2-mercaptoethanol | Life technologies | Cat# 21985023 |
| Fetal Calf Serum | VWR | Cat# 97068-085 |
| HAT supplement | Life technologies | Cat# 21060017 |
| 0.25% Trypsin EDTA | Life technologies | Cat# 25200114 |
| Fibronectin | Millipore | Cat# FC010 |
| Gelatin | Millipore | Cat# 104070 |
| Penicillin/Streptomycin | Life technologies | Cat# 15140163 |
| Mitomycin C | Sigma | Cat# M4287 |
| Deposited Data | ||
| CellProfiler pipelines, ERK-KTR C:N measurements from 3D time-lapse movies, R and Matlab scripts for data analysis | This paper | https://github.com/therealkatlab/Simon2020 |
| Confocal microscopy images | This paper | 10.6084/m9.figshare.127 94810 |
| Experimental Models: Cell Lines | ||
| Mouse: Embryonic stem cell line HprtERK-KTR-mClover | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: R26nls-mKate2 | (Susaki, et al., 2014) | N/A |
| Mouse: HprtERK-KTR-mClover | This paper | JAX#035333 |
| Mouse: CD1 | Charles River Laboratory | Cat# 022 |
| Oligonucleotides | ||
| mKate2_F: 5’-AGATCTGGTACTCGTATGGTGAGCGA GCTGATT-3’ | (Susaki, et al., 2014) | N/A |
| mKate2_R: 5’-TGTGCCTGTAAGCTTTCTTCTGTGCCC CAGTTTGCT-3’ | (Susaki, et al., 2014) | N/A |
| Software and Algorithms | ||
| Imaris 9.1.2 | Bitplane (Oxford Instruments) | https://imaris.oxinst.com/ |
| ImageJ | (Schneider, et al., 2012) | https://imapei.nih.pov/ii/ |
| R Studio 1.0.143 | RStudio, Inc. | https://rstudio.com/ |
| Matlab R2014a/R2018a | MathWorks | https://www.mathworks.com/products/matlab.html |
| CellProfiler 2.1 | (Lamprecht, et al., 2007) | https://cellprofiler.org/ |
| MINS | (Lou, et al., 2014) | http://katlab-tools.org/ |
| ZEN | Carl Zeiss Microsystems | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Other | ||
| Glass-bottom dish | MakTek | Cat# P35G-1.5-14-C |
Image acquisition of fixed samples
Fixed immunostained samples were imaged on a Zeiss LSM880 laser scanning confocal microscope. Embryos were mounted in microdroplets of 5μg/ml Hoechst 33342 in PBS on glass-bottomed dished (Maktek) coated with mineral oil (Sigma). Embryos were imaged along the entire z-axis with 1μm step using an oil-immersion Zeiss EC Plan-Neofluar 40x/NA1.3 with a 0.17mm working distance. Laser power was measured prior to each imaging session and adjusted to maintain consistent power output across experiments to reduce technical variability.
Quantification and statistical analysis
Image processing and quantification
Quantification of the ERK-KTR is outlined in Figure 2A. The ERK-KTR is a ratiometric reporter which shuttles in and out of the nucleus, where the relative intensity between the cytoplasm and nucleus is a read-out of ERK activity. Raw images were converted to single files for each z-plane, time-point and channel. Segmentation and intensity measurements were performed in CellProfiler (Lamprecht, et al., 2007). Image processing, segmentation and calculation of C:N ratio was performed independently for each z-plane and time-point i.e. in 2D. First, the embryo was identified in the ER-KTR channel and used as a mask for objects to ensure no background is included in cytoplasm and nuclear objects, and to measure background mean fluorescent intensity of ERK-KTR. Next, the NLS-mKate2 channel was processed (rescaled intensity, enhance features, gaussian blur), and nuclear segmentation carried out using a three-class Otsu thresholding method. The initial nuclear segmentation was shrunk by 2px and this area used to measure the mean nuclear fluorescent intensity of the raw ERK-KTR. For the cytoplasm, a ring of 6px width around the nucleus with a 1px buffer from the initial nuclear segmentation was used to measure the mean cytoplasmic fluorescent intensity of the raw ERK-KTR. The cytoplasmic ring was restricted to prevent overlap with neighboring cells, and to avoid detection of background pixels at the edge of the embryo. The C:N (cytoplasmic to nuclear ratio) was calculated for each nuclei by dividing the mean background-subtracted cytoplasmic ERK-KTR intensity by the mean background-subtracted nuclear intensity ERK-KTR. This ratio normalizes for the fluorescence decay along the z-axis. The C:N value was calculated per nuclei at each z-plane and time point. To generate a 3D+time image representation, each nuclear object was color-coded by its C:N value as a 16-bit image, and then each z-plane and time-point concatenated into a TIFF stack of C:N value images. In rare cases, there is under-segmentation of dense ICM cells. However, these clumped cells are automatically filtered out based on a size threshold for objects. For these under segmented cells, a second CellProfiler pipeline using two-class Otsu thresholding method, which better segments dense ICM cells, was used for C:N measurements. Calculations and image processing of C:N was performed using CellProfiler outputs in Matlab R2018a (Mathworks) and ImageJ (Schneider, et al., 2012).
