

Abstract
A hallmark of secondary hemophagocytic lymphohistiocytosis (sHLH), a severe form of cytokine storm syndrome (CSS), is the emergence of overactivated macrophages that engulf healthy host blood cells, i.e., hemophagocytosis, and contribute to the dysregulated inflammation-driven pathology. Here we show that depleting SIRPα (SIRPα−/−) in mice during TLR9-driven inflammation exacerbates and accelerates the onset of fulminant sHLH, in which systemic hemophagocytosis, hypercytokinemia, consumptive cytopenias, hyperferritinemia and other HLH hallmarks were apparent. In contrast, mice expressing SIRPα, including those deficient of the SIRPα ligand CD47 (CD47−/−), do not phenocopy SIRPα deficiency and fail to fully develop sHLH, albeit TLR9-inflamed wild-type and CD47−/− mice exhibited hemophagocytosis, anemia and splenomegaly. Although IFNγ is largely considered a driver of HLH pathology, IFNγ neutralization did not preclude the precipitation of sHLH in TLR9-inflamed SIRPα−/− mice, whereas macrophage depletion attenuated sHLH in SIRPα−/− mice. Mechanistic studies confirmed that SIRPα not only restrains macrophages from acquiring a hemophagocytic phenotype, but also tempers their pro-inflammatory cytokine and ferritin secretion by negatively regulating Erk1/2 and p38 activation downstream of TLR9 signaling. In addition to TLR9 agonists, TLR2, TLR3 or TLR4 agonists, as well as TNFα, IL-6, or IL-17A, but not IFNγ, similarly induced sHLH in SIRPα−/− mice but not SIRPα+ mice. Collectively, our study suggests that SIRPα plays a previously unappreciated role in sHLH/CSS pathogenesis by preventing macrophages from becoming both hemophagocytic and hyperactivated under pro-inflammation.
INTRODUCTION
Cytokine storm syndromes (CSS) such as secondary hemophagocytic lymphohistiocytosis (sHLH) are life-threating disorders associated with bacterial and viral infections and have recently been considered a cause for mortality in persons infected with SARS-CoV-2 (1–4). Also known as macrophage activation syndrome (5, 6), sHLH is associated with hyperactivated macrophages that adopt a hemophagocytic phenotype and thus aberrantly engulf other healthy hematopoietic-lineage cells such as red blood cells (RBC), leukocytes and platelets, often leading to consumptive cytopenias (i.e., anemia, leukopenia and thrombocytopenia) (7–9). The underlying mechanism driving macrophages to become hemophagocytic under inflammatory conditions, however, has yet to be clearly determined (10).
Though an ensemble of proteins regulate macrophage phagocytosis, the inhibitory receptor that is well-known to prevent phagocytosis of healthy host cells is SIRPα (11, 12). The SIRPα-CD47 axis serves as the quintessential ‘don’t-eat-me’ signal to prevent unwanted phagocytosis of healthy host cells (13–16). As an inhibitory receptor highly expressed on myeloid leukocytes including macrophages, monocytes (MC), dendritic cells (DC) and neutrophils (PMN), SIRPα is critical to prohibiting phagocytosis of CD47-expressing host cells such as RBC, leukocytes and platelets (17, 18). Despite the importance of this safety mechanism, the absence of SIRPα or CD47 alone does not confer an autoimmune phenotype, as mice devoid of SIRPα (SIRPα−/−) or CD47 (CD47−/−) are healthy like their wild-type (WT) littermates (8, 19). Providing insight into this paradoxical phenomenon, our prior study revealed that macrophages will not phagocytose healthy host cells unless two criteria are simultaneously met: 1) SIRPα-CD47 inhibition must be removed and 2) pro-inflammatory activation signaling must be present, presumably to activate the uncharacterized macrophage phagocytic receptor recognizing healthy host cells (8). While this bipartite ‘fail-safe’ mechanism has yet to be fully understood, we have shown that various TLR agonists or certain pro-inflammatory cytokines such as IL-1β, IL-6, TNFα or IL-17A, but not IFNγ, are capable of driving the required activation signaling (8, 20).
Aside from phagocytosing healthy blood cells, overactivated macrophages have been reported to contribute to other cardinal features commonly presenting in fulminant sHLH including hyperferritinemia, hypertriglyceridemia and hypercytokinemia, the latter being the putative driver of multi-organ failure and death (5, 8, 21–25). Indeed, previous reports have shown that dysregulation of pro-inflammatory macrophage activation can contribute to HLH/CSS pathogenesis in various models of sterile and non-sterile inflammation (9, 26, 27). Along these lines, SIRPα not only canonically inhibits macrophage phagocytosis but has become increasingly appreciated to also negatively regulate macrophage-driven pro-inflammation, with inflammatory diseases such as colitis, peritonitis and diabetic nephropathy being exacerbated under SIRPα deficiency (8, 28–30). Thus, we hypothesized that SIRPα likely prevents the overactivation of macrophages to the point of becoming hemophagocytic under pro-inflammation and potentially serves as a critical barrier in the macrophage-intrinsic contribution to sHLH/CSS pathogenesis.
MATERIALS & METHODS
Mice.
Mice were 8–11 weeks of age and sex-matched for all studies. Wild-type (C57BL/6J) and CD47−/− (B6.129-CD47tm1Fpl/J) mice were purchased from The Jackson Laboratory. SIRPα−/− mice were established as previously described (8). All mice were housed in a specific-pathogen-free facility. All animal experiments and procedures for handling were approved by the Institutional Animal Care and Use Committee of Georgia State University.
Treatments.
In cytosine guanine dinucleotide (CpG) experiments, WT, SIRPα−/− or CD47−/− mice were intraperitoneally (i.p.) injected with 50 μg of CpG-B (ODN1826; InvivoGen) suspended in 50 μl phosphate-buffered saline (PBS) every other day for a total of three injections and were euthanized 48h (day 6) thereafter for analyses. To neutralize IFNγ, mice were intravenously (i.v.) injected with anti-IFNγ antibody (500 μg/mouse; XMG1.2, Bio X cell) or isotype control (BioLegend) two hours prior to receiving CpG injections. For macrophage depletion experiments, mice were i.p. injected with 100 μl of empty or clodronate-containing liposomes (5 mg/ml, Encapsula Nano Sciences) once a day for two consecutive days, with the initial injection given 48h prior (day −2) to the first CpG injection. In other experiments, WT or SIRPα−/− mice were injected with zymosan A (500 μg/mouse, i.p.; Sigma), poly I:C (10 mg/kg, i.p.; InvivoGen), lipopolysaccharide (LPS) (0.25 mg/kg, i.p.; Sigma, E. coli O111:B4), recombinant TNFα (10 μg/kg, i.v.; PeproTech), recombinant IL-6 (10 μg/kg, i.v.; PeproTech), recombinant IL-17A (10 μg/kg, i.v.; PeproTech) or recombinant IFNγ (10 μg/kg, i.v.; PeproTech) every other day for a total of three injections and were euthanized 48h thereafter for analyses (8).
Hematology.
Disease progression was monitored daily by changes in hemoglobin (Hgb), which was assessed by serial sampling of blood (30–40 μl) via the saphenous vein. Hgb concentration was determined by adding 25 μl of whole blood to diluted Drabkin reagent (Sigma) and then measuring the spectral absorbance of hemiglobincyanide at 540nm. Percent change in Hgb was determined by: (100 - (measured Hgb/initial Hgb) * 100). To analyze peripheral blood at the treatment endpoint, mice were euthanized, and blood was extracted by cardiac puncture into heparinized tubes. 100 μl of whole blood was then used to isolate platelets (31), which were then counted on a Cellometer Auto2000 (Nexcelom). The remaining whole blood was then centrifuged (1000 x g) to isolate the plasma for later quantification of cytokines and other biomarkers. The buffy coat and RBC pellet were resuspended in lysis buffer (150 mM ammonium chloride, 10 mM potassium bicarbonate, and 0.1 mM EDTA) for RBC lysis and the remaining leukocytes were then counted on a Cellometer Auto2000.
Flow cytometry.
To isolate splenocytes, spleens were minced, digested in a cocktail of collagenase (0.17 mg/ml, Sigma) and DNase1 (40 μg/ml, Sigma) in RPMI-1640 at 37°C, passed through a 70-μm cell strainer and then contaminating RBC were removed with lysis buffer and several washes. All staining was done in the presence of rat anti-mouse CD16/CD32 antibody (Fc block, BioLegend) and in the dark at 4°C. To assess phagocytosis of RBC, splenocytes were additionally blocked with purified anti-mouse Ter-119 (BioLegend) prior to cell surface staining with fluorescently labeled antibodies (7, 32, 33). Cells were then stained with antibody cocktails comprising: CD45.1-Pacific Blue (A20), CD11b-APC (M1/70), CD11c-FITC (N418), F4/80-PE (BM8), Ly6C-BV650 (HK1.4) and Ly6G-PerCP (1A8) (all from BioLegend) for myeloid cells. To determine lymphocyte populations, cells were stained for the following markers: CD45.1-Pacific Blue (A20), B220-BV605 (RA3–6B2), CD4-APC (RM4–4), CD8-PE and NK1.1-FITC (PK136) (all antibodies from BioLegend). Cells were then washed and incubated with 7-AAD to determine live cells. To assess hemophagocytosis, cells were again washed and then prepared for intracellular staining with fixation and permeabilization buffer (BD Biosciences), washed in perm/wash buffer (BD Biosciences) and then stained with anti-Ter-119-PE/Cy7 (Ter-119, BioLegend). Data were acquired on a BD LSRFortessa (BD Biosciences) and analyzed using FlowJo 10.6.01 (Tree Star).
