

Summary
Although enhancers play critical roles in cancer, quantifying enhancer activities in clinical samples remains challenging, especially for super-enhancers. Enhancer activities can be inferred from enhancer RNA (eRNA) signals, which requires enhancer transcription loci definition. Only a small proportion of human eRNA loci has been precisely identified, limiting investigations of enhancer-mediated oncogenic mechanisms. Here we characterize super-enhancer regions using aggregated RNA-seq data from large cohorts. Super-enhancers usually contain discrete loci featuring sharp eRNA expression peaks. We identify >300,000 eRNA loci in ~377 Mb super-enhancer regions that are regulated by evolutionarily conserved, well-positioned nucleosomes and are frequently dysregulated in cancer. The eRNAs provide explanatory power for cancer phenotypes beyond that provided by mRNA expression through resolving intratumoral heterogeneity with enhancer cell-type specificity. Our study provides a high-resolution map of eRNA loci through which super-enhancer activities can be quantified by RNA-seq and a user-friendly data portal, enabling a broad range of biomedical investigations.
eTOC Blurb
Chen and Liang provide a high-resolution map of eRNA loci through which super-enhancer activities can be conveniently quantified by RNA-seq. The eRNA signals in cancer samples are clinically relevant and provide additional explanatory power for cancer phenotypes beyond those provided by mRNAs through resolving intra-tumor heterogeneity with enhancer cell-type specificity.
Graphical Abstract
Introduction
Enhancers are key non-coding DNA sequences regulating their target genes (Pennacchio et al., 2013). It has been an evolving concept since the early identification of the Simian virus 40 (SV40) DNA sequence enhancing local gene expression in the 1980s (Banerji et al., 1981), followed by the discovery of endogenous locus control regions (LCRs) (Levings and Bungert, 2002). The chromatin modification of H3K4me1 and H3K27ac are known to be effective markers for enhancer identification (Creyghton et al., 2010; Heintzman et al., 2007), and later more epigenetic markers, such as H3.3 and H2A.Z, were reported to be associated with enhancer functions (Goldberg et al., 2010; Jin et al., 2009; Lawrence et al., 2016). With many more enhancer elements characterized in the human genome (ENOCDE Consortium, 2012), the concepts of “stretch-enhancer” and “super-enhancer” were then proposed to refer to large genomic domains with enriched enhancer activity (Parker et al., 2013; Whyte et al., 2013). Identified by highly enriched ChIP-seq signals (Hnisz et al., 2013), super-enhancers are typically over 10 kb and characterized by the extensive intensity or strength of given enhancer markers, such as H3K27ac (Hnisz et al., 2013; Loven et al., 2013; Whyte et al., 2013). They tend to be bound by a large panel of transcription factors (TFs) related to cell fate determination (Pott and Lieb, 2015). Using a combination of high-throughput assays, the ENCODE project has identified the genome-wide DNA regions with enhancer-like features in >100 cell types (ENODE Consortium, 2012). On activation, enhancers open the local chromatin and expose the DNA motifs to attract TFs that can further recruit RNA polymerases (usually RNA Pol II) to generate enhancer RNAs (eRNAs) (Heinz et al., 2015; Murakawa et al., 2016). First identified in neuronal tissues (De Santa et al., 2010; Kim et al., 2010), expressed enhancers were systematically annotated (~65,000 ones) by the FANTOM project in ~400 human tissues and cell types using the CAGE-seq technique targeting the molecules with 5′-cap (Andersson et al., 2014). While some studies show strong evidence that eRNAs are functional in gene regulation by some master TFs and repressors such as p53, estrogen receptors, and Rev-Erbs (Lam et al., 2013; Li et al., 2013; Melo et al., 2013), what proportion of the eRNAs are functional or merely the byproducts of enhancer activation (Catarino and Stark, 2018; Kim et al., 2015) is still an open question.
The critical roles of enhancers in cancer development and tumor response have been increasingly recognized (Bahr et al., 2018; Mack et al., 2018; Takeda et al., 2018). But quantifying enhancer activities in clinical tumor samples remains challenging in practice. RNA-seq data are a convenient, rich information resource for eRNA quantification and enhancer activity approximation (Buenrostro et al., 2013; Chen et al., 2018a; Murakawa et al., 2016). In particular, it works well based on the CAGE-defined enhancers because of their precise eRNA location annotation (Chen et al., 2018a; Chen et al., 2018b; De Santa et al., 2010). Based on CAGE-defined enhancers annotated by FANTOM, using RNA-seq data from The Cancer Genome Atlas (TCGA), we recently demonstrated the utility of the eRNA signals in predicting patient survival and regulation of therapeutic targets (Chen et al., 2018a). However, the CAGE-defined enhancer annotation only covers a small fraction of eRNA loci (~15,000) (Andersson et al., 2014; ENODE Consortium, 2012). Moreover, unlike RNA-seq, CAGE-seq cannot be easily applied to large cohorts of tumor samples such as TCGA, thereby limiting the power of connecting eRNAs with clinical phenotypes. Therefore, it is highly valuable to systematically identify the precise eRNA loci that can be measured by routine RNA-seq data.
To depict a more comprehensive set of eRNA loci beyond those already annotated by FANTOM, we focused on the super-enhancer regions defined by Hnisz et al. (Hnisz et al., 2013). Although the concept of “super-enhancers” is still open to discussion (Pott and Lieb, 2015), we investigated them for the following scientific and technical reasons. First, these super-enhancer regions cover >370 Mb DNA sequence in length and represent regions with the most enriched regulatory signals in the human genome. Dysregulation of super-enhancers is frequently associated with various developmental diseases, including cancer (Alam et al., 2020; Hnisz et al., 2013; Loven et al., 2013; Whyte et al., 2013), indicating their potential biomedical significance (Shin, 2018). Second, compared with typical enhancers, super-enhancers show much stronger signals of RNA Pol II binding (Hnisz et al., 2013), implying that their eRNAs may be more actively transcribed and could potentially be detected by TCGA mRNA-seq data even without CAGE-seq based annotation (Chen et al., 2018a). Third, compared to typical enhancers (~200 bp), super-enhancers are much longer (usually >10 kb) (Pott and Lieb, 2015). Thus, background transcription noise across the super-enhancer bodies substantially confounds the real enhancer activation signals (Berretta and Morillon, 2009; Nagalakshmi et al., 2008; Thompson and Parker, 2007), necessitating the precise separation of the eRNAs from nearby genomic regions (Pott and Lieb, 2015). Finally, unlike other super-enhancers proposed later, these super-enhancer regions have been systematically annotated in a variety of tissues and cell types (86 in total) using consistent H3K27ac ChIP-seq profiles (Hnisz et al., 2013), thereby providing a reliable recurrent frequency of super-enhancer activation across tissues.
We hypothesize that the precise eRNA loci within super-enhancers can be resolved by analyzing aggregated RNA-seq profiles that combine the RNA-seq data of many individual samples. This is because eRNA reads associated with real enhancer activity recurrently accumulate, whereas background transcription noise tends to occur stochastically. The large number of RNA-seq reads obtained would compensate for the statistical power compromised by the low eRNA expression level typically observed in a single sample. Further, the large sample size would help distinguish differential activation of neighbor eRNA loci within a super-enhancer. With the precise eRNA loci thus defined, it would then be possible to quantify the sample-specific eRNA levels using routine RNA-seq data, thereby enabling a broad range of biomedical investigations of super-enhancer activities, especially in clinical samples.
Results
Recurrent eRNA expression peaks in super-enhancers
Figure 1 shows an overview of our study. Since super-enhancers are tissue-specific and collectively constitute up to ~377 Mb of mappable non-coding DNA sequences (Table S1) (Hnisz et al., 2013), we first focused on a subset of 1,531 (out of ~58,000) core super-enhancers (~5 Mb) that were consistently identified and activated in >20 (out of 86) tissue/cell types (Hnisz et al., 2013) (Table S2) for exploratory analysis. To confirm their RNA Pol II binding activities, we examined the association of their eRNAs with H3K27ac in 140 cell lines and observed a positive correlation in the vast majority (>80%) of these core super-enhancers (Figure S1A-E). Using TCGA RNA-seq data (>10,000 samples of 32 cancer types), we generated the aggregated RNA-seq profile for each cancer type to elucidate the eRNA transcriptional landscape in super-enhancers, integrated them with the nucleosome profiling data of 29 tissue/cell types, validated the patterns using GTEx RNA-seq data (~10,000 samples of 31 normal tissues) and FANTOM CAGE-seq enhancer data (>250 human cell lines), and inferred the underlying principles for identifying precise eRNA loci. Second, we applied the rules discerned from this core set to the whole super-enhancer set (~377 Mb) to construct a fine map of >300,000 eRNA loci. Third, through a case study of tumor response to immunotherapy, we demonstrated the power of such a map for eRNA analysis in explaining complex genotype-phenotype relationships. Finally, we performed a pan-cancer analysis of these eRNA loci by integrating other TCGA molecular data and built a user-friendly data portal, The Cancer eRNA Atlas (TCeA), for the scientific community to use all the resources generated in this study.
Figure 1. Overview of this study.

