

Abstract
Oncolytic viruses (OVs) are novel anti-tumor agents with the ability to selectively infect and kill tumor cells while sparing normal tissue. Beyond tumor cytolysis, OVs are capable of priming an anti-tumor immune response via lysis and cross-presentation of locally expressed endogenous tumor antigens, acting as an “endovaccine.” The effectiveness of OVs, similar to other immunotherapies, can be hampered by an immunosuppressive tumor microenvironment. In this study, we modified a previously generated oncolytic herpes simplex virus (oHSV) retargeted to the human HER2 (hHER2) tumor molecule and encoding murine interleukin-12 (mIL-12), by insertion of a second immunomodulatory molecule, murine granulocyte-macrophage colony-stimulating factor (mGM-CSF), to maximize therapeutic efficacy. We assessed the efficacy of this double-armed virus (R-123) compared to singly expressing GM-CSF and IL-12 oHSVs in tumor-bearing mice. While monotherapies were poorly effective, combination with α-PD1 enhanced the anti-tumor response, with the highest efficacy of 100% response rate achieved by the combination of R-123 and α-PD1. Efficacy was T cell-dependent, and the induced immunity was long lasting and able to reject a second contralateral tumor. Importantly, systemic delivery of R-123 combined with α-PD1 was effective in inhibiting the development of tumor metastasis. As such, this approach could have a significant therapeutic impact paving the way for further development of this platform in cancer immunotherapy.
Keywords: oncolytic virus, endovaccine, cancer, cytokines, immune checkpoint, retargeted Herpes virus, IL-12, GM-CSF
Graphical Abstract
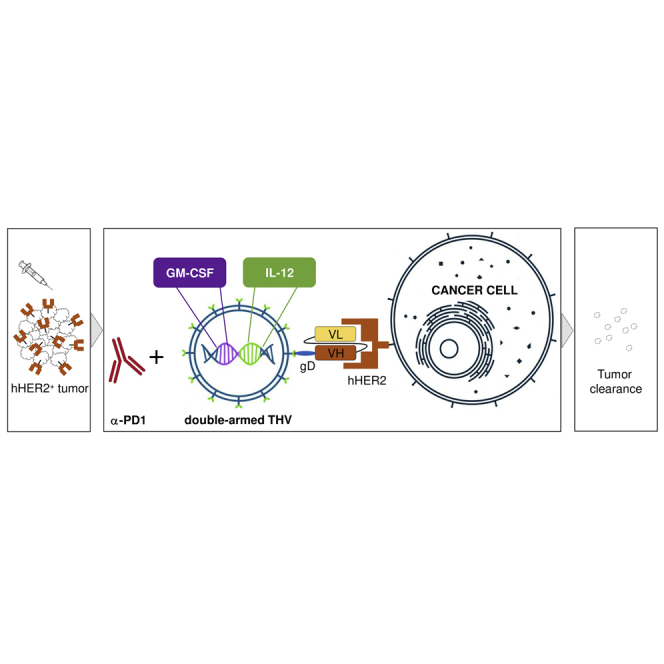
Fully virulent tumor retargeted HSV oncolytic viruses (THVs) are novel immunotherapeutic agents with increased specificity, safety, and potency. De Lucia et al. propose the use of a hHER2 THV expressing IL-12 and GM-CSF as a strategy to potentiate anti-tumor efficacy in combination with anti-PD1, opening future perspectives for local and systemic treatment.