Histology and microscopy.
Freshly isolated spleens and livers were embedded in OCT compound, flash frozen in liquid nitrogen and then stored at −80°C. Tissues were cryostat sectioned at 6–10 μm and then immediately fixed in 4% paraformaldehyde (PFA). Hematoxylin and eosin (H&E) stains were performed for histological analysis of hemophagocytosis in bone marrow smears, spleens and livers. For adoptive transfer experiments, mice were first i.v. injected with ~109 CFSE-stained RBC and after 1h, mice were i.p. injected with CpG (50 μg) or PBS (8). After 12h, mice were euthanized and their spleens removed. In some experiments, spleens were digested and splenocytes were isolated for flow cytometry to quantify the percentage of hemophagocytic (CFSE+) RPM. For immunofluorescent (IF) staining, sections were blocked with 5% BSA and incubated with PE-conjugated rat-anti-mouse F4/80 (BM8; BioLegend). DAPI was used to stain nuclei. For immunohistochemistry (IHC) analyses, spleen tissue sections were treated with 0.3% H2O2, blocked with 10% BSA and then stained with biotin-conjugated rat anti-mouse F4/80 (BM8; BioLegend) at 4°C for 18h. After washing, slides were incubated with HRP-conjugated streptavidin (Thermo Fisher) and exposed to DAB (Thermo Fisher). Thereafter, sections were H&E counterstained. All images were acquired with a BZ-X700 All-In-One Fluorescent Microscope (Keyence).
Macrophage phagocytosis assay.
Bone marrow-derived macrophages (BMM) were generated as previously described (8, 34). To assess phagocytic activity, BMM were treated with PBS, CpG (1 μg/ml), Poly I:C (100 ng/ml), LPS (20 ng/ml), TNFα (20 ng/ml) or IFNγ (20 ng/ml) for 12h, and then were washed and incubated with peripheral blood cells (PBC) for 30m at 37°C. BMM-PBC co-cultures were then washed, extracellular RBC were lysed and then cells were visualized by microscopy. For IF staining, PBC were labeled with CFSE prior to co-culture with BMM. BMM were then labeled with PE-conjugated anti-F4/80 and fixed with 4% PFA. After fixation, nuclei were stained with DAPI and cells were visualized by fluorescent microscopy.
Cytokine and biomarker quantification.
12h following a single injection of PBS, CpG or LPS, plasma levels of IL-1β, IL-6, IL-10, IL-12, TNFα, GM-CSF, or MCP-1 were determined by standard enzyme-linked immunosorbent assay (ELISA) using capture, detection and HRP-conjugated antibodies (all from BioLegend). Similarly, free IL-18 in plasma was assayed using the Mouse IL-18 ELISA Kit (MBL). Plasma levels of triglyceride, ferritin and soluble CD25 (IL-2Rα) were determined using the Triglyceride Assay Kit (Abcam), Mouse Ferritin ELISA Kit (Abcam) and Mouse CD25/IL-2R alpha DuoSet ELISA (R&D Systems), respectively. To assay BMM cytokine or ferritin production, BMM were treated with PBS or CpG (1 μg/ml) ± mCD47.ex for 12h. Thereafter, cell-free BMM-conditioned medium was assessed by ELISA.
Soluble murine CD47 extracellular domain.
The AP-tag2 plasmid containing the extracellular domain of murine CD47 (mCD47.ex) and alkaline phosphatase (AP) was a generous gift of V. Narayanan (University of Pittsburgh School of Medicine) (35). After transfecting COS cells, recombinant mCD47.ex fusion proteins were produced, affinity purified and stored in PBS as previously described (18, 35–38). Prior to use, the binding capacity of mCD47.ex was confirmed by binding to murine SIRPα extracellular domain fusion protein (mSIRPα.ex-Fc) as well as adhesion to macrophages in a CD47-SIRPα-dependent manner.
SDS-PAGE and immunoblot.
BMM were either non-treated or treated with CpG (1 μg/ml) in the presence or absence of mCD47.ex for 30min at 37°C. Thereafter, BMM were washed and then incubated with ice-cold lysis buffer containing 25mM Tris-HCl, pH 7.4, 150mM NaCl, 1% Triton X-100, protease inhibitors (Protease Inhibitor Cocktail, Sigma-Aldrich), phosphatase inhibitors (Phosphatase Inhibitor Cocktail 1 and 2, Sigma), 3mM PMSF and 2mM pervanadate. After centrifugation at 12,000rpm for 5min, cell lysates were collected and then heated at 95°C for 3min in SDS-PAGE sample buffer. After electrophoresis in acrylamide gels, proteins were transferred onto nitrocellulose, followed by blocking with 3% BSA and probed for: SIRPα (clone P84, BioLegend); phospho-Erk1/2 (D13.14.4E); phospho-p38 (D3F9); and beta-actin (13E5) (all from Cell Signaling Technology). Densitometric analyses were performed using Image J (NIH).
Statistical analysis
All graphs and statistical analyses were generated and performed using Prism 6.0 (GraphPad Software). Statistical significance was calculated using Student’s t test (k = 2) or analysis of variance (ANOVA; one- or two-way) (k > 2). For post-hoc analyses, Dunn’s or Tukey’s HSD test was used to determine statistical significance among multiple comparisons, with an experiment-wise error rate of 0.05. When P < 0.05, samples were considered statistically significant. At least three independent experiments were performed for each set of data, which were represented as individual values, mean ± SEM or both.
RESULTS
SIRPα deficiency exacerbates and accelerates the onset of TLR9-induced sHLH
As a TLR9 agonist, cytosine guanine dinucleotide (CpG) is both a relevant TLR stimulus in sHLH and a viable trigger for activating SIRPα−/− phagocytes to engulf healthy host cells (8, 39). To examine the role of SIRPα in TLR9-induced sHLH, we repeatedly injected WT or SIRPα−/− mice with CpG. The development of sHLH was monitored by measuring changes in hemoglobin (Hgb). As shown (Fig. 1A, left panel), whereas WT mice did not exhibit anemia, i.e., ≥ 20% reduction in Hgb, until 24h after the third injection of CpG, SIRPα−/− mice experienced a ~25% drop in Hgb after merely one dose of CpG. With every additional dose of CpG, SIRPα−/− mice became increasingly more anemic and decreased their Hgb in excess of 60% within 48h after three injections (day 6), which led us to terminate the study (Fig. 1A). By day 6, CpG-treated WT mice were only slightly anemic, whereas SIRPα−/− mice had severe pancytopenia (Fig. 1A–1B). Assaying other pathological features of sHLH, we found that plasma ferritin, triglycerides and soluble CD25/IL-2Rα (sCD25) were highly elevated in CpG-treated SIRPα−/− mice, whereas WT mice only had a slight increase in sCD25 (Fig. 1C–1E) (1). Screening resected organs found that WT mice had normal livers and only moderate splenomegaly (Fig. 1F–1G). However, CpG-treated SIRPα−/− mice had severe hepatosplenomegaly, accounting for ≥ 10% of their body weight. Histological examination of CpG-treated spleens revealed the splenic white pulp architecture remained uniform in WT mice but was greatly disrupted in SIRPα−/− mice (Fig. S2A). Indeed, splenic leukocyte expansion was commensurate to the severity of splenomegaly, with CpG-treated SIRPα−/− mice having 2–3-fold more CD45+ splenocytes than WT mice (Fig. S2C). Healthy SIRPα−/− mice had slightly lower frequencies of DC and CD4 T cells (Fig. 1H and S2C), corroborating previous findings (8, 40, 41). After CpG treatment, SIRPα−/− mice had lower splenic frequencies of B cells and CD4 T cells than WT mice; however, adjusting for cell count mitigated these differences, as SIRPα−/− mice actually had 20% more B cells and similar a quantity of CD4 cells as WT mice (Fig. S2C). Whereas neither the frequency nor quantity of splenic myeloid leukocytes increased in CpG-treated WT mice, PMN, MC and RPM had increased 2–3-fold in SIRPα−/− mice (Fig. 1H). Thus, myeloid cell and CD8 T cell expansion appeared to account for the reduced B cell frequency in CpG-treated SIRPα−/− spleens (Fig. S2E).