Principle discovery: we focused on a subset of 1,531 core super-enhancers (~5 Mb) to study their transcriptional patterns by integrating ENCODE ChIP-seq data, TCGA RNA-seq data, and published MNase-seq profiles. Global identification: based on the proposed model in which well-positioned nucleosomes mediate eRNA transcription, we generalized our analysis to the whole set of super-enhancers (~377) to annotate >300 eRNA loci in super-enhancers. Application: with the global map of eRNA loci, we assessed the utility of eRNA signals in explaining the response to immunotherapy, eQTL analysis, pan-cancer analysis, and built a user-friendly data portal for community use. See also Figure S1, Table S1 and Table S2.
To learn the rules for pinpointing eRNA loci, we calculated the eRNA expression levels for all tandem 10 bp windows of DNA in the 5 Mb core super-enhancer set using an aggregated RNA-seq dataset for each of the 32 cancer types (>10,000 TCGA samples in total). Looking at the transcriptional landscape of super-enhancers, we noticed highly recurrent sharp eRNA expression peaks across super-enhancer bodies, as illustrated by a ~670 bp region in Figure 2A. This is one of the super-enhancer regions most consistently identified in nearly half (39 out of 86) of the tissue/cell types (Figure S2A), and also one of the most widely expressed regions across cancer types (Figure 2A-B and S2B-C). In 28 of the 32 cancer types, we observed five sharp eRNA expression peaks with lengths of only a few dozen base-pairs near the 70th, 210th, 390th, 520th, and 640th nucleotide positions. Interestingly, not only the location of the eRNA peaks but also their relative heights (expression levels) showed recurrent patterns, leading to four distinct clusters of the 32 cancer types (Figure 2B). Such an eRNA pattern was further confirmed by the independent GTEx dataset of 31 normal tissues (Figure 2C and S2D-E) (GTEx Consortium, 2017).
Figure 2. Recurrent eRNA expression peaks in super-enhancers.

(A) The eRNA expression on chr3:50,265,725-50,266,396 in 4 cancer types representing the four clusters (C1-C4) of the 32 TCGA cancer types. Each bar represents a 10 bp window in the 672 bp region. The y-axis shows the RPKM in the 10 bp windows, and the x-axis represents the relative genomic coordinates (bp) in the region. The loci on the 70th, 210th, 390th, 520th, and 640th bp have local maximum RPKMs. These and their flanking 20 bp regions are highlighted. (B) A heatmap showing unsupervised clustering of the 32 cancer types based on the relative eRNA expression of all 10 bp windows in this 672 bp region. The eRNA expression levels (RPKM) are normalized into Z-scores within the columns of the heatmap. (C) The number of TCGA cancer types (top) or GTEx tissue types (bottom) with local maximum RPKM values of sliding 200 bp windows in this region. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD/READ, colon/rectum adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma. See also Figure S2.
To systematically identify such eRNA peaks, we searched the 1,531 core super-enhancers for local maximums of expression levels in all possible 200 bp windows and identified a total of 29,828 eRNA expression peaks recurrent in at least three TCGA cancer types (FDR = 0.12, permutation analysis; FDR <0.01 when recurrent in more than three cancer types, Figure S3A). Interestingly, an overwhelming proportion of these eRNA peaks showed a length of only ~100 bp, which then quickly decreased to the baseline (Figure 3A). This pattern of a short pulse held true when the maximum search range was extended to 400 or 600 bp (Figure S3B-C), or using the GTEx dataset (Figure 3B and S3D-E). A typical peak (the median of 29,828 peaks) has a maximum expression level that quickly drops by ~2.3fold as close as 50 bp on either side (Figure 3C). Further, we detected no enrichment of splicing motifs on the body or boundaries of these peaks, in contrast to the strong signals observed on intron-exon junctions (Figure S3F-G). We observed a strong signal of tandem TF binding motifs on either boundary of the eRNA peaks on both strands (Figure 3D), supporting that they resulted from transcriptional initiation rather than RNA splicing, similar to that of the FANTOM eRNA loci with precise transcription start sites (5′-cap). These results suggest that there are biologically meaningful eRNA loci in super-enhancers, and they generate discrete, recurrent, short eRNAs that can be readily detected by RNA-seq.
Figure 3. Characteristics of the super-enhancer eRNA expression peaks.

(A, B) The mean eRNA expression level on the flanking 200 bp sequences of 29,828 recurrent peaks identified in the 1,531 core super-enhancers in 32 TCGA cancer types (A) or 31 GTEx tissue types (B). For each cancer/tissue type, the 29,828 400 bp sequences were aligned with the peaks at the center (0 bp). Each point represents a 10 bp window. Mean RPKM was calculated for all 29,828 10 bp windows with the same relative positions to the peaks (indicated by the x-axis). The resulting mean RPKMs were normalized to Z-scores (indicated by the y-axis) for each cancer/tissue type. (C) The consensus profile of a typical eRNA expression peak in the TCGA dataset. The curve represents the median RPKM of the 29,828 peaks in 32 cancer types. (D) The density of the TF binding site (TFBS) motifs identified within the flanking 100 bp sequences of the 29,828 peaks. The 29,828 200 bp sequences were aligned with the peaks at the center (0 bp). Motifs on these DNA sequences were identified by the FIMO software with q <0.01. The y-value of the orange (or grey) curve represents the number of motif start/end sites identified at the same position relative to the peak (x-axis) on the DNA strand indicated by blue arrows. The phase difference, as indicated by the green arrows, between the orange and the grey curves, represents the enrichment of TFBS motifs at these locations. See also Figure S3.
eRNA expression is regulated by well-positioned nucleosomes
The eRNA peaks we observed were as short as ~100 bp (Figure 2A and 3C), and this length is close to a 147 bp DNA unit occupied by a typical nucleosome, the unit of chromatin organization. Since the nucleosome dynamics is a critical feature of TF binding on DNA motifs (He et al., 2010; West et al., 2014), we hypothesized that the eRNA peaks in super-enhancers are shaped by changes in chromatin organization at the nucleosome level in response to the super-enhancer activation by TF binding. When the super-enhancer is silent, binding motifs are protected from being accessed by TFs through promiscuous interactions. Upon activation, the nucleosome is disassembled, making the motifs available for TF recognition, which would then initiate transcription and generate the observed eRNAs. After activation, the enhancer sequence released from the TFs would soon be reclaimed by the nucleosome for protection (Jin et al., 2009; Mueller et al., 2017). The “state switch” of these nucleosomes are likely to release one unit of 147 bp DNA, thereby explaining the short (~100 bp) and sharp shape of the eRNA peaks (Figure 3A). Notably, even though the core super-enhancers we studied are likely to have effects in multiple tissues, they are still tissue-specific and should, therefore, be silent in the majority of the tissues (they are expected to be active in >20% of the tissue types only). As a result, nucleosomes should occupy the eRNA loci in most tissues surveyed, thereby allowing the detection of well-positioned nucleosomes when examining nucleosome binding signals across tissues. When the super-enhancer is activated, the situation can be more complicated. Upon transcription initiation, the nucleosome occupying an eRNA locus would have to be replaced by a TF (Brahma and Henikoff, 2019; He et al., 2010). However, the TF on this “open” DNA competes with intruding nucleosomes that often contain the histone variants H3.3 and H2A.Z (Jin et al., 2009; Mirny, 2010; Wasson and Hartemink, 2009) in a tissue-specific manner (Goldberg et al., 2010). The nucleosome turnover at this site leads to its observation as a fragile nucleosome (Brahma and Henikoff, 2019), depending on a variety of factors such as MNase digestion time (Brahma and Henikoff, 2019), replication stage (Ramachandran and Henikoff, 2016), and salt concentration (Jin et al., 2009). Recent studies have shown that the widespread nucleosome turnover in the actively transcribed gene bodies or enhancers is generally rapid enough so that no change of nucleosome occupation signal could be observed (Brahma and Henikoff, 2019; Mueller et al., 2017).
To test this hypothesis, we collected the MNase-seq data of 29 tissue/cell types (Figure S4A) and aligned the nucleosome signals flanking the 29,828 peaks. We indeed observed well-positioned nucleosomes on the eRNA expression peaks in all 29 human tissues (Figure 4A and S4B), and even in sperms where nucleosomes are highly sparse (Figure 4B) (Hammoud et al., 2009). The occupancy of these nucleosomes is conserved across macro-evolution, with similar occupancy observed in five tissues from pig, mouse, and human (Figure 4A, middle and bottom panel) with available MNase-seq data (Jiang et al., 2018), indicating the functional importance of nucleosome occupancy at these positions. Thus, our analysis reveals the epigenetic regulation mediated by nucleosome binding on the transcriptional initiation of eRNAs in super-enhancers (summarized in Figure 4C), similar to that observed for gene transcription start sites (TSS).
Figure 4. A dynamic nucleosome model of eRNA peaks in super-enhancers.

(A) The normalized nucleosome intensities (MNase-seq signals) in the flanking 500 bp sequence of the 29,828 recurrent super-enhancer eRNA peaks in 27 human tissue/cell types, and five mouse and pig tissues. For each tissue/cell type, the 29,828 1 kb sequences were aligned with the eRNA peaks at the center (0 bp). Each point represents a 10 bp window. The mean number of mapped MNase-seq reads was calculated for all the 29,828 10 bp windows with the same relative positions to the peaks (indicated by the x-axis). The resulting mean signals were normalized into Z-scores for each tissue/cell type, as indicated by the y-axis. (B) The normalized nucleosome intensities on two human sperm samples calculated similarly. (C) A schematic representation of the impact of a dynamic nucleosome on super-enhancer eRNA peaks. See also Figure S4.