Introduction
Oncolytic virotherapy is emerging as a promising approach for the treatment of different cancer types.1,2 This approach takes advantage of the intrinsic ability of viruses to infect, replicate, and kill the target cells and at the same time to spread to other cells. The anti-tumor activity of oncolytic viruses (OVs) is not only a mere consequence of the direct cytopathic effect of the virus, but it is mainly linked to the induction of an immune response against the tumor. Because of the combined tumor killing via oncolysis and the ability to potently stimulate multiple arms of the immune system, OVs can be considered in situ tumor vaccines (“endovaccines”). The key desirable features of any OV are specificity, safety, and potency. Specificity is usually conferred by attenuating mutations, which cause preferential replication of the virus in tumor cells and not in healthy cells. This is, for example, the case of the talimogene laherparepvec (T-VEC), a type I herpes simplex virus (HSV), currently the only US Food and Drug Administration (FDA)-approved OV for the local treatment of metastatic melanoma.3,4 T-VEC is an attenuated HSV deleted of ICP34.5, a gene that plays a critical role in viral replication and in overcoming the host anti-viral responses.5 In normal cells, activation of the double-stranded RNA-dependent protein kinase PKR is one of the most effective cellular responses to block virus propagation. ICP34.5 protein overcomes the anti-viral responses by blocking PKR and thus allowing for viral replication. In many cancer cells, the PKR pathway is not active. Therefore, HSV lacking ICP34.5 can preferentially replicate in cancer cells and not in healthy cells.6 However, effective replication is not guaranteed in all tumor cells since some may still have an active PKR pathway. An alternative strategy to achieve cancer selectivity, independently of the intrinsic defects of tumor cells, consists in detargeting the viruses from their natural receptors and retargeting viral tropism to cancer-specific receptors via an encoded single-chain variable fragment (scFv) expressed on the surface of the virus.7,8 Such modifications preserve the full lytic capacity of wild-type (WT) HSV and overcome the limits of virus attenuation, hence enhancing potency in all cancer cells, in addition to gaining specificity. Most importantly, because retargeted oHSVs infect no other cells than the specifically targeted tumor cells, their safety profile is very good. Our previous preclinical work has demonstrated that systemic administration of HER2-retargeted HSV is safe in nude mice, by virtue of viral surface modifications causing tropism detargeting from the natural receptors, and retargeting to HER2.9 More recently, we confirmed the good safety profile also in immunocompetent mice.10
A key feature for a successful OV-based therapy is the capacity to induce an immunological response. OVs function as a “kick start” for an anti-tumor response. However, one of the major obstacles for effective T cell function is posed by the highly immunosuppressive tumor microenvironment (TME). OVs can counteract immunosuppression by encoding several immunostimulatory molecules, including checkpoint inhibitors, cytokines, and chemokines, to restore a more favorable inflammatory TME and potentiating the immunological response.11 A number of studies have indeed shown that OVs genetically armed with immunostimulatory molecules exert more robust therapeutic effects than do their non-engineered counterparts.12, 13, 14 The clinically approved HSV (T-VEC) also encodes for a stimulatory factor (granulocyte-macrophage colony-stimulating factor [GM-CSF]) to enhance the anti-tumor response by local recruitment of dendritic cells for antigen presentation.15 Despite the great therapeutic potential, OVs are still not powerful enough, especially in the situation of scarcely immunogenic tumors or in a highly suppressive microenvironment. Combinations of OVs and immune checkpoint blockade therapy have strong synergistic effects. In patients, combined treatment of T-VEC with ipilimumab or pembrolizumab resulted in higher efficacy than did either therapy alone.16,17 In this study, we propose the use of a next-generation targeted herpes virus (THV), which combines the following features: (1) WT backbone for efficient replication and killing of any tumor cells, (2) detargeting from natural receptors for improved safety, (3) retargeting to the tumor-specific antigen human HER2 (hHER2) to gain cancer specificity, and (4) an improved potency by encoding two immunomodulators (GM-CSF and IL-12) for induction of an effective adaptive immunological response. By using a stringent mouse model of tumors non-responsive to α-PD1 treatment and a stringent therapeutic setting of advanced large tumors, we showed that intratumoral THV treatment turned α-PD1 non-responsive tumors to responsive. The highest efficacy was achieved when using the “armed” virus. Intravenous delivery (i.v.) has the potential to reach both the primary tumor and metastatic lesions even when they are not clinically detectable. By exploiting the safety improvements gained with the retargeting to tumor cells of a not attenuated virus,9,10 we explored systemic treatment with THV. We demonstrated that a virus with such features is suitable for systemic delivery. The THV can target the metastatic tumor masses after i.v. injection, exerting a significant anti-tumor activity, also in a contest of pre-existing neutralizing immunity against HSV. This work establishes the proof of principle that immunologically potent THVs armed with multiple cytokines can cure large established tumors when combined with α-PD1 and become effective upon local and systemic delivery.