Fig. 1. SIRPα deficiency exacerbates and accelerates the onset of TLR9-induced sHLH.

A-I) Wild-type (WT) or SIRPα−/− mice were challenged with three intraperitoneal (i.p.) injections of CpG-B (ODN 1864; 50 μg/mouse; n = 9/group) or PBS (100 μl/mouse; n = 5/group) on days 0, 2 and 4 and were euthanized on day 6 for analyses. A) Disease severity was monitored by measuring daily changes in hemoglobin (left). A-B) Following euthanization on day 6: anemia (A; right), leukopenia (B; left) and thrombocytopenia (B; right) were quantified by peripheral blood analyses; C-F) additional blood markers of plasma ferritin (C), triglycerides (D) and soluble CD25 (E) were determined by ELISA. F-G) Hepatosplenomegaly. Livers (F) and spleens (G) were weighed and normalized to their respective total body weight. Representative image of splenomegaly (G; lower). H) Myeloid population frequencies (left) and counts (right) among CD45+ splenocytes were determined by flow cytometry: CD11b-F4/80+ red pulp macrophages (RPM), CD11b+Ly6G-Ly6C+ monocytes (MC), CD11b+Ly6Ghi neutrophil (PMN) and CD11c+ dendritic cells (DC) (Fig. S1: gating strategy). I) Concentrations of plasma cytokines (IL-1β, IL-6, IL-12, TNFα, MCP-1 and GM-CSF) from WT or SIRPα−/− mice 12h after receiving an injection of PBS or CpG. The data represent twelve independent experiments and all data were measured in triplicate. Each symbol represents an individual mouse, with the mean ± SEM also shown. Two-way ANOVA and Tukey’s HSD post-hoc analyses were used determine statistical significance among multiple comparisons: not significant (ns) = P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Being a CSS, sHLH is typified by elevated pro-inflammatory cytokines, with overactivated macrophages being a major source (42–45). As an absence of SIRPα worsens disease conditions (8, 16, 28–30, 46), we assessed whether SIRPα deficiency would exacerbate CpG-induced hypercytokinemia. Healthy WT and SIRPα−/− mice mostly exhibited similar cytokine levels; however, under TLR9 stimulation, SIRPα−/− mice had 2–4-fold higher levels of IL-1β, IL-6, IL-12, IL-18 and TNFα than WT mice (Fig. 1I & S2G). The exacerbated phenotype in CpG-treated SIRPα−/− mice was not due to a stunted anti-inflammatory response, as both mice produced comparable amounts of IL-10 after CpG treatment (Fig. S2G). Next, we assayed cytokines associated with granulopoiesis and myeloid leukocyte chemotaxis to determine what led to the expanded myeloid population in CpG-treated SIRPα−/− spleens (47–49). Similar to IL-10, GM-CSF was low in SIRPα−/− mice prior to TLR9 signaling, but after CpG, circulating GM-CSF significantly increased. The chemoattractant MCP-1 (CCL2) was also produced almost 3-fold greater in SIRPα−/− mice than in WT mice responding to TLR9 agonism. Collectively, these data suggest that an absence of SIRPα enhances both the kinetics and severity of sHLH onset induced by TLR9-driven inflammation (9, 50).
Absence of SIRPα during TLR9 stimulation induces hemophagocytic leukocytes
Our previous study showed that acute inflammation in SIRPα−/− mice led to erythrophagocytosis, consumptive anemia and splenomegaly (8), thus we next assessed the extent of hemophagocytosis within the spleen following TLR9 stimulation. To quantify splenic hemophagocytes, splenic leukocytes (CD45+) were intracellularly stained for the RBC-associated antigen Ter-119 (7, 32, 33, 51). Regardless of genotype, about 10–20% of RPM were Ter-119+ under healthy conditions (Fig. 2A–2B), corroborating previous reports (7, 33). However, whereas the frequency of Ter-119+ RPM did not significantly increase in WT mice following CpG treatment, over 50% of RPM were Ter-119+ in the absence of SIRPα. Since CpG-treated SIRPα−/− spleens were highly infiltrated with other myeloid leukocytes that innately express SIRPα, we also assessed the frequency of Ter-119+ cells among MC, PMN and DC (32). Without inflammatory stimulation, no differences in Ter-119+ MC, PMN and DC frequency were detected. After CpG treatment, however, all SIRPα−/− myeloid cells exhibited robust hemophagocytosis (Fig. 2A–2B), whereas the quantity of Ter-119+ cells did not significantly increase in WT mice. On average, CpG-treated SIRPα−/− mice spleens comprised 18-fold more Ter-119+ leukocytes than CpG-treated WT mice (Fig. 2B). Indeed, hematoxylin and eosin staining of spleen and liver tissue sections, as well as bone marrow smears, were insufficient to detect hemophagocytosis in CpG-treated WT mice (not shown), whereas hemophagocytosis was apparent in tissues isolated from SIRPα−/− mice (Fig. 2C). To determine whether this difference in hemophagocytosis may occur after just one CpG injection, we adoptively transferred fluorescence-labeled (CFSE) RBC with CpG or PBS concurrently injected (8). In mice receiving CpG, 7% of RPM were CFSE+ in WT mice whereas 40% were CFSE+ in SIRPα−/− mice (Fig. 2D). Immunofluorescent staining of spleen sections from CpG-treated mice supported our flow cytometry data (Fig. 2E, inset), with the frequency of CFSE+F4/80+ macrophages (yellow) being much more apparent in spleens of CpG-treated SIRPα−/− mice.
Fig. 2. Absence of SIRPα during TLR9 stimulation induces hemophagocytic leukocytes.

A) Frequencies of hemophagocytic CD45+ splenocytes (F4/80+CD11b- RPM, CD11b+Ly6G-Ly6C+ MC, CD11b+Ly6Ghi PMN and CD11c+ DC) were determined by intracellular staining for RBC (Ter-119+). Isotype controls are shown in Fig. S3A. B) Frequencies (left), individual counts (middle) and total counts (right) of hemophagocytes from PBS- or CpG-treated mice (n = 5/group). C) Representative images of hemophagocytes within the bone marrow, spleen and liver of CpG-treated SIRPα−/− mice as determined by hematoxylin and eosin staining (scale bar: 10 μm) (n = 3/group). D) Histogram frequency of CD45+CD11b-F4/80+ RPM with intracellular RBC (CFSE+). E) Representative image of immunofluorescent stained spleen tissue sections after RBC (109) transfer in CpG-treated WT and SIRPα−/− mice; (scale bar: 100 μm). The data represent eight independent experiments and each symbol represents an individual mouse, with the mean ± SEM also shown. Two-way ANOVA and Tukey’s HSD post-hoc analyses were used to determine statistical significance among multiple comparisons: ns = P > 0.05; ****P < 0.0001.
CD47 deficiency does not phenocopy SIRPα deficiency under TLR9 agonism
Serving as a ligand for SIRPα, CD47 is a critical self-recognition molecule expressed on healthy host cells, which are normally not phagocytosed (19). One study previously reported that serum from HLH patients can downregulate CD47 on hematopoietic stem cells (HSC), which led macrophages to phagocytose HLH serum-treated HSC in vitro. However, the question remains: do CpG-treated CD47−/− mice similarly develop fulminant sHLH as in SIRPα−/− mice? Following CpG treatment, Hgb sharply dropped in CD47−/− mice, which largely mimicked the kinetics and severity in CpG-treated SIRPα−/− mice (Fig. 3A). However, unlike CpG-treated SIRPα−/− mice, CD47−/− mice did not develop leukopenia or thrombocytopenia (Fig. 3B). CpG-treated CD47−/− mice had slightly elevated ferritin, albeit not comparably as severe in SIRPα−/− mice (Fig. 3C). Furthermore, both plasma triglycerides and sCD25 were not highly elevated in CpG-treated CD47−/− mice (Fig. 3D–3E). As anticipated, CD47−/− mice developed splenomegaly after TLR9 stimulation (Fig. 3F and S2B) (8); however, unlike SIRPα-deficiency, an absence of CD47 only led to moderate expansion of RPM and a slight increase in splenic infiltration of PMN (Fig. 3G, S2D and S2F). Despite exhibiting a noticeable increase in Ter-119+ RPM, CpG-treated CD47−/− spleens comprised exceptionally less hemophagocytes than SIRPα−/− spleens, which had nearly 5-fold more Ter-119+ cells than CD47−/− mice (Fig. 3H and S2H-S2I). Assessing plasma cytokines found that CD47−/− mice were appreciably less inflamed than SIRPα−/− mice (Fig. 3I). By calculating H-Scores (1), we found that CpG-treated CD47−/− and WT exhibited similar degrees of severity and both had H-Scores of 58 (Fig. 7), equating to a < 1% probability of having HLH. In stark contrast, CpG-treated SIRPα−/− mice had an H-Score of 186, indicating a 70–80% probability of having HLH. These data suggest that SIRPα negatively regulates inflammation in a CD47-independent manner and that while CD47 deficiency is sufficient to develop hemophagocytosis, anemia and splenomegaly, it does not fully mimic SIRPα deficiency with respect to fully developing sHLH.