Global identification of eRNA loci in super-enhancers
From the above analysis, we made two key observations related to the eRNA loci in super-enhancers: (i) a super-enhancer usually contains multiple eRNA loci generating short eRNA species <100 bp; and (ii) these eRNA loci tend to coincide with well-positioned nucleosomes. We, therefore, generalized our analysis to the whole set of super-enhancers (~377 Mb), which would otherwise suffer greatly from the noise of global transcription background without precise enhancer locus annotation.
We first identified the loci with local maximum RPKM for all possible 140 bp windows in the super-enhancers in each TCGA cancer type, generating a total of >4 million eRNA peak positions. We then calculated the nucleosome signals on the flanking 140 bp for each peak position in 27 MNase-seq datasets (the two sperm samples were excluded). To characterize the major patterns of nucleosome binding, we performed principal component analysis (PCA) on these signals (Figure 5A and S5A-C). The first three principal components (PCs) collectively explained ~85% of the total variations. PC1 effectively reflected the averaged local MNase-seq signal intensity on the flanking 140 bp regions (Figure S5D; Pearson’s R = 0.96; p <2×10−16). PC2 and PC3 were independent of PC1 and from each other (Figure S5E-G). They represented the phase of nucleosome positioning near the eRNA peak. Specifically, PC2 and PC3 divided the 4 million peaks into three groups (shown in different colors in Figure 5A): PC2 represented the relative occupancy of nucleosome up- or down-stream of the peak position (Figure 5B-C) while a negative PC3 indicated a synchronization between nucleosome occupancy and eRNA expression signal (Figure 5D, Figure S5H-I). Thus, a negative PC3 value was a good indicator of an eRNA expression peak aligned with a well-positioned nucleosome, the eRNA locus of our interest. Strikingly, a negative PC3 exhibited a strong correlation with the probability of a common SNP in the eRNA peaks being a GTEx eQTL (R = −0.93; p = 5.2×10−5; Figure 5E), indicating the functional significance of the eRNA peaks coinciding with well-positioned nucleosomes. In contrast, PC1 was a much worse indicator than PC3, emphasizing the importance of nucleosome positioning rather than the affinity of the local sequence to nucleosomes (R = −0.39; p = 0.03; Figure 5F). As true eRNA signals, rather than transcriptional noise, should be synchronized across multiple tissues where they function, we found that PC3 was strongly associated with either the probability of the region being identified as a super-enhancer in the original 86 tissue/cell types (R = −0.89; p = 3.7×10−6; Figure 5G) or the recurrent frequency identified in the 32 TCGA cancer types (R = −0.90; p = 6.7×10−8; Figure 5H).
Figure 5. Identification of eRNA loci in the genome-wide super-enhancer regions.

(A) Principal component analysis (PCA) of nucleosome positioning on the flanking 140 bp sequences around ~4 million loci of local maximum eRNA RPKMs. The color represents the distance between the position with the maximum MNase-seq signal and the position of the local maximum eRNA RPKMs. Only the first 10,000 loci are plotted for convenient visualization. (B, C, and D) The sliding mean of mapped MNase-seq reads in all 27 nucleosome profiles in the flanking 140 bp DNA (each side) were plotted for loci meeting the indicated criterion: PC2<0/PC3>0 (B), PC2>0/PC3>0 (C), or PC3<0 (D). The sequences meeting the indicated criterion were aligned with the loci of the local maximum eRNA RPKMs at the center (0 bp). Each point represents a 10 bp window. The mean number of mapped MNase-seq reads was calculated for all 10 bp windows with the same relative positions to the peaks (indicated by the x-axis). (E, F) The correlation of the PC3 (E) or PC1 (F) quantile with the relative probability for a common SNP in its flanking 20 bp region to be identified as a GTEx eQTL. For loci with indicated PC3/PC1 quantile, this probability was calculated by dividing the number of GTEx eQTLs in the region with the total number of common SNPs (minor allele frequency >20% in 1000 Genome Project) in the region. The resulting probabilities were normalized by dividing them with the minimum value of all quantiles (y-axis). (G, H) The correlation of the PC3 quantile with the frequency for the loci to be identified as a super-enhancer in the original 86 tissue/cell types (G) or with the recurrence frequency as local maximum RPKM in 32 TCGA cancer types (H). (I) The correlation of the relative height of a locus with the relative probability for a common SNP in its flanking 20 bp to be identified as a GTEx eQTL. (J) The correlation of the relative height of a locus with the frequency for the loci to be identified as a super-enhancer in the original 86 tissue/cell types. (K) The distribution of eRNA peaks in super-enhancer regions on chr22 (top panel), with each dot representing one of the ~7,000 peaks on the chromosome (the second panel). A ~660 kb block containing the first five super-enhancers is zoomed in on the third panel. The order of the 162 eRNAs in these regions (fourth panel) and a heatmap representing the pairwise correlation (Spearman’ Rho) among the 162 super-enhancer eRNA peaks in the region across 66 TCGA cancer subtypes (bottom panel). See also Figure S5, S6, and Table S3.
As described above, the eRNA expression peak also tends to form sharp and short transcription pulse ~100 bp in length, which can be measured as the relative peak height, by comparing the peak expression with those of the two “gulfs” ~50 bp away from the peak (Figure 3C). We found this value to be strongly correlated with the probability of a common SNP in the eRNA peaks being a GTEx eQTL signal (R = −0.996; p <2×10−16; Figure 5I) or the probability of the peak region being a super-enhancer in the 86 tissue/cell types (R = 0.86; p = 8.9×10−7; Figure 5J). Therefore, we decided to integrate the eRNA peaks with well-positioned nucleosomes to identify eRNA loci systematically.
Based on permutation analysis of eRNA loci, we developed two criteria to identify candidate eRNA loci by searching the 4 million eRNA peaks for those (i) coincident with well-positioned nucleosomes (PC3 <0), and (ii) with sharp eRNA expression peaks (relative peak height >0.05). We identified >300,000 such eRNA loci (FDR <0.1, permutation analysis) in the ~377 Mb super-enhancer regions (Table S3). This procedure strongly enriched recurrent loci (Figure S5J-K, ranging from ~50,000 to ~120,000 loci per cancer type) that retained high tissue specificity compared to protein-coding genes (Figure S5L). This procedure also identified many more such loci in the super-enhancers than in other regulatory regions or non-regulatory sequences (Figure S5M), consistent with the enrichment of RNA Pol II ChIP-seq signals in the super-enhancers. As visualized on chromosome 22, dense eRNA loci were observed across the super-enhancer regions (Figure 5K, top and middle panels). Interestingly, both heterogeneous and highly co-activating modules were frequently found in genomic neighborhoods. For example, the blue super-enhancer region formed two distinct clusters, whereas a highly complex co-activation pattern was observed across the structure of the green super-enhancer (Figure 5K; bottom panel).
We then evaluated the quality of these eRNA loci in several aspects. First, to confirm if the identified eRNA loci represent the true transcription initiation, rather than transcriptional noise, we analyzed the average enhancer CAGE-seq signal around the eRNA loci (Andersson et al., 2014). Indeed, there was a sharp peak of enhancer transcription initiation signal detected by CAGE-seq at ~60 bp upstream of the eRNA loci on either the Watson or Crick strand (Figure S6A-B). The ~120 bp distance between the two CAGE-seq peaks flanking the eRNA loci is consistent with the role of well-positioned nucleosomes we proposed (Figure 4C). Although the read depth of CAGE-seq data was much lower than that of the RNA-seq data, the flanking CAGE-seq peaks showed an enrichment relative to the genomic background (Figure S6C-D). Despite the local enrichment of CAGE-seq signals near our eRNA loci (Figure S6A-B), it should be emphasized that ~77% of the ~300,000 eRNA loci are at least 2 kb away from any FANTOM-annotated enhancer (Figure S6E), highlighting the discovery power of our approach for novel eRNA loci. Second, we called well-positioned nucleosomes in 15 paired-end MNase-seq samples (Chen et al., 2013) and found >60% of the eRNA loci to be occupied by well-positioned nucleosomes in ≥5 samples studied (Figure S6F), indicating the robustness of this pattern. Taken together, using the key features identified in the core super-enhancers, we systematically identified candidate eRNA loci in the whole set of super-enhancers. These eRNA loci feature well-positioned nucleosomes (as in gene TSS (Jiang and Pugh, 2009)) and are enriched in eQTL associations, suggesting that these eRNA loci are functionally important and represent quantifiable units for studying super-enhancer activities using routine RNA-seq data.