Results
Generation of IL-12- and GM-CSF-Armed hHER2-Retargeted oHSV
By using a hHER2-retargeted oHSV, we have recently demonstrated higher anti-tumor efficacy when the virus is armed with murine interleukin-12 (mIL-12) relative to the unarmed virus.10 In an effort to further enhance tumor cytotoxicity and improve the immune effector response, we have engineered the hHER2-retargeted IL-12-armed oHSV R-11518 to express a further immunostimulatory cytokine, GM-CSF. IL-12 and GM-CSF were respectively inserted into the US1–US2 intergenic region and between the UL26 and UL27 intergenic region. The resulting double-armed virus, named R-123, was compared to the single-armed IL-12 virus (R-115), the single-armed GM-CSF virus (R-121), and to the unarmed counterpart (R-113) (Figure 1A). The R-121 virus was obtained by inserting the GM-CSF-encoding cassette into the UL26–UL27 intergenic region.
Figure 1.

Schematic and In Vitro Characterization of Armed hHER2-Retargeted THVs
(A) Representation of THV genomes. R-113, R-115, R-121, and R-123 are de-targeted from HSV natural receptors through a deletion of the aa 6–38 in gD, replaced with the scFv to HER2 for retargeting. R-115 carries the mIL-12 gene in the US1–US2 intergenic region, R-121 carries the mGM-CSF gene in the UL26–UL27 intergenic region, while R-123 carries both in the same genome positions. (B) SK-OV-3 cells were infected at an MOI of 0.1 with R-113, R-115, R-121, or R-123. mIL-12 or mGM-CSF production was quantified in the supernatant by ELISA after 24 and 48 h of infection. The results are presented as the mean of two independent experiments + SEM. (C) Viral yield of R-113, R-115, R-121, and R-123. Titration was performed in SK-OV-3 after 24 and 48 h of infection of SK-OV-3 cells. Data are shown as the mean of three independent experiments + SEM. Statistics were generated using an unpaired Mann-Whitney nonparametric test. ns, not significant.
To confirm transgene expression and production of the two encoded cytokines, supernatants from virus-infected hHER2+ cell cultures (SK-OV-3) were analyzed by enzyme-linked immunosorbent assay (ELISA) 24 and 48 h post-infection. The amounts of mIL-12 and GM-CSF produced by double-armed virus versus both single-armed viruses were very similar, indicating that the double insertion of the two transgenes did not affect the expression (Figure 1B). The four viruses R-113, R-115, R-121, and R-123 were finally compared in terms of viral growth and production by measuring the virus yield as plaque-forming units (PFU)/mL upon infection of the HER2-expressing SK-OV-3 cells. The average yield measured was found to be similar (Figure 1C).
Monotherapy with Cytokine-Armed THVs Is Not Sufficiently Effective to Eradicate Large Established Tumors
To evaluate the anti-tumor activity of the hHER2-retargeted oHSVs, we made use of a stably transduced Lewis lung carcinoma murine cell line expressing hHER2 (HER2-LLC1) for in vivo studies.10 The syngeneic C57BL/6 mice transgenic for hHER2 were used as the host mouse model, as no immune responses are generated against the transgenic product upon tumor implantation.10,19 By using the very same mouse model, we have demonstrated effective and superior efficacy of retargeted R-115 oHSV armed with IL-12 versus the unarmed counterpart in a therapeutic setting in which the treatment was performed 3 or 10 days after tumor implantation, considered early and late treatments, respectively.10 In order to be more stringent, we used for this study an advanced setting consisting of mice all bearing large tumors at the beginning of treatment. We have previously shown that the effectiveness of the immunotherapies can be better evaluated in a setting of advanced disease and that some therapies that are effective upon early treatments might fail in situations of advanced disease, given the highly immunosuppressive TME.20 Therefore, in the current study, mice were challenged with a subcutaneous (s.c.) injection of HER2-LLC1 cells. Tumors were allowed to grow until they reached an average volume of ≈110 mm3, in order to recapitulate a late therapeutic setting of large tumors. At this stage (day 0), treatment with R-113, R-115, R-121, or R-123 started. The THVs were injected intratumorally every 2–3 days for a total of five injections at a dose of 108 PFU each (Figure 2A). In this situation, the unarmed R-113 and the single-armed R-121 or R-115 virus had very limited efficacy when administered alone (10%–20%) (Figure 2B). A trend versus a better anti-tumor response was observed when tumor-bearing mice were treated with the double-armed R-123 virus, which elicited tumor regression in about 40% of the animals (Figure 2B).