Fig. 3. CD47 deficiency does not phenocopy SIRPα deficiency under TLR9 agonism.

A-I) SIRPα−/− or CD47−/− mice were injected three times with CpG (n = 5/group) or PBS (n = 5/group) on days 0, 2 and 4 and euthanized on day 6 for analyses. A) Daily changes in hemoglobin (left). Following euthanization on day 6: anemia (A; right), leukopenia (B; left) and thrombocytopenia (B; right) were quantified and additional blood markers of plasma ferritin (C), triglycerides (D) and soluble CD25 (E) were determined. F-H) Spleens were isolated to determine splenomegaly (F), the frequency of myeloid leukocytes (G) and the frequency (H; left) and (H; right) count of splenic hemophagocytes. I) Plasma cytokines (MCP-1, GM-CSF, IL-6, and TNFα) assayed 12h following a single CpG or PBS injection. The data represent nine independent experiments and all data were measured in triplicate. Each symbol represents an individual mouse, with the mean ± SEM also shown. Two-way ANOVA and Tukey’s HSD post-hoc analyses were used to determine statistical significance among multiple comparisons: ns = P > 0.05; *P < 0.05; ***P < 0.001; ****P < 0.0001.
Fig. 7. Summary of HLH phenotype in mice with varied treatments.
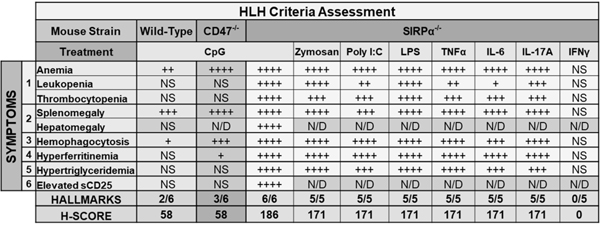
Six or five hallmarks were assessed in CpG-treated or other inflammatory stimuli-treated mice, respectively, in accordance with the HLH-2004 diagnostic criteria. An H-score ≥169 accurately classifies 90% of individuals and corresponds to a sensitivity of 93% and specificity of 86% for HLH diagnosis. NS, not significant = P > 0.05; + = P < 0.05; ++ = P < 0.01; +++ = P < 0.001; ++++ P < 0.0001; ND, not determined.
Macrophage depletion but not IFNγ neutralization ameliorates TLR9-induced sHLH in SIRPα−/− mice
Hypercytokinemia is largely considered to precede multi-organ failure and death in CSS/HLH. Accordingly, current efforts to treat HLH/CSS primarily revolve around neutralization of cytokines considered putative drivers of the immunopathology. IFNγ is one such cytokine that is considered critical to manifesting immunopathological features of HLH (25, 39, 50), and indeed, previous studies have shown that neutralization IFNγ may reverse sHLH symptoms in the TLR9-sHLH mouse model, even when exacerbated by co-administering antagonistic IL-10 receptor antibodies (39, 50, 52, 53). Thus, we assessed whether IFNγ neutralization could similarly prove to be therapeutic in CpG-treated SIRPα−/− mice by co-administering anti-IFNγ antibody (αIFNγ; XMG1.2) or isotype control (IgG ctl.) (Fig. 4). To our surprise, IFNγ neutralization was largely ineffective in preventing sHLH onset in SIRPα−/− mice, suggesting sHLH pathogenesis under SIRPα deficiency is not dependent upon IFNγ.
Fig. 4. Macrophage depletion but not IFNγ neutralization precludes TLR9-induced sHLH in SIRPα−/− mice.

SIRPα−/− mice were treated with PBS (n = 5) or CpG co-administered with IgG isotype control (n = 5) or αIFNγ (XMG1.2; n = 5) on days 0, 2 and 4 and euthanized on day 6 for analyses. To deplete macrophages empty (EL; n = 5) or clodronate-containing liposomes (CL2MDP; n = 5) were administered prior (day −2 and −1) to CpG treatment. A) Daily changes in hemoglobin (left). Following euthanization on day 6: A-E) anemia (A; right), leukopenia (B; left) and thrombocytopenia (B; right) were quantified and additional blood markers of plasma ferritin (C), triglycerides (D) and soluble CD25 (E) were determined. F-H) Spleens were isolated to determine splenomegaly (F), the frequency of myeloid leukocytes (G) and the frequency (H; left) and the count (H; middle and left) of hemophagocytes. I) Plasma cytokines (IL-1β, IL-6, IL-12, TNFα and GM-CSF) assayed 12h following a single injection of PBS, CpG + IgG isotype control, CpG + αIFNγ or CpG alone after EL or CL2MDP treatment. The data represent thirteen independent experiments and all data were measured in triplicate. Each symbol represents an individual mouse, with the mean ± SEM also shown. One-way ANOVA and Dunn’s post-hoc analyses were used determine statistical significance among multiple comparisons: not significant (ns) = P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Our prior study implicated macrophages as drivers of anemia in inflamed SIRPα−/− mice (8), thus we assessed the therapeutic efficacy of macrophage depletion by administering empty (EL) or clodrosome-containing liposomes (CL2MDP) prior to (day −2 and −1) CpG treatment (26, 27, 54–57). In contrast to IFNγ neutralization, macrophage depletion by CL2MDP precluded sHLH in CpG-treated SIRPα−/− mice. CL2MDP prevented the rapid onset of severe pancytopenia, with CpG-treated SIRPα−/− mice only exhibiting a 10% drop in Hgb by day 6 (Fig. 4A–4B). Furthermore, macrophage depletion drastically reduced plasma ferritin and triglyceride levels and slightly lowered sCD25 levels as well (Fig. 4C–4E). Assessing resected spleens, CL2MDP effectively prevented splenomegaly and also led to a significant reduction in myeloid splenocyte frequency and count (Fig. 4F–4H & S3B). While CL2MDP did not significantly affect the frequency of Ter-119+ splenocytes, adjusting for cell count revealed a marked reduction in the quantity of splenic hemophagocytes (Fig. 4I). Notably, CL2MDP largely abated hypercytokinemia, albeit GM-CSF remained moderately elevated (Fig. 4J). These data suggest that phagocytes, chiefly macrophages, are primarily responsible for TLR9-driven sHLH pathology in SIRPα−/− mice.
SIRPα negatively regulates TLR9-driven Erk1/2 and p38 activation to inhibit macrophage function
Given CL2DMP treatment abrogated sHLH onset in CpG-treated SIRPα−/− mice, we sought to determine whether SIRPα directly affects TLR9 signaling in macrophages. As anticipated, SIRPα inhibited CpG-treated WT and CD47−/− bone marrow-derived macrophages (BMM) from phagocytosing WT (CD47+) peripheral blood cells (PBC) (Fig. 5A). Indeed, fluorescent-labeled CpG-activated WT or CD47−/− BMM (red) failed to phagocytose CFSE-positive PBC (green), whereas similarly treated SIRPα−/− BMM were highly phagocytic after CpG treatment and engulfed many PBC, including RBC and nucleated cells (arrows). Aside from being more hemophagocytic, SIRPα−/− BMM were adept at secreting pro-inflammatory cytokines (Fig. 5B). To determine the effect of SIRPα on BMM cytokine production, WT, CD47−/− and SIRPα−/− BMM were treated with CpG in the presence or absence of CD47 ligation via murine CD47 extracellular domain fusion protein (mCD47.ex) (18, 35–38). In the absence of mCD47.ex, CpG-treated WT and CD47−/− BMM produced on average 2-fold less IL-1β, IL-6, IL-12 and TNFα than SIRPα−/− BMM (Fig. 5B). Conversely, SIRPα−/− BMM produced markedly less IL-10 than WT or CD47−/− BMM in response to TLR9 signaling. Given that previous studies have shown macrophages are a major source of extracellular ferritin (24, 58, 59), BMM-conditioned culture medium was also assayed for extracellular ferritin (Fig. 5C). Indeed, SIRPα−/− BMM secreted significantly more ferritin after CpG treatment than WT or CD47−/− BMM. Furthermore, the disparity between WT or CD47−/− BMM and SIRPα−/− BMM insofar as their capacities to secrete pro-inflammatory cytokines and ferritin was magnified in the presence CD47 ligation (+ mCD47.ex) (Fig. 5B–5C). Along these lines, CpG-treated WT and CD47−/− BMM produced even more anti-inflammatory cytokine IL-10 in the presence of mCD47.ex, whereas SIRPα−/− BMM production of IL-10 remained limited under TLR9 signaling. Given that CD47-SIRPα signaling appeared to impact BMM cytokine production in response to TLR9 stimulation, we examined potential differences in macrophage TLR9 signaling in the presence or absence of CD47-SIRPα signaling (Fig. 5D). The extent to which Erk1/2 (p44/42) and p38 (p38) were phosphorylated was mostly similar among BMM after CpG treatment alone. However, in the presence of CD47 ligation (+ mCD47.ex) and thus strong SIRPα signaling, CpG-treated WT and CD47−/− BMM exhibited a level of phosphorylation Erk1/2 and p38 that paralleled that of non-treated BMM, whereas SIRPα−/− BMM were unaffected and maintained MAPK activation. Together, these data suggest that SIRPα tempers TLR9 signaling in macrophages by inhibiting MAPK.