eRNA expression provides extra quantitative power for clinical phenotypes
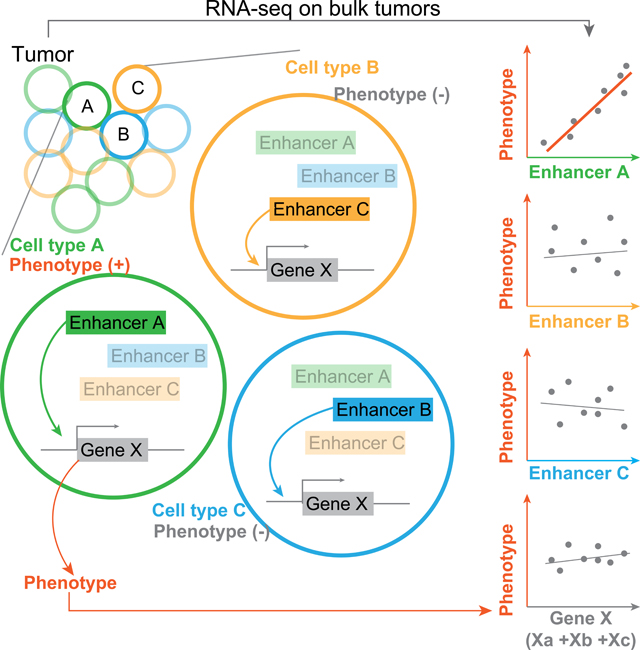
Since the effects of enhancers must ultimately converge on their target genes, one key question is whether and how the eRNA loci we detected can provide additional quantitative power in dissecting genotype-phenotype relationships (Chen et al., 2018c). With a comprehensive catalog of eRNA loci (>300,000), we hypothesize that eRNA signals better explain quantitative traits than gene expression because of the tissue or cell-type specificity of the super-enhancers, which can be illustrated as follows. In a hypothetical case, the cell-type-specific enhancers A, B, and C are regulators of the gene X in cell types A, B, and C, respectively (Figure 6A). However, only the cell type A contributes to a quantitative trait through the activity of gene X. As a result, only the activation level of enhancer A should be strongly associated with the trait, while the predictive power of gene X is compromised by including the expression signals of gene X in cell types B and C (which have no effects on the phenotype of interest). This scenario is particularly relevant in the RNA-seq analysis of clinical tumor samples in two aspects. First, according to a recent CAGE-based study, a much larger proportion of eRNAs (>30%) is highly cell-type-specific compared to mRNAs (<5%). Super-enhancers are even more cell-type specific, and their activation often represents cell lineages in H3K27ac ChIP-seq studies (Hnisz et al., 2013). Second, clinical tissue samples (e.g., solid tumors) usually are a mixture of various cell types (Marusyk et al., 2020). The eRNA peaks identified in this study are mostly restricted to a few cancer types (Figure S5K), distinct from the protein-coding genes in the same dataset (Figure S5L), which are readily detectable in many tissues. Therefore, the expression level of protein-coding genes from bulk RNA-seq data largely reflects the average signals across different cell types within a tumor sample, whereas the eRNA levels likely retain more cell-type-specific signals. As a result, differentially expressed eRNA signals could reasonably outperform differentially expressed mRNAs in quantitative power for complex traits determined by one or a few specific cell types, such as immunotherapy responses and endocrine resistance (Augello et al., 2019; Hanker et al., 2020).
Figure 6. The eRNA loci in super-enhancers provide additional explanatory power for immunotherapeutic response by resolving tumor heterogeneity.

(A) A cartoon model illustrating how cell-type-specific enhancers can increase the explanatory power of quantitative traits by resolving tumor heterogeneity. Left: a hypothetical bulk tumor consists of three different cell types in which enhancer A, B, or C controls the expression of gene X, respectively; and only cell type C contributes to a phenotype of interest. Right: distinct correlation patterns when correlating different eRNA or mRNA signals with the phenotype. (B) The p-value distribution of coding genes through the differential analysis in a cohort of 28 melanomas with different responses to anti-PD1 immunotherapy (q <0.1). (C) The p-value distribution of eRNAs through the differential analysis in the same cohort as in B (q <0.05; ANOVA test). (D) Unsupervised clustering based on the RPKM values of the 164 eRNA loci across the 28 tumors. The RPKMs were normalized by each eRNA peak (row) in the 28 tumors. (E) GSEA results of the 36 genes targeted by at least one GTEx eQTL in the 164 eRNA locus regions (defined as the flanking 50 bp DNA on either side of the eRNA loci). (F) The combined expression level (sum of log2RPKM) of the 8 non-redundant genes in E exhibits prognostic power in a cohort of 42 patients receiving anti-CTLA-4 immunotherapy (p = 0.037; one-sided HF-test). See also Figure S7 and Table S4.
To support the concept, we studied the value of our eRNA loci in predicting tumor response to cancer immunotherapy. In a cohort of 28 melanoma tumors differentially responding to the anti-PD1 immunotherapy (Hugo et al., 2017), the response to which requires the interactions among T cells, tumor cells, and an array of other cells in the microenvironment, we found that none of the ~20,000 coding genes showed significant differential expression (Figure 6B; q <0.1), as originally reported (Hugo et al., 2017). In sharp contrast, when the same statistics were applied to the ~40,000 eRNA loci identified in TCGA melanoma dataset, we detected 164 eRNAs with differential expression at the level of q <0.05 (Figure 6C, Table S4). Interestingly, all of these 164 eRNA loci showed consistent hyperactivation in the fully responding group (Figure 6D), even though they were evenly distributed across the genome (Figure S7A). To understand the biological theme of these eRNA signals, we identified their 36 target genes by eQTLs (as annotated by GTEx) and performed gene set enrichment analysis (GSEA). This small gene set showed significant enrichment for the genes downregulated in exhausted CD8+ T cells and those over-expressed in expanding CD8+ T cells (Figure 6E), suggesting that the activation of the 164 super-enhancer eRNAs (and hence the 36 genes) is important in the functional CD8+ T cells. Consistently, the 164 eRNAs were expressed in the primary T-cells while being largely undetectable in several homogeneous cell lines regardless of tissue of origins (Figure S7B). To further support this finding, the combined expression level of these 164 eRNAs was correlated with a T cell dysfunction gene signature (R = −0.35; n = 310; p = 6.6×10−10; Figure S7C), and a T cell exclusion signature (R = −0.15; n = 310; p = 7×10−3; Figure S7D) obtained from a recent gene-based study where the predictive model was developed based on dozens of cancer patient cohorts (Jiang et al., 2018). Furthermore, the two GSEA gene signatures were associated with patient survival in a cohort of 42 melanoma patients receiving anti-CTLA4 immunotherapy (Van Allen et al., 2015). These results provide a vivid example of how the eRNA signals can provide additional insights beyond mRNA expression analysis by resolving intra-tumor heterogeneity through their cell-type specificity (CD8+ T cell in this case).
eRNA loci are dysregulated and show clinical relevance in cancer
To study super-enhancer dysregulation in human cancers, we first compared the expression levels of the ~300,000 eRNA loci between tumors and normal samples in 12 cancer types with ≥20 tumor-normal sample pairs and observed a global activation of super-enhancers in many cancers (Figure 7A), similar to that of typical enhancers we recently reported (Chen et al., 2018a). Interestingly, a substantial portion of the eRNA loci was affected by the driver events of focal copy-number amplification, ~4-fold more likely than being affected by driver deletion events (Figure 7B). This pattern was in sharp contrast to the contributions of copy-number driver events affecting protein-coding genes, for which focal deletions were ~1.2-fold more likely to occur (Figure 7C). The same pattern held true when recurrent events across multiple cancer types were merged (Figure 7D).
Figure 7. Pan-cancer analysis of the eRNA loci in super-enhancers.

(A) Differential expression of eRNA loci between tumor and normal samples in 12 cancer types with >20 tumor-normal pairs; * p<0.01 (paired t-test). (B) Numbers of driver focal somatic copy number alterations (SCNA) affecting at least one eRNA locus but no protein-coding genes. Driver focal SCNAs were detected by GISTIC. (C) The number of driver focal SCNAs affecting the protein-coding genes. (D) The same analysis as in (B) except that the overlapping focal SCNAs identified in different cancer types were merged. (E) Differential CpG methylations in eRNA loci between tumor and normal samples in 10 cancer types with methylation profiles of >10 tumor-normal pairs available. Hypo/hypermethylation in tumors, compared to normal, is indicated as red/blue. Only significant changes are colored (q<0.01; paired t-test). Probes with absolute changes >20% in at least one cancer type were included in the unsupervised clustering. (F-G) The number of differential expressions for eRNA loci, including significant hypo- (F) or hyper- (G) methylation changes as defined in (E) (>20% and q<0.01). Differential expression changes were determined by paired t-test. (H) The number of eRNA loci with prognostic power on overall survival, progression-free interval, or disease-specific survival in 32 TCGA cancer types (q<0.05; log-rank test).
CpG methylation plays a critical role in controlling its nearby regulatory elements (Skvortsova et al., 2019). We found ~4,000 eRNA loci containing at least one CpG methylation probe of the Human Methylation 450k array used in TCGA project. In these probes, 1,187 (>30%) CpG dinucleotides showed significant changes at the DNA methylation level (Figure 7E; >20% absolute changes; FDR <0.01; paired t-test). These changes can be clearly divided into two clusters (hypo- and hypermethylation) with near consistency across different cancer types (Figure 7E). These methylation changes were associated with the expression changes of the 360 eRNA loci (Figure 7F-G; log2Fold-change >2; FDR <0.05; paired t-test) in the same patients, of which 174 (~50%) events were the deactivation of eRNA loci with hypermethylated CpGs inside (Figure 7G). We observed another 93 activation events on eRNA loci containing hypomethylated CpGs (Figure 7F). These results support that the hypermethylation of within-peak CpGs is an important indicator of super-enhancer deactivation during tumorigenesis (Skvortsova et al., 2019). Finally, we found ~50,000 eRNA loci (or ~62,000 associations) whose expression levels were associated with clinical outcomes, such as patient survival time, in at least one cancer type (Figure 7H), supporting their functional and clinical relevance.