Figure 2.

Treatment with THVs as a Stand-Alone Is Poorly Effective in a Therapeutic Setting of Large Established Tumors
(A) Schematic of the experimental setting. HER2-LLC1 tumor cells were implanted into hHER2-transgenic/tolerant mice by s.c. injection. Treatments started in randomized established tumors (mean = 110 mm3), day 0. Five intratumoral injections of R-113, R-121, R-115, or R-123 (108 PFU/injection) were performed. (B) Tumor volumes over time are shown. Each line represents the growth pattern of individual tumors (n = 8–9). Dashed or solid lines represent non-responders or responders, respectively. Percentages on the graphs indicate the rate of response as sum of complete and partial response (≥40% tumor shrinkage). Data shown are representative of two independent experiments. Statistical analysis was performed by a mid-p exact test. ∗p < 0.05.
Combination of α-PD1 and Double-Armed IL-12 and GM-CSF THV Induces Strong Anti-tumor Activity
To improve the therapeutic efficacy of the THV-based oncotherapy, the combination with α-PD1 was considered. Five viral injections were performed into the tumor mass, together with α-PD1 treatment according to the schedule depicted in Figure 3A. While monotherapies with either unarmed and armed THVs were moderately effective (Figure 2B), the combined treatment of R-113, R-115, R-121, or R-123 with α-PD1 led to a significant improvement in efficacy, and the percentage of tumor response was observed in 60%, 75%, 50%, and 100% of treated mice, respectively (Figure 3B). The most potent effect was observed in the R-123 and α-PD1 arm, with all mice responding to the combination therapy. These results suggest that (1) combination of OVs with checkpoint inhibitors is required to eradicate large tumors, and (2) the inclusion of the two encoded cytokines contributes to ameliorate the potency of the oHSV.
Figure 3.

Double-Armed IL-12 and GM-CSF R-123 Is Highly Effective in Combination with α-PD1
(A) Schematic of treatments. Mice were inoculated s.c. with HER2-LLC1 cells, randomized into six groups when tumors reached 110 mm3 as volume mean and treated with α-mPD1 antibody every 3–4 days until day 17, as stand-alone therapy or in combination with 108 PFU/injection of R-113, R-115, R-121, or R-123. (B) Growth of vehicle, α-PD1, or combo-treated tumors. Each line represents an individual HER2-LLC1 tumor (n = 8-15) followed over time (dashed lines for non-responders and solid lines for responders). Percentages on the graphs indicate the rate of response as sum of complete and partial response (≥40% tumor shrinkage). Data are representative of three independent experiments. Statistical analysis was performed by a mid-p exact test. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
The Anti-tumor Activity of R-123/α-PD1 Is T Cell-Mediated
To investigate the mechanism of anti-tumor responses, mice undergoing complete tumor regression upon combined treatment were rechallenged with a second contralateral tumor inoculum. Protection from tumor rechallenge was observed in 100% of mice, indicative of a long-lasting adaptive T cell memory response (Figure 4A). To evaluate the role of the immune system on the effectiveness of R-123 and α-PD1, a series of experiments were performed in several conditions of immune cells depletion.
Figure 4.