Fig. 5. SIRPα negatively regulates TLR9-driven Erk1/2 and p38 activation to inhibit macrophage function.

A) Bone marrow-derived macrophages (BMM) were generated from WT, CD47−/− or SIRPα−/− mice, treated with CpG (1 μg/ml) and then co-cultured with peripheral blood cells (PBC) isolated from healthy WT mice. Representative microscopy images of immunofluorescent stained BMM (PE-F4/80; red) and PBC (CFSE; green), with nuclei stained by DAPI (blue). The data represent three independent experiments and each phagocytosis assay was performed in triplicate. B-C) WT, CD47−/− and SIRPα−/− BMM were treated with CpG (1 μg/ml) in the presence or absence of CD47 ligation (± mCD47.ex) for 12h and cell-free medium was then assayed by ELISA to quantify IL-1β, IL-6, IL-10, IL-12, TNFα (B) and ferritin (C) secretion. The data represent four independent experiments and each experimental group was performed in triplicate. The data are presented as mean + SEM. Two-way ANOVA and Tukey’s HSD post-hoc analyses were used determine statistical significance among multiple comparisons: not significant (ns) = P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. D) Representative immunoblot analyses of SIRPα, p38 phosphorylation (p-p38), Erk1/2 phosphorylation (p-p44/42) and beta-actin protein abundance in WT, CD47−/− and SIRPα−/− BMM that were non-treated, treated with mCD47.ex alone or treated with CpG (1 μg/ml) in the presence or absence of CD47 ligation (± mCD47.ex) for 30 minutes. All immunoblots are representative of five independent experiments. Densitometric analysis was used to determine the relative change in phosphorylated p44/42 and p38 by normalizing against beta-actin.
Activation of SIRPα−/− macrophages by various inflammatory factors confers sHLH-like disease
Given other TLR agonists and certain cytokines fulfill the requisite activation signaling for phagocytosis of healthy host cells (8), we treated mice with zymosan A (TLR2), poly I:C (TLR3), LPS (TLR4), TNFα, IL-6 or IL-17A. One study has shown that IFNγ alone induces a partial HLH phenotype in mice (7), thus we also assessed IFNγ-treated mice. Following zymosan, LPS or IFNγ treatment, WT mice developed mild anemia (Fig. S4A). LPS treatment also induced splenomegaly and elevated ferritin (Fig. S4D and S4E). Otherwise, all treatments failed to develop sHLH symptoms in WT mice. In contrast, treating SIRPα−/− mice with any inflammatory stimuli, except IFNγ, led to an sHLH phenotype as that induced by CpG (Fig. 6). All hemophagocytosis-activating factors conferred a moderate to severe drop in Hgb (Fig. 6A). While all treatments induced severe thrombocytopenia, poly I:C, TNFα and IL-6 only induced mild/moderate leukopenia (Fig. 6B–6C). All treatments, except IFNγ, led to severe splenomegaly (Fig. 6D), hyperferritinemia (Fig. 6E) and hypertriglyceridemia (Fig. 6F). Immunohistochemical staining against F4/80 in spleen sections and in vitro phagocytosis assays with poly I:C, LPS or TNFα, but not IFNγ, demonstrated the exceptional capacity of activated SIRPα−/− phagocytes to uptake RBC (Fig. 6G). Quantification of plasma cytokines in LPS-treated SIRPα−/− mice again demonstrated markedly worse hypercytokinemia under SIRPα deficiency (Fig. 6H). H-scores were calculated and summarized in Figure 7. Collectively, these data indicate the importance of SIRPα in not only suppressing a hemophagocytic phenotype but also preventing the development of sHLH/CSS under various inflammatory conditions.
Fig. 6. Activation of SIRPα−/− macrophages by various inflammatory factors confers sHLH-like disease.

SIRPα−/− mice (n = 5 mice per treatment) were injected with PBS, zymosan A (500 μg/mouse, i.p.), poly I:C (10 mg/kg, i.p.), LPS (0.25 mg/kg, i.p.), recombinant TNFα (10 μg/kg, i.v.), recombinant IL-6 (10 μg/kg, i.v.), recombinant IL-17A (10 μg/kg, i.v.) or recombinant IFNγ (10 μg/kg, i.v.) on days 0, 2 and 4 were then euthanized on day 6 for analyses. Peripheral blood markers for HLH were assessed: anemia (A), leukopenia (B), thrombocytopenia (C), hyperferritinemia (E) and hypertriglyceridemia (F). D) Spleens were also excised and weighed to determine splenomegaly. Representative images of SIRPα−/− BMM treated with poly I:C (100 ng/ml), LPS (20 ng/ml), TNFα (20 ng/ml) or IFNγ (20 ng/ml) for 12h and then were incubated with RBC isolated from healthy mice (G; upper). Representative images of immunohistochemical staining against F4/80 among spleens isolated from poly I:C-, LPS-, TNFα- or IFNγ-treated SIRPα−/− mice (G; lower). H) 12h after one injection of LPS, the concentration of IL-1β, IL-6, IL-10, TNFα and GM-CSF were quantified. The data represent sixteen independent experiments. Each symbol represents an individual mouse. One-way ANOVA and Dunn’s post-hoc analyses (A-F) or Student’s t test (H) were used to determined statistical significance among multiple comparisons: NS = P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
DISCUSSION
The appearance of aberrant histiocytes phagocytosing healthy host cells in sHLH is largely attributed to the hypercytokinemic milieu (60, 61). However, the mechanism by which inflammatory signaling overactivates macrophages and other myeloid leukocytes to the point of manifesting a hemophagocytic phenotype remains unclear. This study identifies SIRPα as a critical deterrent against the induction of hemophagocytic myeloid leukocytes under inflammation and also the development of sHLH/CSS. We found that activation of SIRPα−/− leukocytes by TLR agonists (TLR2, TLR3, TLR4 or TLR9) or pro-inflammatory cytokines (TNFα, IL-6 or IL-17A, but not IFNγ) leads to severe pancytopenia and other hallmarks of sHLH (1, 6). While HLH-associated cytokines downregulate CD47 on HSC, allowing macrophages to phagocytose HSC in vitro (62), we found that TLR9-driven inflammation in CD47−/− mice, unlike SIRPα−/− mice, does not induce sHLH, but rather only leads to hemophagocytosis, anemia and splenomegaly (Fig. 3 and Fig. 7). Conceivably, the differences between SIRPα and CD47 in cellular expression and function underly the inability of CD47−/− mice to fully develop severe sHLH under TLR9 agonism as in SIRPα−/− mice (63, 64). sHLH symptoms associated with macrophage activation, such as hypercytokinemia, hypertriglyceridemia and hyperferritinemia, were less severe in CpG-treated CD47−/− mice than in SIRPα−/− mice (21, 24, 25). This disparity likely manifests due to the differing roles of SIRPα and CD47 in regulating macrophage activation, as we and others have shown that SIRPα tempers the pro-inflammatory macrophage phenotype (Fig. 5) (8, 29). Collectively, these observations suggest that there are CD47-independent SIRPα regulatory mechanisms that remain to be clarified.