Cell-tracking of embryos was performed manually in Imaris (Bitplane) on the mKate2 channel using the Spots function. 3D+time images of C:N values were imported into Imaris to perform measurements. Spots of diameter 5μm were placed inside each tracked nucleus to sample at least two Z-slices (2μm step). Not all ICM cells could be tracked in each embryo due to cells moving out of frame, or difficulty tracking daughter cells after loss of nuclear NLS-mKate2 upon nuclear envelope breakdown at mitosis.
Time-lapse movies and fixed immunostained images of the same embryo were manually registered in Imaris. mitotic, apoptotic, distinctly shaped or sized cells, and abutting mural TE cells were annotated and used as landmarks to aid manual registration and orient embryos in 3D.
To perform cell-counts of live-imaged embryos and lineage assignment of fixed immunostained embryos, a Matlab based 3D nuclear segmentation software tool, MINS was used as described previously (Saiz, et al., 2016a; Lou, et al., 2014). Correction for fluorescence decay along the z-axis was performed by fitting linear regressions to the log fluorescence values as a function of the z – axis and correcting using an empirical Bayes method, as detailed in (Saiz, et al., 2016b).
Lineage identities were assigned as follows. Outer TE cells were manually annotated in Imaris by their position; overlying the cavity as mural TE and overlying ICM as polar TE. ICM in fixed images were assigned based on NANOG and GATA6 expression using k-means clustering method, used previously (Saiz, et al., 2016b), where GATA6+NANOG+ marks double positive progenitors, NANOG+ marks EPI and GATA6+ marks PrE. Lineage identities of fixed immunostained embryos were then used to annotate corresponding cells in registered final frame in the time-lapse movie of the same embryo. Any ambiguous ICM cells (inc. double negative cells) that could not be registered with the corresponding fixed image were not assigned a lineage identity and excluded from analysis.
To generate supplemental movies of time-lapse imaged embryos, images were registered between time points to compensate for embryo movement during the experiment. For this, we used the descriptor based series registration plug-in in ImageJ (Preibisch, et al., 2010), with NLS mKate2 as the registration channel.
Data processing
All plots were generated using R Studio version 1.0.143 and Matlab R2018a. During mitosis there is breakdown of the nuclear envelope and thus, no shuttling of KTR. Therefore, time-points when a cell was undergoing mitosis were manually labelled based on NLS-mKate2 localization, and excluded from analysis. Apoptotic cells were also removed, and only cell tracks of length equal to or greater than 5 frames were included in the analysis. Parent and daughter cells before and after mitosis were treated independently.
Statistical power calculations were not used to pre-determine sample size. Figure legends and panels provide statistical details for each experiment, including p-values, the exact value and representation of n, definition of data points, and details of data representation. For comparison of ERK-KTR C:N values between different lineages (Figure 4F, Supplemental Figure 4D), we used a linear mixed effects model to account for embryo-fixed effects. If there was a significant effect (p<0.05, ANOVA test) of lineage in the linear mixed effects model, we performed and reported p-values for post-hoc Tukey test for pairwise comparisons between lineages. For other pairwise comparisons, we used the non-parametric Wilxocon test to compare between the means for each embryo (n reported in figure legends). When multiple groups were tested, we applied a Bonferroni correction for multiple comparisons. Boxplots in all figures are as follow: top and bottom edges of boxes represent third and first quartiles, middle lines mark the median, top and bottom whiskers extend from first and third quartile to lowest and highest value within 1.5 * inter-quartile range.