The Anti-tumor Activity of R-123/α-PD1 Is T Cell-Mediated
(A) Complete responders upon R-123/α-PD1 combo therapy received a second contralateral tumor challenge (day 40). Tumor growth was monitored until day 100. (B) Efficacy of R-123/α-PD1 following depletion of CD4+ or CD8+ T cells or NK cells. Mice bearing large HER2-LLC1-established tumors were treated with the combination R-123/α-PD1 according to the schematic in Figure 3A together with anti-mCD8, anti-mCD4, or anti-mNK antibody. Each line represents an individual tumor (dashed for non-responders, solid for responders); n = 8–9. Percentages on the graphs indicate the rate of response as sum of complete and partial response (≥40% tumor shrinkage). Statistical analysis was performed by a mid-p exact test. ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. (C) Levels of T cells in the blood of R-123/α-PD1-treated mice (day 10). Each symbol represents an individual sample (black, untreated mice; red, treated mice); n = 6–8. Bars show the mean of two independent experiments with SEM. Statistics were generated using an unpaired Mann-Whitney nonparametric test. ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001.
Selective depletion of CD8+ or CD4+ T cells affected in a significant manner the capability of the combined treatment of R-123 and α-PD1 to cure large s.c. tumors. In particular, CD4+ T cell depletion caused an almost complete abrogation of the anti-tumor effect, highlighting a major contribution of this lymphocyte population to the treatment efficacy. Depletion of CD8+ T cells also impacted the efficacy of the treatment, demonstrating that OV-mediated anti-tumor immunity depends on T cells (Figure 4B). We further depleted interferon (IFN)-γ, showing that the anti-tumor effect of R-123 and α-PD1 is IFN-γ-mediated (Figure S1). The contribution of the innate arm to the therapeutic effect was also explored by in vivo depletion of NK cells, showing in this case a minor impact on the response rate (Figure 4B). Interestingly, the combined treatment with R-123 and α-PD1 induced an overall increase in the number of T cells, including both CD4+ and CD8+ T cells present in the blood of treated mice versus untreated control mice (Figure 4C).
Systemic Delivery of Armed THV in Combination with α-PD1 Effectively Controls Lung Metastatic Nodules
OVs currently in various stages of clinical trials, or in clinical practice, e.g., T-VEC, are often administered to cancer patients by the intratumoral route, mainly because of safety reasons, consequent to the lack of specific targeting to cancer cells. There is intense interest in systemically deliverable OVs in order to treat patients with inaccessible tumors and particularly for those with metastatic diseases.
To explore the capability of hHER2-retargeted oHSVs to reach the target tumor and exert their anti-tumor activity after systemic administration, we considered a therapeutic setting of tumors diffuse to lungs, considered as a model of metastatic disease. LLC1 cells are capable of developing cancerous pulmonary nodules when injected by tail vein. Therefore, HER2-LLC1 cells were administered through the mouse tail vein (day −3). Three days later, the viral and checkpoint inhibitor treatments started. The i.v. administration of R-123 was performed in combination with α-PD1. 2 weeks after the beginning of treatments, mouse lungs were harvested and analyzed for the development of tumor nodules (Figure 5A). Treatment with R-123 in combination with α-PD1 was highly effective in reducing the number of lung nodules, and it resulted in a significantly higher anti-tumor activity as compared to α-PD1 alone; the latter exhibited similar efficacy as the vehicle (Figure 5B). Beyond confirming the strong anti-tumor efficacy of the double cytokine-armed R-123, the model highlighted the possibility of exploring the systemic route of administration in the context of retargeted OVs. Pre-existing anti-herpes immunity is prevalent in the human population and may reduce the effectiveness of a systemic oncolytic treatment. Thus, the efficacy of the i.v. treatment in combination with α-PD1 was assessed in HSV-immunized mice. Animals were pre-immunized with a recombinant HSV-1 (F)-derived virus21 using the prime-boost schedule outlined in Figure 5C. As expected, immunization of mice with HSV led to development of neutralizing antibodies (NAbs) to HSV as determined by an in vitro viral neutralization assay (Figure 5D). To evaluate the effect of immunization on the activity of THV to prevent lung metastases formation, HSV-naive and HSV-immunized animals were inoculated i.v. with the HER2-LLC1 and then treated with R-123 in combination with α-PD1. Prior immunization with HSV did not impact the efficacy of treatment, as evidenced by the similar reduction of lung nodule formation observed in both HSV-naive and HSV immune-treated groups (Figure 5E).