Whereas SIRPα canonically suppresses phagocytosis of healthy host cells, several studies suggest that SIRPα also negatively regulates pro-inflammation, as SIRPα deficiency exacerbates disease conditions (8, 28–30). Similarly, SIRPα deficiency worsened and accelerated the onset of TLR9-driven sHLH (Fig. 1 and Fig. 2). Comparable severity of the sHLH phenotype has been shown to also manifest in CpG-treated mice with intact SIRPα signaling if they are deficient in IL-18BP, which opposes IL-18 signaling (50). However, unlike other sHLH models, TLR9-challenged SIRPα−/− mice rapidly exhibited severe anemia, hemophagocytosis and hypercytokinemia following the initial CpG injection, which likely points to the involvement of innate immunity in sHLH pathology therein. Wang et al showed that a specific sequence of pathogen sensing, i.e., viral (TLR3) to bacterial (TLR4) challenge, leads to metabolic dysregulation in macrophages and subsequently induces a highly lethal hyperinflammatory state resembling HLH secondary to endotoxic shock (9). Similarly, Mahajan et al has shown that dysregulation of lipid signaling in macrophages can also confer macrophage-intrinsic sHLH/CSS pathology (26, 27). In parallel, our data suggests that macrophages, and likely other myeloid leukocytes, also become dysregulated and hyperinflammatory in the absence of SIRPα-dependent inhibition (Fig. 4 and Fig. 5), leading to severe and rapid onset of macrophage-intrinsic sHLH/CSS pathology. Indeed, activating SIRPα−/− macrophages with TLR agonists or pro-inflammatory cytokines led to exuberant hemophagocytosis and macrophage-intrinsic fulminant sHLH (Fig. 6 and Fig. 7), whereas depleting macrophages prevented sHLH onset in TLR9-inflamed SIRPα−/− mice (Fig. 4). Mechanistic studies suggest that this phenotype was partially due to SIRPα inhibiting TLR9-driven macrophage activation by negatively regulating MAPK pathways (Fig. 5), thus removing SIRPα appears to ‘prime’ macrophages and facilitates their acquisition of a hyperinflammatory hemophagocytic state. Other studies have similarly shown that SIRPα regulates M1 and M2 macrophage polarization by modulating PI3K-Akt, MAPK and NF-kB pathways (29, 46). However, given that CL2MDP has been shown to be non-specific (55, 65–67), further investigation is required to ascertain a definitive role for macrophages over other phagocytes in HLH/CSS pathogenesis in SIRPα−/− mice.
The pro-inflammatory cytokine IFNγ is considered a key molecule driving inflammation in HLH and is intimately associated with hemophagocytosis and anemia of inflammation (7, 52, 68–70). For example, two HLH patients had not developed hemophagocytosis owing to an IFNγ-receptor deficiency (71). Indeed, IFNγ administration alone is sufficient to induce hemophagocytic RPM in WT mice, leading to consumptive pancytopenia (7). To that end, researchers have endeavored to treat HLH by neutralizing IFNγ, which has demonstrated exceptional efficacy in various settings (39, 50, 68). In contrast, IFNγ neutralization was non-therapeutic in SIRPα−/− mice (Fig. 4) and IFNγ treatment failed to induce any aspects of sHLH in SIRPα−/− mice (Fig. 6 and Fig. 7), corroborating our previous study showing that, while many pro-inflammatory cytokines and TLR agonists may drive SIRPα−/− macrophages to become hemophagocytic, IFNγ has no such effect (8). Thus, future studies are necessary to determine the role of IFNγ in driving macrophages to become hemophagocytic if not for providing the putative activation signaling (8). Supporting IFNγ-independent mechanisms in HLH onset, Albeituni et al compared the therapeutic efficacies of αIFNγ and ruxolitinib – a JAK1/2 inhibitor – in murine models of HLH and found that ruxolitinib was superior, as it reduced neutrophil expansion and tissue infiltration (53, 72, 73). In parallel, an absence of SIRPα under TLR9-driven inflammation greatly increased granulopoiesis and PMN tissue infiltration, which may partially underly the exceptionally severe sHLH phenotype in CpG-treated SIRPα−/− mice and the inefficacy of αIFNγ (Fig. 5). Interestingly, CL2MDP depletion reduced circulating GM-CSF and also the frequency and number of splenic PMN in CpG-treated SIRPα−/− mice (Fig. 6). Furthermore, we show the mechanism by which macrophages and other myeloid leukocytes may become hemophagocytic and drive HLH-like disease under SIRPα deficiency is partially redundant, as an array of TLR agonists and pro-inflammatory cytokines are capable of providing the activation signaling (Fig. 6 and Fig. 7). This heterogenous capacity to drive myeloid leukocytes toward a hemophagocytic phenotype likely underscores why hemophagocytosis is neither specific nor sensitive to HLH or CSS in general and may also lend an explanation as to why CSS/HLH patients differentially respond to current cytokine neutralization therapies (1, 53, 74–78).
Although our studies reveal a link between SIRPα and the nascence of both hemophagocytes and HLH-like disease, a major question remains unanswered: do events preceding the onset of HLH/CSS comprise a phase during which SIRPα becomes downregulated or lost? Another puzzle is whether SIRPα can be downregulated under physiological or pathological conditions? Although the first question is currently unanswered and certainly demands further investigation, some studies have shown particular TLR-driven inflammation or disease conditions such as diabetic nephropathy lead to or are associated with a loss of SIRPα expression (8, 20, 30, 46). Conceivably, these conditions not only provide the pre-disposing condition, i.e., an absence of SIRPα, but also the necessary inflammatory activation, i.e., TLR or pro-inflammatory cytokine signaling, to drive leukocytes toward a hemophagocytic phenotype and potentially confer HLH/CSS-like disease. Akilesh et al recently showed that chronic TLR7 and TLR9 signaling reprograms myelopoiesis toward differentiating specialized monocytes, referred to as inflammatory hemophagocytes (iHPCs) (33). iHPCs arose in aged mice with constitutively active TLR7 (TLR7.1) or, to a lesser degree, in mice injected with R848 (TLR7 agonist) or CpG every day for 13 or 5 days, respectively. However, these iHPCs appear to represent a separate subset of hemophagocytes following long-term myelopoietic reprogramming (79, 80), and given the stark differences in the kinetics and severity of hemophagocytosis and cytopenias, these iHPCs likely differ from hemophagocytes that arise in the absence of SIRPα. Nevertheless, our findings collectively demonstrate that SIRPα plays an indispensable role in preventing myeloid leukocytes from becoming hemophagocytic under inflammation and may also provide an explanation for why heterogenous inciting inflammatory factors can inevitably culminate in HLH/CSS pathology.
Supplementary Material
KEY POINTS:
SIRPα deficiency enables various inflammatory stimuli to confer sHLH/CSS in mice.
Macrophage depletion, but not IFNγ neutralization, precludes sHLH in SIRPα−/− mice.
SIRPα negatively regulates TLR9 signaling by inhibiting Erk1/2 and p38 activation.
Acknowledgements
The authors thank the staff in the Georgia State University Animal Resources Program for assisting in experiments and animal care. The visual abstract was created using BioRender (www.BioRender.com).
Financial Support
This work was supported in part by the a National Institute of Allergy and Infectious Disease NIH/NIAID Grant (R01AI106839), a National Cancer Institute Grant (R21CA241271), a Georgia Research Alliance (GRA) Venture Development Grant, a Biolocity Innovation & Commercialization grant, a Molecular Basis of Disease Fellowship from Georgia State University (K.K.), an Ahmed T. Abdelal Molecular Genetics and Biotechnology Fellowship from Georgia State University (K.K.) and a Careers in Immunology Fellowship from American Association of Immunologists (Z.B.).