To identify a threshold ERK-KTR C:N value to assign high versus low ERK activity, we compared ICM cells from MEKi and FGF treated embryos. We iteratively tested thresholds at 0.01 steps across the entire range of the dataset to classify values as high (FGF treated) and low (MEKi treated) ERK activity. The best threshold (C:N value = 1.2142) was determined as the median of thresholds most accurately classifying cells as MEKi and FGF treated.
Peaks of ERK activity were classified in Matlab R2018a. Traces were smoothed using smoothdat with a Gaussian weighted moving average filter with window length 3 to reduce noise for peak calling. Peaks were identified using findpeaks. The threshold for the peak prominence was determined empirically based on visually identifiable peaks, and the threshold for the peak maximum was the same as assigning high-low ERK activity (described above).
Supplementary Material
Supplemental Movie 1: Dynamic ERK activity in ESCs (Related to Figure 1B) Time-lapse confocal imaging of mouse embryonic stem cells (ESC) expressing HprtCAG-ERK-KTR-mClover grown in serum/LIF culture conditions. Timestamp shows Hour:Min. Scale bar 20μm.
Supplemental Movie 2: Quantification of ERK-KTR C:N in 3D in the mouse blastocyst. (Related to Figure 2A). Sequential z-slices of ERK-KTR, and 180 degree rotation of 3D max intensity projection (MIP) of ERK-KTR, NLS mKate2 and ERK-KTR C:N values in mid-blastocyst stage hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryo. Heatmap for ERK-KTR C:N as in Supplemental Figure 2A. Scale bar 20μm.
Supplemental Movie 3: ERK-KTR is a read out of FGF/ERK signaling in the mouse blastocyst. (Related to Figure 2C). Time-lapse confocal images of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryos in control, MEKi and FGF culture conditions. Max intensity projections (MIP) and a single z-slice at mid-point through the ICM are shown. ERK-KTR C:N values corresponding to ERK activity is shown as a heatmap as indicated in Figure 2C. Time-lapse interval 5 min for a total of 2 hours. Timestamp shows Hour:Min. Scale bar 20μm.
Supplemental Movie 4: Tracking ERK activity in blastocyst (Related to Figure 3 and Figure 4). Max intensity projection of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryo in 2 hour, 5 min interval time-lapse movie. (LEFT) ERK-KTR (MIDDLE) NLS-mKate2 overlaid with manually tracked spots in Imaris (Bitplane). Color coding indicates lineage identity by the end of the movie. Double positive ICM progenitor (DP): purple, Primitive endoderm (PrE): blue, Epiblast (EPI): red. Polar trophectoderm (TE): light green. Mural TE: dark green. (RIGHT) ERK-KTR C:N showing levels of ERK activity. Heatmap as in Figure 4C. Scale bar 20μm.
Supplemental Movie 5: Retrospective lineage assignment of the ICM (Related to Figure 4). 360 degree rotation of a mid-blastocyst stage embryo registered to assign ICM lineage identity before and after fixation. (LEFT) Max intensity projection (MIP) confocal image of the embryo after being fixed and stained. 3D rendering in Imaris (Bitplane) corresponding to an ICM cell’s lineage identity after k-means clustering of relative NANOG and GATA6 levels (as in Figure 4B) (Saiz, et al., 2016b). Double positive ICM progenitor (DP): purple, Primitive endoderm (PrE): blue, Epiblast (EPI): red. Merged with Hoechst (grey) to show nuclei. (RIGHT) MIP of the live embryo in the final time frame of 2 hour time-lapse movie. 3D rendering corresponding to ICM lineage identity shown (LEFT) merged with NLS mKate2 (grey) to show nuclei. Scale bar 20μm.
Highlights:
Generation of a targeted ERK-KTR mouse line that reports ERK signaling in blastocysts.
FGF4 signals from the ICM spatially pattern the trophectoderm.
Signalling heterogeneity in ICM cells corresponds to distinct lineage identities.