Figure 5.

Systemic Delivery of R-123 Controls Lung Metastasis Development in Combination with α-PD1
(A) Schematic of lung metastatic setting. HER2-LLC1 cells were i.v. injected into the mouse tail vein. Treatments started after 3 days (d0): R-123 was i.v. delivered (five injections at 108 PFU each), combined with α-PD1. At day 14 lung nodules were counted. (B) Number of lung nodules in vehicle-treated (black symbols), α-PD1-treated (gray symbols), or R-123/α-PD1-treated (red symbols) mice. Bars show mean with SEM of two independent experiments; dots represent individual animals (n = 10–20). (C) Prime-boost immunization scheme with WT HSV. For the efficacy experiment, naive versus HSV immune mice were i.v. inoculated with HER2-LLC1 cells. Treatments started after 3 days (d0) and were performed as described in (A). (D) Anti-HSV antibody serum titers determined by plaque reduction neutralization test at week 12 after immunization. The y axis indicates the dilution factor at which the number of viral plaques was reduced by 50% compared to the control sample (virus alone). Bars show mean with SEM. (E) Numbers of lung nodules in untreated mice (black dots ) versus treated mice, HSV naive mice (red dots), or HSV immune mice (blue dots) are shown. Bars show mean with SEM; dots represent individual animals (n = 8–12). Statistics were generated using an unpaired Mann-Whitney nonparametric test. ∗p ≤ 0.05, ∗∗∗∗p ≤ 0.001.
Discussion
OVs are considered highly promising tools to increase the efficacy of a checkpoint inhibitor. In this study, we have developed an approach based on the use of a fully replicative HER2-retargeted oHSV armed with IL-12 and GM-CSF to maximize therapeutic activity. By using a stringent setting of large murine tumors and a model refractory to α-PD1 monotherapy22,23 we demonstrated (1) highly synergistic effect of THV, unarmed and armed ones, with α-PD1, and (2) enhanced efficacy by potentiation of the viral payload to express multiple immunomodulatory agents. Our earlier in vivo studies demonstrated that the efficacy of retargeted oHSV as a stand-alone therapy, even when armed with IL-12 (R-115), is dramatically reduced when moving from an early therapeutic setting to late treatment, suggesting that the hostile microenvironment found in a setting of late treatment (e.g., in the presence of large established tumors) limits the anti-tumor activity of such viruses.10 In the present study, we confirmed the modest effect of unarmed and armed THV, either single or double armed, in mice bearing large tumors, suggesting that a combination with a checkpoint inhibitor might be beneficial to ameliorate the anti-tumor response. The combination of α-PD1 and double-armed IL-12 and GM-CSF (R-123) given intratumorally resulted in a 100% response, 80% of which included complete response with full eradication of advanced tumors. Importantly, note that these results were achieved by using a tumor cell line, HER2-LLC1, that is markedly less permissive to HSV infection than the human cancer cells and the syngeneic C57BL/6 mice, a strain among the most highly resistant to HSV infection.10,24
This model also showed resistance to α-PD1 treatment, as demonstrated by the absence of the therapeutic response upon α-PD1 administration. The resistance of these tumors to α-PD1 therapy was overcome with the addition of retargeted oHSVs, with the maximum therapeutic benefit achieved in the presence of the double-armed cytokine virus. This confirms and extends the notion that the use of OVs, particularly armed viruses, can be applied to optimize immunotherapy and overcome resistance to checkpoint blockade. One of the major limitations of checkpoint blockade includes situations in which the tumor is invisible to the immune system, the so-called “cold” and “excluded” tumors. It is likely that the addition of THV to the checkpoint blockade is able to generate an inflamed “hot” tumor environment, thereby reawakening anti-tumor immune responses, as also demonstrated by our studies,10 as well as by other studies.25 The therapeutic effect of R-123 and α-PD1 was T cell-mediated, likely attributable to their ability to produce IFN-γ, as shown by a significant reduction of anti-tumor efficacy by depleting CD4+ or CD8+ T cells or by blocking IFN-γ. Depletion of natural killer (NK) cells had no impact on the effectiveness of treatment. Indeed, induction of tumor-directed adaptive immune responses has been widely recognized as a key mechanism to achieve long-term therapeutic success in the clinic.4,26