Abbreviations
- SIRPα
Signal regulatory protein alpha
- HLH
hemophagocytic lymphohistiocytosis
- CSS
cytokine storm syndrome
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- MC
monocytes
- DC
dendritic cells
- PMN
neutrophil
- WT
wild-type
- Hgb
hemoglobin
- sCD25
soluble CD25
- HSC
hematopoietic stem cells
- EL
empty liposomes
- CL2MDP
clodronate-containing liposomes
- mCD47.ex
soluble murine CD47 extracellular domain fusion protein
- IL-18BP
interleukin 18 binding protein
- iHPC
inflammatory hemophagocyte
REFERENCES
- 1.Al-Samkari H, and Berliner N. 2018. Hemophagocytic Lymphohistiocytosis. Annual Review of Pathology: Mechanisms of Disease 13: 27–49. [DOI] [PubMed] [Google Scholar]
- 2.Janka GE 2007. Hemophagocytic syndromes. Blood Reviews 21: 245–253. [DOI] [PubMed] [Google Scholar]
- 3.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, and Manson JJ. 2020. COVID-19: consider cytokine storm syndromes and immunosuppression. The Lancet 395: 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao X 2020. COVID-19: immunopathology and its implications for therapy. Nature Reviews Immunology 20: 269–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bracaglia C, Prencipe G, and De Benedetti F. 2017. Macrophage Activation Syndrome: different mechanisms leading to a one clinical syndrome. Pediatr Rheumatol Online J 15: 5–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikiforow S, and Berliner N. 2020. To “Lump” or to “Split” in Macrophage Activation Syndrome and Hemophagocytic Lymphohistiocytosis. 72: 206–209. [DOI] [PubMed] [Google Scholar]
- 7.Zoller EE, Lykens JE, Terrell CE, Aliberti J, Filipovich AH, Henson PM, and Jordan MB. 2011. Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med 208: 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bian Z, Shi L, Guo Y-L, Lv Z, Tang C, Niu S, Tremblay A, Venkataramani M, Culpepper C, Li L, Zhou Z, Mansour A, Zhang Y, Gewirtz A, Kidder K, Zen K, and Liu Y. 2016. Cd47-Sirpα interaction and IL-10 constrain inflammation-induced macrophage phagocytosis of healthy self-cells. Proceedings of the National Academy of Sciences 113: E5434-E5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang A, Pope SD, Weinstein JS, Yu S, Zhang C, Booth CJ, and Medzhitov R. 2019. Specific sequences of infectious challenge lead to secondary hemophagocytic lymphohistiocytosis-like disease in mice. Proceedings of the National Academy of Sciences 116: 2200–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crayne CB, Albeituni S, Nichols KE, and Cron RQ. 2019. The Immunology of Macrophage Activation Syndrome. Frontiers in Immunology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barclay AN, and Van den Berg TK. 2014. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol 32: 25–50. [DOI] [PubMed] [Google Scholar]
- 12.Gordon S 2016. Phagocytosis: An Immunobiologic Process. Immunity 44: 463–475. [DOI] [PubMed] [Google Scholar]
- 13.Lemke G 2019. How macrophages deal with death. Nature Reviews Immunology 19: 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takizawa H, and Manz MG. 2007. Macrophage tolerance: CD47–SIRP-α–mediated signals matter. Nature Immunology 8: 1287–1289. [DOI] [PubMed] [Google Scholar]
- 15.Ide K, Wang H, Tahara H, Liu J, Wang X, Asahara T, Sykes M, Yang Y-G, and Ohdan H. 2007. Role for CD47-SIRPalpha signaling in xenograft rejection by macrophages. Proc Natl Acad Sci U S A 104: 5062–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Londino JD, Gulick D, Isenberg JS, and Mallampalli RK. 2015. Cleavage of Signal Regulatory Protein alpha (SIRPalpha) Enhances Inflammatory Signaling. The Journal of biological chemistry 290: 31113–31125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi T, Li J, Chen H, Wu J, An J, Xu Y, Hu Y, Lowell CA, and Cyster JG. 2015. Splenic Dendritic Cells Survey Red Blood Cells for Missing Self-CD47 to Trigger Adaptive Immune Responses. Immunity 43: 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Buhring HJ, Zen K, Burst SL, Schnell FJ, Williams IR, and Parkos CA. 2002. Signal regulatory protein (SIRPalpha), a cellular ligand for CD47, regulates neutrophil transmigration. J Biol Chem 277: 10028–10036. [DOI] [PubMed] [Google Scholar]
- 19.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, and Lindberg FP. 2000. Role of CD47 as a marker of self on red blood cells. Science 288: 2051–2054. [DOI] [PubMed] [Google Scholar]
- 20.Bennett LF, Liao C, Quickel MD, Yeoh BS, Vijay-Kumar M, Hankey-Giblin P, Prabhu KS, and Paulson RF. 2019. Inflammation induces stress erythropoiesis through heme-dependent activation of SPI-C. Science Signaling 12: eaap7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grom AA, and Mellins ED. 2010. Macrophage activation syndrome: advances towards understanding pathogenesis. Curr Opin Rheumatol 22: 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravelli A, Grom AA, Behrens EM, and Cron RQ. 2012. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes & Immunity 13: 289–298. [DOI] [PubMed] [Google Scholar]
- 23.Lerkvaleekul B, and Vilaiyuk S. 2018. Macrophage activation syndrome: early diagnosis is key. Open Access Rheumatol 10: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Zhang DL, Crooks DR, Sougrat R, Morgenstern A, Galy B, Hentze MW, Lazaro FJ, Rouault TA, and Meyron-Holtz EG. 2010. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 116: 1574–1584. [DOI] [PubMed] [Google Scholar]
- 25.George MR 2014. Hemophagocytic lymphohistiocytosis: review of etiologies and management. Journal of blood medicine 5: 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahajan S, Decker CE, Yang Z, Veis D, Mellins ED, and Faccio R. 2019. Plcγ2/Tmem178 dependent pathway in myeloid cells modulates the pathogenesis of cytokine storm syndrome. J Autoimmun 100: 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahajan S, Mellins ED, and Faccio R. 2020. Diacylglycerol Kinase ζ Regulates Macrophage Responses in Juvenile Arthritis and Cytokine Storm Syndrome Mouse Models. The Journal of Immunology 204: 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zen K, Guo Y, Bian Z, Lv Z, Zhu D, Ohnishi H, Matozaki T, and Liu Y. 2013. Inflammation-induced proteolytic processing of the SIRPα cytoplasmic ITIM in neutrophils propagates a proinflammatory state. Nature Communications 4: 2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi L, Bian Z, and Liu Y. 2017. Dual role of SIRPα in macrophage activation: inhibiting M1 while promoting M2 polarization via selectively activating SHP-1 and SHP-2 signal. The Journal of Immunology 198: 67.12–67.12. [Google Scholar]
- 30.Li L, Liu Y, Li S, Yang R, Zeng C, Rong W, Liang H, Zhang M, Zhu X, Kidder K, Liu Y, Liu Z, and Zen K. 2019. Signal regulatory protein α protects podocytes through promoting autophagic activity. JCI Insight 5: e124747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aurbach K, Spindler M, Haining EJ, Bender M, and Pleines I. 2019. Blood collection, platelet isolation and measurement of platelet count and size in mice-a practical guide. Platelets 30: 698–707. [DOI] [PubMed] [Google Scholar]
- 32.Ohyagi H, Onai N, Sato T, Yotsumoto S, Liu J, Akiba H, Yagita H, Atarashi K, Honda K, Roers A, Müller W, Kurabayashi K, Hosoi-Amaike M, Takahashi N, Hirokawa M, Matsushima K, Sawada K, and Ohteki T. 2013. Monocyte-Derived Dendritic Cells Perform Hemophagocytosis to Fine-Tune Excessive Immune Responses. Immunity 39: 584–598. [DOI] [PubMed] [Google Scholar]
- 33.Akilesh HM, Buechler MB, Duggan JM, Hahn WO, Matta B, Sun X, Gessay G, Whalen E, Mason M, Presnell SR, Elkon KB, Lacy-Hulbert A, Barnes BJ, Pepper M, and Hamerman JA. 2019. Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science 363: eaao5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ha B, Lv Z, Bian Z, Zhang X, Mishra A, and Liu Y. 2013. ‘Clustering’ SIRPalpha into the plasma membrane lipid microdomains is required for activated monocytes and macrophages to mediate effective cell surface interactions with CD47. PloS one 8: e77615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang PH, Lagenaur CF, and Narayanan V. 1999. Integrin-associated protein is a ligand for the P84 neural adhesion molecule. J Biol Chem 274: 559–562. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, Tong Q, Zhou Y, Lee HW, Yang JJ, Buhring HJ, Chen YT, Ha B, Chen CX, Yang Y, and Zen K. 2007. Functional elements on SIRPalpha IgV domain mediate cell surface binding to CD47. J Mol Biol 365: 680–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, O’Connor MB, Mandell KJ, Zen K, Ullrich A, Buhring HJ, and Parkos CA. 2004. Peptide-mediated inhibition of neutrophil transmigration by blocking CD47 interactions with signal regulatory protein alpha. J Immunol 172: 2578–2585. [DOI] [PubMed] [Google Scholar]
- 38.Lv Z, Bian Z, Shi L, Niu S, Ha B, Tremblay A, Li L, Zhang X, Paluszynski J, Liu M, Zen K, and Liu Y. 2015. Loss of Cell Surface CD47 Clustering Formation and Binding Avidity to SIRPalpha Facilitate Apoptotic Cell Clearance by Macrophages. J Immunol 195: 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, Kambayashi T, and Koretzky GA. 2011. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest 121: 2264–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito Y, Iwamura H, Kaneko T, Ohnishi H, Murata Y, Okazawa H, Kanazawa Y, Sato-Hashimoto M, Kobayashi H, Oldenborg PA, Naito M, Kaneko Y, Nojima Y, and Matozaki T. 2010. Regulation by SIRPalpha of dendritic cell homeostasis in lymphoid tissues. Blood 116: 3517–3525. [DOI] [PubMed] [Google Scholar]