PrE exhibit elevated ERK activity, while EPI show sporadic pulses of activity.
ACKNOWLEDGEMENTS
We thank the Transgenic Core Facility at MSKCC for generating chimeric mice; Elvin Feng of the Molecular Cytology Core Facility for help with 3D+t cropping of cells in time-lapse movies; members of the Hadjantonakis and Pe’er labs, and Kathryn Anderson, Jordi Garcia-Ojalvo, Eric Siggia and Phil Soriano for helpful discussions and comments on the manuscript; Michael Pokrass and Sergi Regot for sharing their CellProfiler pipeline and Matlab script to perform 2D C:N calculations which we modified and built on for our analyses. This work was supported by grants from the National Institutes of Health (NIH) to A.-K.H. (R01DK084391, R01HD094868 and P30CA008748). C.S.S. was supported by a training award from NYSTEM (C32599GG). C.S. and D.R. were supported by the Max Plank Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors have no competing interests to declare.
REFERENCES
- Albeck JG, Mills GB, and Brugge JS (2013). Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell 49, 249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Kumagai Y, Sakurai A, Komatsu N, Fujita Y, Shionyu C, and Matsuda M (2013). Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol Cell 52, 529–40. [DOI] [PubMed] [Google Scholar]
- Artus J, Kang M, Cohen-Tannoudji M, and Hadjantonakis AK (2013). PDGF signaling is required for primitive endoderm cell survival in the inner cell mass of the mouse blastocyst. Stem Cells 31, 1932–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azami T, Bassalert C, Allegre N, Valverde Estrella L, Pouchin P, Ema M, and Chazaud C (2019). Regulation of the ERK signalling pathway in the developing mouse blastocyst. Development 146. [DOI] [PubMed] [Google Scholar]
- Behringer R, Gertsenstein M, Nagy KV, and Nagy A (2014). Manipulating the mouse embryo : a laboratory manual (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; ) [Google Scholar]
- Bessonnard S, Coqueran S, Vandormael-Pournin S, Dufour A, Artus J, and Cohen-Tannoudji M (2017). ICM conversion to epiblast by FGF/ERK inhibition is limited in time and requires transcription and protein degradation. Sci Rep 7, 12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessonnard S, De Mot L, Gonze D, Barriol M, Dennis C, Goldbeter A, Dupont G, and Chazaud C (2014). Gata6, Nanog and Erk signaling control cell fate in the inner cell mass through a tristable regulatory network. Development 141, 3637–48. [DOI] [PubMed] [Google Scholar]
- Bessonnard S, Vandormael-Pournin S, Coqueran S, Cohen-Tannoudji M, and Artus J (2019). PDGF Signaling in Primitive Endoderm Cell Survival Is Mediated by PI3K-mTOR Through p53-Independent Mechanism. Stem Cells 37, 888–898. [DOI] [PubMed] [Google Scholar]
- Brewer JR, Mazot P, and Soriano P (2016). Genetic insights into the mechanisms of Fgf signaling. Genes Dev 30, 751–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JR, Molotkov A, Mazot P, Hoch RV, and Soriano P (2015). Fgfr1 regulates development through the combinatorial use of signaling proteins. Genes Dev 29, 1863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai N, Patel Y, Jacobson K, McMahon J, McMahon A, and Rappolee DA (1998). FGF is an essential regulator of the fifth cell division in preimplantation mouse embryos. Dev Biol 198, 105–15. [DOI] [PubMed] [Google Scholar]
- Chazaud C, Yamanaka Y, Pawson T, and Rossant J (2006). Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the Grb2-MAPK pathway. Dev Cell 10, 615–24. [DOI] [PubMed] [Google Scholar]
- Chen H, Guo R, Zhang Q, Guo H, Yang M, Wu Z, Gao S, Liu L, and Chen L (2015). Erk signaling is indispensable for genomic stability and self-renewal of mouse embryonic stem cells. Proc Natl Acad Sci U S A 112, E5936–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou N, Weberling A, Strathdee D, Anderson KI, Timpson P, and Zernicka-Goetz M (2019). Morphogenesis of extra-embryonic tissues directs the remodelling of the mouse embryo at implantation. Nat Commun 10, 3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon P, Gelin-Licht R, Ganesan A, Zhang J, and Levchenko A (2016). Single-cell dynamics and variability of MAPK activity in a yeast differentiation pathway. Proc Natl Acad Sci U S A 113, E5896–E5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Belly H, Jones PH, Paluch EK, and Chalut KJ (2019). Membrane tension mediated mechanotransduction drives fate choice in embryonic stem cells. bioRxiv, 798959. [Google Scholar]