To facilitate the community use of our results, we have built a user-friendly data portal, The Cancer eRNA Atlas (https://bioinformatics.mdanderson.org/public-software/tcea). This data portal provides (1) the detailed annotation of mappable non-coding super-enhancer regions (~377 Mb) surveyed in this study; (2) the details of the core super-enhancer regions (~5 Mb); (3) the expression level (RPKM) of >300,000 super-enhancer transcription units in >10,000 TCGA tumor samples, >9,600 GTEx normal samples, and >900 CCLE cell lines; (4) the super-enhancer eRNA loci and genes associated with responses to immunotherapy; (5) the associations of super-enhancer transcription units with clinical outcome, somatic copy-number alteration, and CpG methylation in TCGA datasets; (6) the 3D eRNA-locus/promoter interactions in >50 ChIA-PET or HiC datasets (Wang et al., 2018), resulting in a strong enrichment of positive eRNA/target-gene co-expression (Figure S7E-H); (7) additional super-enhancer regions: we collected ~350 H3K27ac profiles from Cistrome (Liu et al., 2011) and SEdb (Jiang et al., 2019) and annotated the potential eRNA loci in an additional ~350 Mb putative super-enhancer.
Discussion
We recently showed that cohort-based eRNA expression analysis is powerful in studying cancer mechanisms based on CAGE-seq-defined enhancers (Chen et al., 2018a; Chen et al., 2018b). However, it is difficult to apply a similar strategy to super-enhancers, mostly due to their large size (>10 kb in length). By integrating dynamic nucleosomes with eRNA expression signals from aggregated RNA-seq data, we developed a systematic strategy to identify their eRNA loci, wherein the tissue-specific property of super-enhancers is the key. In a tissue A where the super-enhancer is active, and a motif is required by the TFs, the local nucleosome is recognized and labeled by chromatin modifiers and then intentionally opens to allow the TF-DNA binding; whereas in the majority of tissues where the super-enhancer is silent, a well-positioned nucleosome is necessary to prevent the TF-motif contact and suppress unwanted enhancer activation (He et al., 2010; Zhang et al., 2008). This is why we could observe well-positioned nucleosomes coinciding with the sharp transcriptional peaks when integrating eRNA expression and nucleosome position data across different tissues. An alternative model is that during rapid transcription initiation on enhancers, cells can alter DNA accessibility of well-positioned nucleosomes using ATP-powered chromatin remodelers without changing nucleosome occupation (Mueller et al., 2017). Both models suggest the importance of well-positioned nucleosomes on the TF-motifs in enhancers. Strikingly, the constraint on well-positioned nucleosomes is conserved across a billion years of evolution, indicating its essential role in maintaining the overall transcriptional structure of super-enhancers. In contrast to the eRNA loci we detected, background transcription is a nature of genome organization that can be caused by DNA breathing (Chen et al., 2012), replication (Brar et al., 2012), or repair (Michelini et al., 2017). As a result, the low background transcription would amount to substantial noise in large-size regulatory elements such as super-enhancers. Importantly, this transcriptional noise does not have a protective (well-positioned) nucleosome accompanying it in other tissues, or when the DNA is closed (Figure 5E and I), thus, it is more likely to be associated with fuzzy nucleosomes (Mavrich et al., 2008).
One limitation of our study is that the public, consortium RNA-seq datasets (TCGA or GTEx) used are polyA+ selected, and can only capture a subset of eRNA signals. But the aggregated RNA-seq data across hundreds of individual samples help mitigate this limitation and still allow the detection of a large number of eRNA loci. With the eRNA loci thus defined, the most frequently generated polyA+-selected RNA-seq data can be readily used to characterize super-enhancer activities. We expect to detect more eRNA loci using a similar approach on rRNA-depleted RNA-seq data and will revisit this when a large amount of such RNA-seq data become available.
In summary, our study systematically identified >300,000 eRNA loci in ~377 Mb super-enhancer regions, allowing the possibility to quantify the activation of super-enhancers using RNA-seq data. As demonstrated in the case study of cancer immunotherapy, the eRNA levels can largely retain cell-type-specific signals, whereas the mRNA expression levels by bulk RNA-seq data are more likely to be confounded by different cell types within a tumor sample. Thus, the eRNA loci map defined here will increase the power to explain quantitative traits beyond gene expression, thereby opening a new horizon to investigate the biological functions and potential applications of super-enhancers in various developmental and disease processes.
STAR Methods
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Han Liang (hliang1@mdanderson.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
To facilitate the utilization of the results generated in this study by a broad community, we have provided all the information on our super-enhancer analysis in The Cancer eRNA Atlas (TCeA) data portal that can be accessed at https://bioinformatics.mdanderson.org/public-software/tcea. The supplemental datasets include 5 files. Briefly, Table S1 provides the detailed annotation of mappable non-coding super-enhancer regions (~377 Mb) surveyed in this study. Table S2 provides the details of the core super-enhancer regions (~5 Mb). Table S3 provides the information of >300,000 eRNA loci detected in >10,000 TCGA tumor samples. Table S4 provides the eRNA loci and genes associated with responses to immunotherapy. Table S5 provides the sample information used in this study.
Method Details
Annotation of super-enhancers
We obtained the annotation of super-enhancers in a panel of 86 human tissues and cell types from a previous study (Hnisz et al., 2013). In total, a list of 58,283 genomic regions was identified as super-enhancers in at least one tissue or cell type. This annotation was based on UCSC Hg19. We obtained all the exons under the attribute “ensembl_exon_id” from the GRCH37 ENSEMBL archive (https://grch37.ensembl.org/index.html) using the R package “biomaRt” (Durinck et al., 2009) and removed these exons and their flanking 100 bp sequences from the above-mentioned super-enhancer regions to avoid contamination of transcriptional signals with known genes. We then obtained a human genome benchmark (Zook et al., 2014) from ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/analysis/NIST_union_callsets_06172013/union13callableMQonlymerged_addcert_nouncert_excludesimplerep_excludesegdups_excludedecoy_excludeRepSeqSTRs_noCNVs_v2.18_2mindatasets_5minYesNoRatio.bed.gz, that excluded the genomic regions that were ambiguous for mutation calling or short read mapping. Only the non-coding super-enhancer regions within the genome benchmark regions were considered for further eRNA expression analysis in this study to control for mappability. Following these steps, we generated a list of 65,728 genomic regions of super-enhancers (some of the original 58,283 super-enhancers were divided into multiple regions) for our eRNA expression analysis (Table S1). To identify a set of core super-enhancer regions with activities in multiple tissues and cell types, we divided the above sequences into non-overlapping 10 bp windows and compared all these windows with the original super-enhancer annotations in the 86 tissue and cell types to obtain their tissue specificity information. We obtained 1,531 genomic regions with super-enhancer activities in >20 of the 86 tissue and cell types (Table S2).
Analysis of eRNA and ChIP-seq H3K27ac signals
For the H3K27ac data, we obtained two datasets from ENCODE (n = 50, most of which are normal human tissues and primary cells) (ENCODE Consortium, 2012) and the Cistrome database (n = 90, all of which are cancer cell lines) (Mei et al., 2017). The corresponding RNA-seq data for these samples were obtained from the ENCODE (n = 50) and CCLE (n = 90) database (Barretina et al., 2012), respectively. The identifiers of these samples in the corresponding database are provided in Table S5. Notably, the Cistrome database provides mapped bigwig files in the genome version of UCSC Hg38, and we converted the 1,531 super-enhancer regions from Hg19 to Hg38 using UCSC liftOver (Casper et al., 2018). For the ENCODE ChIP-seq samples, the level of H3K27ac on each super-enhancer was defined as the average fold change over the control as provided in the ENCODE bigwig file (Table S5). We used the average signal in the super-enhancer regions from the ENCODE RNA-seq bigwig files as the readout of eRNA expression. For the Cistrome H3K27ac dataset, we called the signal on each super-enhancer and then normalized it using the average signal across the whole genome to measure its H3K27ac level in a given sample. The corresponding RNA-seq bam files of the 90 cancer cell lines were obtained from CCLE (the bam file ids are provided in Table S5) (Barretina et al., 2012). The RPM of each of the 1,531 regions was calculated as the intensity of its eRNA expression. Since the ENCODE dataset and the Cistrome dataset are very different in terms of genomic coordinates, signal determination, normalization methods, and tissues of origin, we did not combine the two datasets and calculated the Spearman’s Rhos separately to measure the correlation between an eRNA and its local H3K27ac level for each super-enhancer in either dataset (Figure S1A-B). To evaluate the broadness of the positive correlations, we used a one-sided t-test on the Spearman’s Rhos to calculate p values (so that a large negative Rho would not pass the t-test). For each super-enhancer, the p values generated from the two datasets were combined into one using the Fisher’s method (Fisher, 1928) before being subjected to a q-value computation (Figure S1C) (Storey and Tibshirani, 2003). We then estimated the pi0, or the percentage of true null H0 (no positive correlation between the eRNA and H3K27ac) based on the distribution of the resulting combined p-values of the 1,531 super-enhancers using the R package “qvalue” (Figure S1D) (Storey and Tibshirani, 2003), Phison’s LFDR (Phipson, 2013), and Nettleton’s method (Nettleton et al., 2006).