- 41.Saito Y, Respatika D, Komori S, Washio K, Nishimura T, Kotani T, Murata Y, Okazawa H, Ohnishi H, Kaneko Y, Yui K, Yasutomo K, Nishigori C, Nojima Y, and Matozaki T. 2017. SIRPα+ dendritic cells regulate homeostasis of fibroblastic reticular cells via TNF receptor ligands in the adult spleen. Proceedings of the National Academy of Sciences 114: E10151-E10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schulert GS, and Grom AA. 2014. Macrophage activation syndrome and cytokine-directed therapies. Best Pract Res Clin Rheumatol 28: 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Canna SW, and Behrens EM. 2012. Making sense of the cytokine storm: a conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin North Am 59: 329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Behrens EM, and Koretzky GA. 2017. Review: Cytokine Storm Syndrome: Looking Toward the Precision Medicine Era. 69: 1135–1143. [DOI] [PubMed] [Google Scholar]
- 45.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, DiMattia MA, Zaal KJM, Sanchez GAM, Kim H, Chapelle D, Plass N, Huang Y, Villarino AV, Biancotto A, Fleisher TA, Duncan JA, O’Shea JJ, Benseler S, Grom A, Deng Z, Laxer RM, and Goldbach-Mansky R. 2014. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nature Genetics 46: 1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kong X-N, Yan H-X, Chen L, Dong L-W, Yang W, Liu Q, Yu L-X, Huang D-D, Liu S-Q, Liu H, Wu M-C, and Wang H-Y. 2007. LPS-induced down-regulation of signal regulatory protein {alpha} contributes to innate immune activation in macrophages. The Journal of experimental medicine 204: 2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kantari C, Pederzoli-Ribeil M, and Witko-Sarsat V. 2008. The role of neutrophils and monocytes in innate immunity. Contrib Microbiol 15: 118–146. [DOI] [PubMed] [Google Scholar]
- 48.Bian Z, Shi L, Venkataramani M, Abdelaal AM, Culpepper C, Kidder K, Liang H, Zen K, and Liu Y. 2018. Tumor conditions induce bone marrow expansion of granulocytic, but not monocytic, immunosuppressive leukocytes with increased CXCR2 expression in mice. Eur J Immunol 48: 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khanna-Gupta A, and Berliner N. 2018. Granulocytopoiesis and monocytopoiesis In Hematology. Elsevier; 321–333. e321. [Google Scholar]
- 50.Girard-Guyonvarc’h C, Palomo J, Martin P, Rodriguez E, Troccaz S, Palmer G, and Gabay C. 2018. Unopposed IL-18 signaling leads to severe TLR9-induced macrophage activation syndrome in mice. Blood 131: 1430–1441. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Zhang J, Ginzburg Y, Li H, Xue F, De Franceschi L, Chasis JA, Mohandas N, and An X. 2013. Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood 121: e43–e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canna SW, Wrobel J, Chu N, Kreiger PA, Paessler M, and Behrens EM. 2013. Interferon-gamma mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum 65: 1764–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Albeituni S, Verbist KC, Tedrick PE, Tillman H, Picarsic J, Bassett R, and Nichols KE. 2019. Mechanisms of action of ruxolitinib in murine models of hemophagocytic lymphohistiocytosis. Blood 134: 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirby AC, Beattie L, Maroof A, van Rooijen N, and Kaye PM. 2009. SIGNR1-negative red pulp macrophages protect against acute streptococcal sepsis after Leishmania donovani-induced loss of marginal zone macrophages. Am J Pathol 175: 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ciavarra RP, Taylor L, Greene AR, Yousefieh N, Horeth D, van Rooijen N, Steel C, Gregory B, Birkenbach M, and Sekellick M. 2005. Impact of macrophage and dendritic cell subset elimination on antiviral immunity, viral clearance and production of type 1 interferon. Virology 342: 177–189. [DOI] [PubMed] [Google Scholar]
- 56.Bu L, Gao M, Qu S, and Liu D. 2013. Intraperitoneal injection of clodronate liposomes eliminates visceral adipose macrophages and blocks high-fat diet-induced weight gain and development of insulin resistance. AAPS J 15: 1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biewenga J, van der Ende MB, Krist LFG, Borst A, Ghufron M, and van Rooijen N. 1995. Macrophage depletion in the rat after intraperitoneal administration of liposome-encapsulated clodronate: Depletion kinetics and accelerated repopulation of peritoneal and omental macrophages by administration of freund’s adjuvant. Cell and Tissue Research 280: 189–196. [DOI] [PubMed] [Google Scholar]
- 58.Truman-Rosentsvit M, Berenbaum D, Spektor L, Cohen LA, Belizowsky-Moshe S, Lifshitz L, Ma J, Li W, Kesselman E, Abutbul-Ionita I, Danino D, Gutierrez L, Li H, Li K, Lou H, Regoni M, Poli M, Glaser F, Rouault TA, and Meyron-Holtz EG. 2018. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 131: 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosário C, Zandman-Goddard G, Meyron-Holtz EG, D’Cruz DP, and Shoenfeld Y. 2013. The Hyperferritinemic Syndrome: macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Medicine 11: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hayden A, Park S, Giustini D, Lee AY, and Chen LY. 2016. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev 30: 411–420. [DOI] [PubMed] [Google Scholar]
- 61.Schulert GS, and Grom AA. 2015. Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annu Rev Med 66: 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuriyama T, Takenaka K, Kohno K, Yamauchi T, Daitoku S, Yoshimoto G, Kikushige Y, Kishimoto J, Abe Y, Harada N, Miyamoto T, Iwasaki H, Teshima T, and Akashi K. 2012. Engulfment of hematopoietic stem cells caused by down-regulation of CD47 is critical in the pathogenesis of hemophagocytic lymphohistiocytosis. Blood 120: 4058–4067. [DOI] [PubMed] [Google Scholar]
- 63.Oldenborg P-A 2013. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematology 2013: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barclay AN, and Brown MH. 2006. The SIRP family of receptors and immune regulation. Nat Rev Immunol 6: 457–464. [DOI] [PubMed] [Google Scholar]
- 65.Danenberg Haim D, Fishbein I, Gao J, Mönkkönen J, Reich R, Gati I, Moerman E, and Golomb G. 2002. Macrophage Depletion by Clodronate-Containing Liposomes Reduces Neointimal Formation After Balloon Injury in Rats and Rabbits. Circulation 106: 599–605. [DOI] [PubMed] [Google Scholar]
- 66.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjällman AHM, Ballmer-Hofer K, and Schwendener RA. 2006. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer 95: 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee S, Rosen S, Van Rooijen PMD,N, and Noble-Haeusslein L 2011. Prevention of Both Neutrophil and Monocyte Recruitment Promotes Recovery after Spinal Cord Injury. Journal of neurotrauma 28: 1893–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lounder DT, Bin Q, de Min C, and Jordan MB. 2019. Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Adv 3: 47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burn TN, Weaver L, Rood JE, Chu N, Bodansky A, Kreiger PA, and Behrens EM. 2019. Genetic Deficiency of Interferon-gamma Reveals Interferon-gamma-Independent Manifestations of Murine Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Das R, Guan P, Sprague L, Verbist K, Tedrick P, An QA, Cheng C, Kurachi M, Levine R, Wherry EJ, Canna SW, Behrens EM, and Nichols KE. 2016. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood 127: 1666–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tesi B, Sieni E, Neves C, Romano F, Cetica V, Cordeiro AI, Chiang S, Schlums H, Galli L, Avenali S, Tondo A, Canessa C, Henter JI, Nordenskjold M, Hsu AP, Holland SM, Neves JF, Azzari C, and Bryceson YT. 2015. Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN-gamma receptor deficiency. J Allergy Clin Immunol 135: 1638–1641. [DOI] [PubMed] [Google Scholar]
- 72.Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, and de Saint Basile G. 2016. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood 128: 60–71. [DOI] [PubMed] [Google Scholar]
- 73.Ahmed A, Merrill SA, Alsawah F, Bockenstedt P, Campagnaro E, Devata S, Gitlin SD, Kaminski M, Cusick A, Phillips T, Sood S, Talpaz M, Quiery A, Boonstra PS, and Wilcox RA. 2019. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. The Lancet Haematology 6: e630-e637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu D, and Yang XO. 2020. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. Journal of Microbiology, Immunology and Infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu X, Han M, Li T, Sun W, Wang D, Fu B, Zhou Y, Zheng X, Yang Y, Li X, Zhang X, Pan A, and Wei H. 2020. Effective treatment of severe COVID-19 patients with tocilizumab. Proceedings of the National Academy of Sciences 117: 10970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang C, Wu Z, Li J-W, Zhao H, and Wang G-Q. 2020. Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. International Journal of Antimicrobial Agents 55: 105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chinn IK, Eckstein OS, Peckham-Gregory EC, Goldberg BR, Forbes LR, Nicholas SK, Mace EM, Vogel TP, Abhyankar HA, Diaz MI, Heslop HE, Krance RA, Martinez CA, Nguyen TC, Bashir DA, Goldman JR, Stray-Pedersen A, Pedroza LA, Poli MC, Aldave-Becerra JC, McGhee SA, Al-Herz W, Chamdin A, Coban-Akdemir ZH, Jhangiani SN, Muzny DM, Cao TN, Hong DN, Gibbs RA, Lupski JR, Orange JS, McClain KL, and Allen CE. 2018. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood 132: 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, and O’Shea JJ. 2017. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 17: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grigoriou M, Banos A, Filia A, Pavlidis P, Giannouli S, Karali V, Nikolopoulos D, Pieta A, Bertsias G, Verginis P, Mitroulis I, and Boumpas DT. 2020. Transcriptome reprogramming and myeloid skewing in haematopoietic stem and progenitor cells in systemic lupus erythematosus. Annals of the rheumatic diseases 79: 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yvan-Charvet L, and Ng LG. 2019. Granulopoiesis and Neutrophil Homeostasis: A Metabolic, Daily Balancing Act. Trends in Immunology 40: 598–612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.