- de la Cova C, Townley R, Regot S, and Greenwald I (2017). A Real-Time Biosensor for ERK Activity Reveals Signaling Dynamics during C. elegans Cell Fate Specification. Dev Cell 42, 542–553 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mot L, Gonze D, Bessonnard S, Chazaud C, Goldbeter A, and Dupont G (2016). Cell Fate Specification Based on Tristability in the Inner Cell Mass of Mouse Blastocysts. Biophys J 110, 710–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deathridge J, Antolovic V, Parsons M, and Chubb JR (2019). Live imaging of ERK signalling dynamics in differentiating mouse embryonic stem cells. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetschman T, Gregg RG, Maeda N, Hooper ML, Melton DW, Thompson S, and Smithies O (1987). Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330, 576–8. [DOI] [PubMed] [Google Scholar]
- Durdu S, Iskar M, Revenu C, Schieber N, Kunze A, Bork P, Schwab Y, and Gilmour D (2014). Luminal signalling links cell communication to tissue architecture during organogenesis. Nature 515, 120–4. [DOI] [PubMed] [Google Scholar]
- Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, and Goldfarb M (1995). Requirement of FGF-4 for postimplantation mouse development. Science 267, 246–9. [DOI] [PubMed] [Google Scholar]
- Frias-Aldeguer J, Kip M, Vivié J, Li L, Alemany A, Korving J, Darmis F, van Oudenaarden A, Geijsen N, and Rivron NC (2020). Embryonic signals perpetuate polar-like trophoblast stem cells and pattern the blastocyst axis. bioRxiv, 510362. [Google Scholar]
- Gardner RL (2000). Flow of cells from polar to mural trophectoderm is polarized in the mouse blastocyst. Hum Reprod 15, 694–701. [DOI] [PubMed] [Google Scholar]
- Gillies TE, Pargett M, Minguet M, Davies AE, and Albeck JG (2017). Linear Integration of ERK Activity Predominates over Persistence Detection in Fra-1 Regulation. Cell Syst 5, 549–563 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin SN, and Papaioannou VE (2003). Paracrine action of FGF4 during periimplantation development maintains trophectoderm and primitive endoderm. Genesis 36, 40–7. [DOI] [PubMed] [Google Scholar]
- Grabarek JB, Zyzynska K, Saiz N, Piliszek A, Frankenberg S, Nichols J, Hadjantonakis AK, and Plusa B (2012). Differential plasticity of epiblast and primitive endoderm precursors within the ICM of the early mouse embryo. Development 139, 129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND, and Robson P (2010). Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell 18, 675–85. [DOI] [PubMed] [Google Scholar]
- Hadjantonakis AK, and Papaioannou VE (2004). Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Biotechnol 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratsuka T, Fujita Y, Naoki H, Aoki K, Kamioka Y, and Matsuda M (2015). Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. Elife 4, e05178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper M, Hardy K, Handyside A, Hunter S, and Monk M (1987). HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature 326, 292–5. [DOI] [PubMed] [Google Scholar]
- Kang M, Garg V, and Hadjantonakis AK (2017). Lineage Establishment and Progression within the Inner Cell Mass of the Mouse Blastocyst Requires FGFR1 and FGFR2. Dev Cell 41, 496–510 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Piliszek A, Artus J, and Hadjantonakis AK (2013). FGF4 is required for lineage restriction and salt-and-pepper distribution of primitive endoderm factors but not their initial expression in the mouse. Development 140, 267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitadate Y, Jorg DJ, Tokue M, Maruyama A, Ichikawa R, Tsuchiya S, Segi-Nishida E, Nakagawa T, Uchida A, Kimura-Yoshida C, et al. (2019). Competition for Mitogens Regulates Spermatogenic Stem Cell Homeostasis in an Open Niche. Cell Stem Cell 24, 79–92 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawchuk D, Honma-Yamanaka N, Anani S, and Yamanaka Y (2013). FGF4 is a limiting factor controlling the proportions of primitive endoderm and epiblast in the ICM of the mouse blastocyst. Dev Biol 384, 65–71. [DOI] [PubMed] [Google Scholar]