Super-enhancer eRNA expression profiles
For tissue/cancer type (or subtype) level expression profiling, we first combined the 10,004 (including 720 normal samples and 9,284 tumors; see sample information in Table S5) TCGA bam files into merged bam files of 32 cancer types or the 66 cancer subtypes (Cancer Genome Atlas Research et al., 2013). For each of the 10 bp windows on the 1,531 super-enhancer regions, we calculated the RPKM and classified a window as “expressed” in a given cancer type if at least one read with mapping quality >20 was observed in ≥5 samples and >5% of the total samples of that cancer type. The 672 bp long genomic region of chr3:50,265,725–50,266,396 (Hg19) was selected for illustration in Figure 2A since (i) it was identified as a super-enhancer in nearly half (39/86) of the tissue/cell types, and (ii) nearly all of the positions in this region were expressed in >30 cancer types.
To identify eRNA peaks in the core super-enhancer regions, we searched the 1,531 regions, using a window size of 200 bp and a step length of 10 bp, for the local maximum RPKM in each of the 32 cancer types. Two positions of local maximum RPKMs (in different cancer types) were merged if they were <20 bp away from each other. To merge two peaks, the peak with a local maximum value in more cancer types determined the position of the merged peak. If the two positions had the same number of supporting cancer types, the one with a higher RPKM value determined the position of the merged peak. A position identified as the local maximum RPKMs in ≥3 of the 32 cancer types was considered as a transcriptional peak on the core super-enhancer. To develop this cutoff, we removed the reads mapped to the top 10% positions with the highest RPKM in the 5 Mb core super-enhancers (to avoid the bias introduced by positions with extremely high expression levels) and randomly assigned new positions to the other reads. The permutated reads were used as input for peak identification in the same way as described above. We used the 5% quantile of the RPKM of the resultant peaks as a cutoff. Peaks from real data with a lower RPKM value than the cutoff were defined as false identification. The FDR of peaks recurrent in a given number of cancer types were thus calculated. More than 96% of the peaks recurrent in ≥3 of the 32 cancer types showed higher RPKM than the peaks identified using permutated reads (Figure S3A). The 200 bp window size was changed to 400 bp or 600 bp in Figure 3 to assess any potential technical bias. Using this strategy, we generated a list of 29,828 transcriptional peaks in the ~5 Mb region of the 1,531 core super-enhancers (related to Figure 2, 3, and S3). To identify eRNA expression peaks in the general super-enhancer regions (n = 65,728; ~377 Mb, see details in Table S1), we computed the RPKM values of all the non-redundant 10 bp windows. The sequence range for local maximum RPKM search was set to 140 bp as it is the length of a typical nucleosome. A total of 4,355,962 loci were identified as local maximum RPKMs (of the 140 bp sequence) in at least one cancer type. For each locus, we calculated (i) the PC3 using the nucleosome positioning around it as described below and (ii) the relative peak height, which was computed as the local maximum RPKM minus the RPKM at its ± 50 bp loci, whichever was smaller (Figure 3C). If an identified locus had the local maximum RPKM in more than one cancer type, we used the second largest relative peak height of these cancer types for this locus to avoid potential outlier effects. Among the ~4 million loci of local maximum RPKMs, we selected 302,951 loci with PC3 <0 and the relative peak height of >0.05 (in RPKM) as the final super-enhancer eRNA peaks. Notably, two peaks <20 bp away from each other (in different cancer types) were merged as described above. In total, we identified 302,951 loci (Table S3). To estimate the FDR of eRNA location identification with the cutoff of PC3<0 and the peak height >0.05 (in RPKM), we used a similar strategy as that for the 5 Mb core super-enhancer regions. Specifically, we removed the top 10% positions with the highest RPKM values and randomly assigned new positions to the other reads. The permutated reads were used as input for local maximum RPKM identification. The PC3 and the peak height of the resulted positions of local maximum RPKMs were calculated as that for the real data. We identified ~26,000 peaks passing the two criteria in three permutation analysis and estimated the eRNA location identification FDR to be ~0.086 (FDR <0.1). The peak on chromosome 22 was displayed using the R package “karyoploteR”(Gel and Serra, 2017). Chr22 was selected since it is the smallest chromosome and thus provides convenient visualization.
For sample-level expression profiling, the expression level for each of the 302,951 peaks was defined as the RPKM in its flanking 20 bp region. For all the 302,951 super-enhancer peaks, we computed their expression levels in 10,004 TCGA and 9,664 GTEx RNA-seq samples (Consortium, 2017), respectively. For the analysis of rRNA-depleted RNA-seq data, we obtained the data from GEO (GSE69360) (Choy et al., 2015). The raw reads were mapped to the reference genome hg19 using Tophat2.0 with default settings (Trapnell et al., 2012). The expression level of each eRNA was then defined in the same way as for the TCGA eRNA analysis.
Motif discovery and chromatin organization in super-enhancer eRNA peaks
We used the FIMO software (Bailey et al., 2009) with default settings to identify all the DNA motifs annotated by the Mononucleotide human motifs database (Kulakovskiy et al., 2016) in the flanking 200 bp region of the 29,828 core super-enhancer peaks (related to Figure 3D-E). The FIMO outputs with FDR <0.01 were considered as valid motifs. We identified the splicing factor motifs by submitting the DNA sequences of interest to the SFmap online server with default settings (http://sfmap.technion.ac.il/) (Paz et al., 2010).
For nucleosome analysis, we collected a panel of 29 MNase-seq profiles of various human tissues and cell types (including two sperm samples) (ENCODE Consortium, 2012; Descostes et al., 2014; Diermeier et al., 2014; Du et al., 2017; Gaffney et al., 2012; Gaidatzis et al., 2014; Gomez et al., 2016; Hammoud et al., 2009; Hu et al., 2011; Jiang et al., 2018; Jung et al., 2012; Kelly et al., 2012; Kfir et al., 2015; Lavender et al., 2016; Shah et al., 2018; West et al., 2014; Yazdi et al., 2015; Zhang et al., 2016). The raw reads for all these samples were downloaded from the SRA database (the SRR run IDs are provided in Table S5). Long reads were trimmed to 50 bp to make the samples more comparable in terms of mappability. All reads were mapped onto the human genome hg19 using Bowtie2 with default settings (Langmead and Salzberg, 2012). Only reads with mapping quality >10 were kept for further analysis. For a read mapped to the genomic locus of X, we extended the read and considered the region between X+40 to X+110 as being occupied by a nucleosome confidently (Zhang et al., 2008). The normalized read number (Z-score) on each genomic locus was considered as the readout of nucleosome occupancy (related to Figure 4 and S3). For evolutionary analysis, we converted the genomic locus on hg19 to mouse (UCSC genome version mm9) and pig (UCSC genome version susScr3) using the UCSC liftOver software (Jiang et al., 2018). For the PCA, we computed, across 27 human samples (the two sperms were excluded), the mean nucleosome signal of all 10 bp windows within the flanking 140 bp region relative to each of the ~4 million loci of local maximum RPKM, generating a matrix of 29 × 4,355,962 as the input. The first three components, PC1, PC2, and PC3, explained 52.3%, 18.2%, and 13.3% of the total variation (summed up to be 83.9%), respectively, and were kept for further analysis (related to Figure 5 and S4). Although the PCA included all the 4 million peaks, only the first 10,000 are displayed in Figure 5A and Figure S4A-C for convenient visualization. The GTEx eQTLs were obtained from the GTEx data portal (GTEx_Analysis_v7_eQTL.tar.gz) under the link https://gtexportal.org/home/datasets (GTEx Consortium, 2017). For each quantile in Figure 5E, F and I, we defined the probability for a common SNP to be a GTEx eQTL as the number of GTEx eQTLs in the loci’s flanking 20 bp divided by the total number of common SNPs (minor allele frequency >20% in 1000 Genome Project dataset) in the same regions. For the analysis of CAGE-seq signal flanking the eRNA loci, we collected 266 CAGE-seq datasets (in ctss format) of human cell lines from http://fantom.gsc.riken.jp/5/datafiles/latest/basic/human.cell_line.hCAGE/, and 512 human primary cells from http://fantom.gsc.riken.jp/5/datafiles/latest/basic/human.primary_cell.hCAGE/ as listed in Table S5. Since the ctss files contained the 5′-end of CAGE-seq reads from both FANTOM promoters and enhancers, we selected those from FANTOM eRNAs using reads mapped to the FANTOM enhancers (http://fantom.gsc.riken.jp/5/datafiles/latest/extra/Enhancers/human_permissive_enhancers_phase_1_and_2.bed.gz) and their flanking 200 bp regions. The resultant reads were compared with the 302,951 eRNA loci identified in this study, and the relative distances of each read to all of its nearby eRNA loci (<1 kb) were calculated. We counted reads within 1 kb distance of two (or more) eRNA loci multiple times for all the eRNA loci. The 302,951 eRNA loci and their flanking 1 kb DNA were then aligned with the loci of the eRNA peak at the center. We then counted the number of reads mapped to all the 10 bp tandem windows within these aligned sequences. Windows with the same relative distance to all the eRNA loci were combined for the calculation of the CAGE-seq signal. The relative CAGE-seq signal was then defined as the number of reads in a given window normalized by the average number of all 10 bp windows in the aligned sequences (Figure S5A-B).