- Kudo T, Jeknic S, Macklin DN, Akhter S, Hughey JJ, Regot S, and Covert MW (2018). Live-cell measurements of kinase activity in single cells using translocation reporters. Nat Protoc 13, 155–169. [DOI] [PubMed] [Google Scholar]
- Kurowski A, Molotkov A, and Soriano P (2019). FGFR1 regulates trophectoderm development and facilitates blastocyst implantation. Dev Biol 446, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AJ, St-Pierre F, Gong Y, Marshall JD, Cranfill PJ, Baird MA, McKeown MR, Wiedenmann J, Davidson MW, Schnitzer MJ, et al. (2012). Improving FRET dynamic range with bright green and red fluorescent proteins. Nat Methods 9, 1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht MR, Sabatini DM, and Carpenter AE (2007). CellProfiler: free, versatile software for automated biological image analysis. Biotechniques 42, 71–5. [DOI] [PubMed] [Google Scholar]
- Lanner F, and Rossant J (2010). The role of FGF/Erk signaling in pluripotent cells. Development 137, 3351–60. [DOI] [PubMed] [Google Scholar]
- Levine JH, Lin Y, and Elowitz MB (2013). Functional roles of pulsing in genetic circuits. Science 342, 1193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, and Elowitz MB (2019). Communication codes in developmental signaling pathways. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Kang M, Xenopoulos P, Munoz-Descalzo S, and Hadjantonakis AK (2014). A rapid and efficient 2D/3D nuclear segmentation method for analysis of early mouse embryo and stem cell image data. Stem Cell Reports 2, 382–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo I, and Kimura-Yoshida C (2014). Extracellular distribution of diffusible growth factors controlled by heparan sulfate proteoglycans during mammalian embryogenesis. Philos Trans R Soc Lond B Biol Sci 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihajlovic AI, and Bruce AW (2017). The first cell-fate decision of mouse preimplantation embryo development: integrating cell position and polarity. Open Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov A, Mazot P, Brewer JR, Cinalli RM, and Soriano P (2017). Distinct Requirements for FGFR1 and FGFR2 in Primitive Endoderm Development and Exit from Pluripotency. Dev Cell 41, 511–526 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov A, and Soriano P (2018). Distinct mechanisms for PDGF and FGF signaling in primitive endoderm development. Dev Biol 442, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Valon L, Levillayer F, and Levayer R (2019). Competition for Space Induces Cell Elimination through Compaction-Driven ERK Downregulation. Curr Biol 29, 23–34 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgani SM, Saiz N, Garg V, Raina D, Simon CS, Kang M, Arias AM, Nichols J, Schroter C, and Hadjantonakis AK (2018). A Sprouty4 reporter to monitor FGF/ERK signaling activity in ESCs and mice. Dev Biol 441, 104–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LO, Smith S, Chen RH, Fingar DC, and Blenis J (2002). Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol 4, 556–64. [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, and Behringer R (2008). Karyotyping mouse cells. CSH Protoc 2008, pdb prot4706. [DOI] [PubMed] [Google Scholar]
- Nandagopal N, Santat LA, LeBon L, Sprinzak D, Bronner ME, and Elowitz MB (2018). Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 172, 869–880 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Silva J, Roode M, and Smith A (2009). Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development 136, 3215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, and Miyazaki J (1991). Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–9. [DOI] [PubMed] [Google Scholar]
- Nowotschin S, Setty M, Kuo YY, Liu V, Garg V, Sharma R, Simon CS, Saiz N, Gardner R, Boutet SC, et al. (2019). The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi Y, Huber W, Tsumura A, Kang M, Xenopoulos P, Kurimoto K, Oles AK, Arauzo-Bravo MJ, Saitou M, Hadjantonakis AK, et al. (2014). Cell-to-cell expression variability followed by signal reinforcement progressively segregates early mouse lineages. Nat Cell Biol 16, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM (2000). FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays 22, 108–12. [DOI] [PubMed] [Google Scholar]
- Patel AL, and Shvartsman SY (2018). Outstanding questions in developmental ERK signaling. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plusa B, Piliszek A, Frankenberg S, Artus J, and Hadjantonakis AK (2008). Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development 135, 3081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Schindelin J, and Tomancak P (2010). Software for bead-based registration of selective plane illumination microscopy data. Nat Methods 7, 418–9. [DOI] [PubMed] [Google Scholar]
- Purvis JE, and Lahav G (2013). Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina D, Stanoev A, Bahadori A, Protzek M, Koseska A, and Schröter C (2020). Cell-cell communication through FGF4 generates and maintains robust proportions of differentiated cell fates in embryonic stem cells. bioRxiv, 2020.02.14.949701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regot S, Hughey JJ, Bajar BT, Carrasco S, and Covert MW (2014). High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes J, Chen JY, Stewart-Ornstein J, Karhohs KW, Mock CS, and Lahav G (2018). Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol Cell 71, 581–591 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan AQ, Chan CJ, Graner F, and Hiiragi T (2019). Lumen Expansion Facilitates Epiblast-Primitive Endoderm Fate Specification during Mouse Blastocyst Formation. Dev Cell 51, 684–697 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Chung M, Dobrzynski M, Fey D, Blum Y, Sik Lee S, Peter M, Kholodenko BN, Li Jeon N, and Pertz O (2016). Frequency modulation of ERK activation dynamics rewires cell fate. Mol Syst Biol 12, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz N, Kang M, Schrode N, Lou X, and Hadjantonakis AK (2016a). Quantitative Analysis of Protein Expression to Study Lineage Specification in Mouse Preimplantation Embryos. J Vis Exp, 53654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz N, Mora-Bitria L, Rahman S, George H, Herder JP, García-Ojalvo J, and Hadjantonakis AK (2020). Growth factor-mediated coupling between lineage size and cell fate choice underlies robustness of mammalian development. bioRxiv, 2019.12.27.889006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz N, Williams KM, Seshan VE, and Hadjantonakis AK (2016b). Asynchronous fate decisions by single cells collectively ensure consistent lineage composition in the mouse blastocyst. Nat Commun 7, 13463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SD, Verveer PJ, and Bastiaens PI (2007). Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol 9, 324–30. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrode N, Saiz N, Di Talia S, and Hadjantonakis AK (2014). GATA6 levels modulate primitive endoderm cell fate choice and timing in the mouse blastocyst. Dev Cell 29, 454–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa K, Kimura-Yoshida C, Nagai N, Mukai K, Matsubara K, Watanabe H, Matsuda Y, Mochida K, and Matsuo I (2011). Cell surface heparan sulfate chains regulate local reception of FGF signaling in the mouse embryo. Dev Cell 21, 257–72. [DOI] [PubMed] [Google Scholar]
- Simon CS, Hadjantonakis AK, and Schroter C (2018a). Making lineage decisions with biological noise: Lessons from the early mouse embryo. Wiley Interdiscip Rev Dev Biol 7, e319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon CS, Zhang L, Wu T, Cai W, Saiz N, Nowotschin S, Cai CL, and Hadjantonakis AK (2018b). A Gata4 nuclear GFP transcriptional reporter to study endoderm and cardiac development in the mouse. Biol Open 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad P, Gunther S, Reichmann J, Krzic U, Balazs B, de Medeiros G, Norlin N, Hiiragi T, Hufnagel L, and Ellenberg J (2016). Inverted light-sheet microscope for imaging mouse pre implantation development. Nat Methods 13, 139–42. [DOI] [PubMed] [Google Scholar]
- Susaki EA, Tainaka K, Perrin D, Kishino F, Tawara T, Watanabe TM, Yokoyama C, Onoe H, Eguchi M, Yamaguchi S, et al. (2014). Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–39. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, and Rossant J (1998). Promotion of trophoblast stem cell proliferation by FGF4. Science 282, 2072–5. [DOI] [PubMed] [Google Scholar]