For 15 (out of 29) non-sperm, paired-end MNase-seq profiles, we called individual nucleosomes using DANPOS2.0 (Chen et al., 2013). The pair-end reads were mapped to the reference genome hg19 using Bowtie2 (Langmead and Salzberg, 2012) with default settings. The bowtie results were then converted into bed files before being sorted, using the bamtobed function in bedtools (Quinlan and Hall, 2010). The bed files were input to DANPOS2.0 using danpos.py with default settings. We defined an eRNA locus to overlap with a nucleosome if the “center” parameter from the DANPOS2.0 result was within the ± 20 bp region of that eRNA locus (Figure S6G).
eRNA expression analysis for tumor response to immunotherapy
We obtained the raw reads of 28 melanoma tumors receiving anti-PD1 immunotherapy from the SRA database (see the SRR IDs in Table S5)(Hugo et al., 2017). The reads were mapped to the human genome (UCSC hg19) using Bowtie2 with default settings (Langmead and Salzberg, 2012). Only reads with mapping quality >20 were kept for further analysis. We found 37,651 super-enhancer eRNA loci identified in the TCGA melanoma cancer type (SKCM) with detectable expression in ≥14 (50%) of the 28 tumors. Each of these eRNAs was subjected to ANOVA test among the three groups with differential responses to the anti-PD1 immunotherapy. The p-values were converted to q-values to adjust for multiple comparisons. The 164 eRNA loci with q <0.05 are provided in Table S4 and displayed using the R package “karyoploteR” (Gel and Serra, 2017). The gene-level expressions of these 28 tumors were obtained from the original study. We found 36 GTEx eQTLs on the 164 eRNA loci associated with 36 genes (Table S4). There were no common SNPs/GTEx eQTLs within the other 128 eRNA loci, and thus, they were not included in the enrichment analysis. The 36 downstream genes of these eQTLs were subjected to GSEA using the GSEA online server (http://software.broadinstitute.org/gsea/index.jsp) with default settings (Subramanian et al., 2005). The TIDE scores of the 310 TCGA SKCM tumors for immunotherapy response prediction were obtained from http://tide.dfci.harvard.edu/ (Jiang et al., 2018). For the cohort of 42 patients receiving anti-CTLA-4 immunotherapy, raw reads were obtained from the SRA database (see the SRR IDs in Table S5) (Van Allen et al., 2015). The reads were mapped to the human genome (UCSC hg19) using Bowtie2 with default settings. Only reads with mapping quality >20 were kept for further analysis. The log2RPKM of the 8 genes selected from GSEA were calculated and summed up as a combined score for CD8+ T cell functionality. To test whether this score was associated with better survival in the 42 patients, we used the R package “FHtest” to perform a one-sided FH-test on its effect on the one-year survival rate (Oller and Langohr, 2017).
Integrative analysis of eRNA loci with other TCGA molecular and clinical data
For somatic copy-number analysis, we surveyed a list of 138,781 focal somatic copy-number alterations (SCNA) identified with the GISTIC software with an FDR <0.05 (Mermel et al., 2011) by the TCGA Pan-Cancer analysis consortium (Cancer Genome Atlas Research et al., 2013). We considered SCNAs without overlaps with any genes (or ncRNA) annotated by the Human GENCODE database v18 (https://www.gencodegenes.org/human/releases.html) as non-coding driver events (Frankish et al., 2018). We found 3,678/1,047 non-coding amplification/deletion SCNAs involving at least one eRNA locus identified in this study. A full list of these events is provided in our data portal.
For CpG DNA methylation analysis, we selected 3,919 eRNA loci containing at least one CpG probe in the Human Methylation 450K array used in TCGA project. A total of 8,430 TCGA samples have both methylation and super-enhancer peak expression data available. This 3,919×8,430 matrix is provided in our data portal. For differential CpG methylation analysis, we considered the 10 cancer types with >10 tumor-normal paired samples and used paired t-test to determine the significance of the methylation changes (ranging from 0% to 100%) between the normal and tumor samples. A CpG methylation change with an FDR <0.01 and an absolute change >20% was considered as a significant hit.
For prognostic analysis, we surveyed only (i) the eRNA loci annotated in a given cancer type (Table S3), and (ii) with detectable expression in >10 samples and >10% of the total samples in that cancer type. For each loci, we used the Cox regression coefficient to measure the association between its expression (determined as either group, RPKM, or log2RPKM) and clinical outcomes (measured as either overall survival [OS], disease-specific survival [DSS], or progression-free interval [PFI]) (Liu et al., 2018). The expression level “group” was a binary parameter generated by dividing the patients into two groups according to the RPKM of the peak of interest, with the lower half (RPKM ≤ median) assigned as 0 and the higher half (RPKM >median) assigned as 1. The resulting p-values were converted into q-values to correct for multiple comparisons (Storey and Tibshirani, 2003). A list of 49,849 eRNA loci associated with any of the clinical outcomes (q <0.05) is provided in our data portal.
Analysis of eRNA-locus/gene co-expression and 3D chromatin interactions
We calculated the co-expression patterns between each eRNA-gene pair across the 32 cancer types and selected those that were consistently co-expressed/reversed in at least 3 cancer types. In each cancer type, we required a p-value with Bonferroni correction to be <0.01 and an absolute Spearman’s Rho to be >0.3. We observed ~161 million eRNA-gene pairs meeting these criteria, among which 72.1% were positively co-expressed. We applied the same cutoffs to the protein-coding genes across the 32 cancer types to identify ~63 million gene-gene interactions, of which about half (55%) were positive correlations. From 3D Genome Browser (Wang et al., 2018), we then obtained the HiC chromatin loops calculated by Peakachu (https://github.com/tariks/peakachu) from 56 HiC datasets. We compared the HiC loops with the co-expressed eRNA-gene pairs and selected 32,298 pairs connected by at least one loop in any of the 56 HiC profiles, of which 96.6% were positively co-expressed (Figure S7D-H).
Supplementary Material
Table S1. The annotation of 65,728 super-enhancer regions subjected to RNA-seq analysis, Related to Figure 1
Table S2. The annotation of 1,531 core super-enhancers, Related to Figure 1
Table S3. The information of 302,951 super-enhancer eRNA loci in the TCGA dataset (BED format), Related to Figure 5
Table S4. The super-enhancer eRNA loci and genes associated with responses to immunotherapy, Related to Figure 6
Table S5. Annotation of datasets used in this study, Related to STAR Methods.
Highlights.
Super-enhancers contain discrete eRNA loci featured by sharp eRNA peaks
Expression of such eRNA loci is regulated by dynamic, well-positioned nucleosomes
eRNA signals confer explanatory power on quantitative traits beyond gene expression
Super-enhancer activities are dysregulated in cancer by diverse mechanisms
Acknowledgments
This study was supported by grants from the U.S. National Institutes of Health (U24CA209851, U01CA217842, P50CA221703 to H.L., and P30CA016672), an MD Anderson Faculty Scholar Award (to H.L.), and the Lorraine Dell Program in Bioinformatics for Personalization of Cancer Medicine (to H.L.). We thank Kamalika Mojumdar, Mei-Ju May Chen and Jun Li for assistance.