- Tosenberger A, Gonze D, Bessonnard S, Cohen-Tannoudji M, Chazaud C, and Dupont G (2017). A multiscale model of early cell lineage specification including cell division. NPJ Syst Biol Appl 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenopoulos P, Kang M, Puliafito A, Di Talia S, and Hadjantonakis AK (2015). Heterogeneities in Nanog Expression Drive Stable Commitment to Pluripotency in the Mouse Blastocyst. Cell Rep 10, 1508–01520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka Y, Lanner F, and Rossant J (2010). FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development 137, 715–24. [DOI] [PubMed] [Google Scholar]
- Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, George EL, and Neel BG (2006). An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell 10, 317–27. [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, and Smith A (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SR, Burkhardt M, Nowak M, Ries J, Petrasek Z, Scholpp S, Schwille P, and Brand M (2009). Fgf8 morphogen gradient forms by a source-sink mechanism with freely diffusing molecules. Nature 461, 533–6. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Huang H, Zhang L, Wu R, Chung CI, Zhang SQ, Torra J, Schepis A, Coughlin SR, Kornberg TB, et al. (2018). Visualizing Dynamics of Cell Signaling In Vivo with a Phase Separation-Based Kinase Reporter. Mol Cell 69, 334–346 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie 1: Dynamic ERK activity in ESCs (Related to Figure 1B) Time-lapse confocal imaging of mouse embryonic stem cells (ESC) expressing HprtCAG-ERK-KTR-mClover grown in serum/LIF culture conditions. Timestamp shows Hour:Min. Scale bar 20μm.
Supplemental Movie 2: Quantification of ERK-KTR C:N in 3D in the mouse blastocyst. (Related to Figure 2A). Sequential z-slices of ERK-KTR, and 180 degree rotation of 3D max intensity projection (MIP) of ERK-KTR, NLS mKate2 and ERK-KTR C:N values in mid-blastocyst stage hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryo. Heatmap for ERK-KTR C:N as in Supplemental Figure 2A. Scale bar 20μm.
Supplemental Movie 3: ERK-KTR is a read out of FGF/ERK signaling in the mouse blastocyst. (Related to Figure 2C). Time-lapse confocal images of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryos in control, MEKi and FGF culture conditions. Max intensity projections (MIP) and a single z-slice at mid-point through the ICM are shown. ERK-KTR C:N values corresponding to ERK activity is shown as a heatmap as indicated in Figure 2C. Time-lapse interval 5 min for a total of 2 hours. Timestamp shows Hour:Min. Scale bar 20μm.
Supplemental Movie 4: Tracking ERK activity in blastocyst (Related to Figure 3 and Figure 4). Max intensity projection of hemizygous HprtERK-KTR and homozygous R26NLS-mKate2 embryo in 2 hour, 5 min interval time-lapse movie. (LEFT) ERK-KTR (MIDDLE) NLS-mKate2 overlaid with manually tracked spots in Imaris (Bitplane). Color coding indicates lineage identity by the end of the movie. Double positive ICM progenitor (DP): purple, Primitive endoderm (PrE): blue, Epiblast (EPI): red. Polar trophectoderm (TE): light green. Mural TE: dark green. (RIGHT) ERK-KTR C:N showing levels of ERK activity. Heatmap as in Figure 4C. Scale bar 20μm.
Supplemental Movie 5: Retrospective lineage assignment of the ICM (Related to Figure 4). 360 degree rotation of a mid-blastocyst stage embryo registered to assign ICM lineage identity before and after fixation. (LEFT) Max intensity projection (MIP) confocal image of the embryo after being fixed and stained. 3D rendering in Imaris (Bitplane) corresponding to an ICM cell’s lineage identity after k-means clustering of relative NANOG and GATA6 levels (as in Figure 4B) (Saiz, et al., 2016b). Double positive ICM progenitor (DP): purple, Primitive endoderm (PrE): blue, Epiblast (EPI): red. Merged with Hoechst (grey) to show nuclei. (RIGHT) MIP of the live embryo in the final time frame of 2 hour time-lapse movie. 3D rendering corresponding to ICM lineage identity shown (LEFT) merged with NLS mKate2 (grey) to show nuclei. Scale bar 20μm.
Data Availability Statement
The dataset and code generated and used in this study are available at Github, https://github.com/therealkatlab/Simon2020. Time-lapse microscopy images generated in this study are available at Figshare, DOI: 10.6084/m9.figshare.12794810.