Footnotes
Declaration of Interests
H.L. is a shareholder and scientific advisor to Precision Scientific Ltd.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, Ambati CR, Amin SB, Gu B, Chen TY, et al. (2020). KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 37, 599–617 e597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. (2014). An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augello MA, Liu D, Deonarine LD, Robinson BD, Huang D, Stelloo S, Blattner M, Doane AS, Wong EWP, Chen Y, et al. (2019). CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell 35, 817–819. [DOI] [PubMed] [Google Scholar]
- Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R, et al. (2018). A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553, 515–520. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, and Schaffner W (1981). Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta J, and Morillon A (2009). Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep 10, 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahma S, and Henikoff S (2019). Epigenome Regulation by Dynamic Nucleosome Unwrapping. Trends Biochem Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, and Weissman JS (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper J, Zweig AS, Villarreal C, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Karolchik D, et al. (2018). The UCSC Genome Browser database: 2018 update. Nucleic Acids Res 46, D762–D769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino RR, and Stark A (2018). Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev 32, 202–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Cancer Genome Atlas Research, N., and Liang H (2018a). A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 173, 386–399 e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Li C, Zhou Z, and Liang H (2018b). Fast-Evolving Human-Specific Neural Enhancers Are Associated with Aging-Related Diseases. Cell Syst 6, 604–611 e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wu CI, and He X (2018c). The Genotype-Phenotype Relationships in the Light of Natural Selection. Mol Biol Evol 35, 525–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, and Li W (2013). DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Chen Z, Chen H, Su Z, Yang J, Lin F, Shi S, and He X (2012). Nucleosomes suppress spontaneous mutations base-specifically in eukaryotes. Science 335, 1235–1238. [DOI] [PubMed] [Google Scholar]
- Choy JY, Boon PL, Bertin N, and Fullwood MJ (2015). A resource of ribosomal RNA-depleted RNA-Seq data from different normal adult and fetal human tissues. Sci Data 2, 150063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, and Natoli G (2010). A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 8, e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descostes N, Heidemann M, Spinelli L, Schuller R, Maqbool MA, Fenouil R, Koch F, Innocenti C, Gut M, Gut I, et al. (2014). Tyrosine phosphorylation of RNA polymerase II CTD is associated with antisense promoter transcription and active enhancers in mammalian cells. Elife 3, e02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diermeier S, Kolovos P, Heizinger L, Schwartz U, Georgomanolis T, Zirkel A, Wedemann G, Grosveld F, Knoch TA, Merkl R, et al. (2014). TNFalpha signalling primes chromatin for NF-kappaB binding and induces rapid and widespread nucleosome repositioning. Genome Biol 15, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Liu Z, Cao X, Chen X, Chen Z, Zhang X, Zhang X, and Jiang C (2017). Nucleosome eviction along with H3K9ac deposition enhances Sox2 binding during human neuroectodermal commitment. Cell Death Differ 24, 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA (1928). Statistical methods for research workers, 2d edn (Edinburgh, London,: Oliver and Boyd; ). [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2018). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, and Pritchard JK (2012). Controls of nucleosome positioning in the human genome. PLoS Genet 8, e1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D, Burger L, Murr R, Lerch A, Dessus-Babus S, Schubeler D, and Stadler MB (2014). DNA sequence explains seemingly disordered methylation levels in partially methylated domains of Mammalian genomes. PLoS Genet 10, e1004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gel B, and Serra E (2017). karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33, 3088–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez NC, Hepperla AJ, Dumitru R, Simon JM, Fang F, and Davis IJ (2016). Widespread Chromatin Accessibility at Repetitive Elements Links Stem Cells with Human Cancer. Cell Rep 17, 1607–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GTEx. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, and Cairns BR (2009). Distinctive chromatin in human sperm packages genes for embryo development. Nature 460, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanker AB, Sudhan DR, and Arteaga CL (2020). Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 37, 496–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al. (2010). Nucleosome dynamics define transcriptional enhancers. Nat Genet 42, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39, 311–318. [DOI] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Schones DE, Cui K, Ybarra R, Northrup D, Tang Q, Gattinoni L, Restifo NP, Huang S, and Zhao K (2011). Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res 21, 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2017). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 168, 542. [DOI] [PubMed] [Google Scholar]
- Jiang C, and Pugh BF (2009). Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Qian F, Bai X, Liu Y, Wang Q, Ai B, Han X, Shi S, Zhang J, Li X, et al. (2019). SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res 47, D235–D243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, and Felsenfeld G (2009). H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet 41, 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung I, Kim SK, Kim M, Han YM, Kim YS, Kim D, and Lee D (2012). H2B monoubiquitylation is a 5’-enriched active transcription mark and correlates with exon-intron structure in human cells. Genome Res 22, 1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, and Jones PA (2012). Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 22, 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfir N, Lev-Maor G, Glaich O, Alajem A, Datta A, Sze SK, Meshorer E, and Ast G (2015). SF3B1 association with chromatin determines splicing outcomes. Cell Rep 11, 618–629. [DOI] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, and Gray JM (2015). Enhancer RNAs: a class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb Perspect Biol 7, a018622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, Vorontsov IE, Yevshin IS, Soboleva AV, Kasianov AS, Ashoor H, BaAlawi W, Bajic VB, Medvedeva YA, Kolpakov FA, and Makeev VJ (2016). HOCOMOCO: expansion and enhancement of the collection of transcription factor binding sites models. Nucleic Acids Res 44, D116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. (2013). Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavender CA, Cannady KR, Hoffman JA, Trotter KW, Gilchrist DA, Bennett BD, Burkholder AB, Burd CJ, Fargo DC, and Archer TK (2016). Downstream Antisense Transcription Predicts Genomic Features That Define the Specific Chromatin Environment at Mammalian Promoters. PLoS Genet 12, e1006224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Daujat S, and Schneider R (2016). Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet 32, 42–56. [DOI] [PubMed] [Google Scholar]
- Levings PP, and Bungert J (2002). The human beta-globin locus control region. Eur J Biochem 269, 1589–1599. [DOI] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. (2013). Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. (2011). Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, Wang X, Erkek S, Federation A, Song A, Lee C, et al. (2018). Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 553, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Janiszewska M, and Polyak K (2020). Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 37, 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, and Pugh BF (2008). A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S, Qin Q, Wu Q, Sun H, Zheng R, Zang C, Zhu M, Wu J, Shi X, Taing L, et al. (2017). Cistrome Data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res 45, D658–D662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Leveille N, Kalluri R, et al. (2013). eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 49, 524–535. [DOI] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini F, Pitchiaya S, Vitelli V, Sharma S, Gioia U, Pessina F, Cabrini M, Wang Y, Capozzo I, Iannelli F, et al. (2017). Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat Cell Biol 19, 1400–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirny LA (2010). Nucleosome-mediated cooperativity between transcription factors. Proc Natl Acad Sci U S A 107, 22534–22539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B, Mieczkowski J, Kundu S, Wang P, Sadreyev R, Tolstorukov MY, and Kingston RE (2017). Widespread changes in nucleosome accessibility without changes in nucleosome occupancy during a rapid transcriptional induction. Genes Dev 31, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakawa Y, Yoshihara M, Kawaji H, Nishikawa M, Zayed H, Suzuki H, Fantom C, and Hayashizaki Y (2016). Enhanced Identification of Transcriptional Enhancers Provides Mechanistic Insights into Diseases. Trends Genet 32, 76–88. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, and Snyder M (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettleton D, Hwang JTG, Caldo RA, and Wise RP (2006). Estimating the number of true null hypotheses from a histogram of p values. J Agr Biol Envir St 11, 337–356. [Google Scholar]
- Oller R, and Langohr K (2017). FHtest: An R Package for the Comparison of Survival Curves with Censored Data. J Stat Softw 81, 1–25. [Google Scholar]
- Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Program NCS, et al. (2013). Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A 110, 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz I, Akerman M, Dror I, Kosti I, and Mandel-Gutfreund Y (2010). SFmap: a web server for motif analysis and prediction of splicing factor binding sites. Nucleic Acids Res 38, W281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Bickmore W, Dean A, Nobrega MA, and Bejerano G (2013). Enhancers: five essential questions. Nat Rev Genet 14, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B (2013). Empirical Bayes Modelling of Expression Profiles and Their Associations. PhD Thesis, University of Melbourne http://repository.unimelb.edu.au/10187/17614. [Google Scholar]
- Pott S, and Lieb JD (2015). What are super-enhancers? Nat Genet 47, 8–12. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, and Henikoff S (2016). Transcriptional Regulators Compete with Nucleosomes Post-replication. Cell 165, 580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Maqbool MA, Yahia Y, El Aabidine AZ, Esnault C, Forne I, Decker TM, Martin D, Schuller R, Krebs S, et al. (2018). Tyrosine-1 of RNA Polymerase II CTD Controls Global Termination of Gene Transcription in Mammals. Mol Cell 69, 48–61 e46. [DOI] [PubMed] [Google Scholar]
- Shin HY (2018). Targeting Super-Enhancers for Disease Treatment and Diagnosis. Mol Cells 41, 506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvortsova K, Masle-Farquhar E, Luu PL, Song JZ, Qu W, Zotenko E, Gould CM, Du Q, Peters TJ, Colino-Sanguino Y, et al. (2019). DNA Hypermethylation Encroachment at CpG Island Borders in Cancer Is Predisposed by H3K4 Monomethylation Patterns. Cancer Cell 35, 297–314 e298. [DOI] [PubMed] [Google Scholar]
- Storey JD, and Tibshirani R (2003). Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda DY, Spisak S, Seo JH, Bell C, O’Connor E, Korthauer K, Ribli D, Csabai I, Solymosi N, Szallasi Z, et al. (2018). A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 174, 422–432 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DM, and Parker R (2007). Cytoplasmic decay of intergenic transcripts in Saccharomyces cerevisiae. Mol Cell Biol 27, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Song F, Zhang B, Zhang L, Xu J, Kuang D, Li D, Choudhary MNK, Li Y, Hu M, et al. (2018). The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol 19, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasson T, and Hartemink AJ (2009). An ensemble model of competitive multi-factor binding of the genome. Genome Res 19, 2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Cook A, Alver BH, Stadtfeld M, Deaton AM, Hochedlinger K, Park PJ, Tolstorukov MY, and Kingston RE (2014). Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat Commun 5, 4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi PG, Pedersen BA, Taylor JF, Khattab OS, Chen YH, Chen Y, Jacobsen SE, and Wang PH (2015). Nucleosome Organization in Human Embryonic Stem Cells. PLoS One 10, e0136314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Li Y, Kulik M, Tiedemann RL, Robertson KD, Dalton S, and Zhao S (2016). Nucleosome positioning changes during human embryonic stem cell differentiation. Epigenetics 11, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shin H, Song JS, Lei Y, and Liu XS (2008). Identifying positioned nucleosomes with epigenetic marks in human from ChIP-Seq. BMC Genomics 9, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, and Salit M (2014). Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol 32, 246–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The annotation of 65,728 super-enhancer regions subjected to RNA-seq analysis, Related to Figure 1
Table S2. The annotation of 1,531 core super-enhancers, Related to Figure 1
Table S3. The information of 302,951 super-enhancer eRNA loci in the TCGA dataset (BED format), Related to Figure 5
Table S4. The super-enhancer eRNA loci and genes associated with responses to immunotherapy, Related to Figure 6
Table S5. Annotation of datasets used in this study, Related to STAR Methods.
Data Availability Statement
To facilitate the utilization of the results generated in this study by a broad community, we have provided all the information on our super-enhancer analysis in The Cancer eRNA Atlas (TCeA) data portal that can be accessed at https://bioinformatics.mdanderson.org/public-software/tcea. The supplemental datasets include 5 files. Briefly, Table S1 provides the detailed annotation of mappable non-coding super-enhancer regions (~377 Mb) surveyed in this study. Table S2 provides the details of the core super-enhancer regions (~5 Mb). Table S3 provides the information of >300,000 eRNA loci detected in >10,000 TCGA tumor samples. Table S4 provides the eRNA loci and genes associated with responses to immunotherapy. Table S5 provides the sample information used in this study.