More recently, a key role of infiltrating CD8+ T cells was documented in patients responding to a combination of T-VEC and pembrolizumab.17 Analysis of tumor biopsies demonstrated that T-VEC treatment increased the presence of CD8+ T cells in 75% of injected lesions and that the increase in CD8+ T cell density appeared to be associated with the response to combination therapy.17 The combined treatment of α-PD1 and R-123 elicited a systemic anti-tumor effect and a long-lasting memory response, as proven by the full protection from the growth of contralateral, distant, untreated tumors. An increase in circulating CD4+ and CD8+ T cell levels was observed upon treatment. A systemic increase in circulating CD4+ and CD8+ T cells was also found in patients upon T-VEC administration, providing evidence for the generation of a systemic anti-tumor response.17
We are currently investigating the induction of LLC1 neoantigen-specific T cell responses after combined treatment to better elucidate the mechanisms of immune-mediated anti-tumor responses. In line with our findings, the use of armed OVs has shown improved therapeutic benefit in clinical trials and preclinical models.11 Several viruses have been armed with IL-12 and tested in different tumor models, including oHSV, with the demonstration of enhanced efficacy.27, 28, 29 Multiple preclinical studies demonstrated that OVs armed with IL-12 yield better anti-tumor effects than does vectored GM-CSF.30,31 Consistent with previous studies assessing the effectiveness of cytokine-armed oHSV,30,31 our armed GM-CSF THV exerted similar efficacy than to that of the parental non-cytokine virus, while IL-12-armed THV showed a very strong anti-tumor activity. The contribution of GM-CSF was revealed when coupling GM-CSF and IL-12 in one single THV. The expression of multiple transgenes from a single oncolytic HSV has been recently reported with a potent antitumor effect and improved therapeutic effect in combination with PD1 blockade in murine models,32 supporting the use of the multiple arming strategy as a potent and versatile approach to develop new oncolytic immunotherapies for clinical use.
Prompted by the rate of response achieved upon local administration of R-123 combined with α-PD1, which is the highest achieved so far using a retargeted oHSV to treat established tumors, we next assessed whether the same approach might also be effective for i.v. delivery. The systemic administration of oHSV represents an appealing route of administration because it allows treatment of diffuse metastases as well as the primary lesions. Several types of OVs have been administered systemically for cancer therapy.33 However, to date, the most frequently used route in clinical studies is local administration, including the approved administration of T-VEC. The intratumoral route is preferred (1) to ensure safety by limiting the risk of replication in normal cells, and (2) to avoid pre-existing neutralizing antibodies that may dampen the efficacy of OVs. Recently, a first-in-human phase 1 study in young cancer patients demonstrated feasibility and tolerability of i.v. administration of an attenuated oHSV, although no evidence of intratumoral virus replication was found after systemic administration.34
In the present study, for the first time, we showed that a retargeted and cytokine-armed oHSV is effective in controlling lung metastases formation when systemically injected, in combination with α-PD1. Systemic administration was safe, as no toxicity was observed in the i.v. treated mice, confirming our previous data.9,10 To further improve the safety of our THV, we have recently generated a new oHSV that combines the feature of retargeting with tumor replication conditioning as an option to be considered for i.v. delivery.35 Neutralizing antibodies are one of the main obstacles for systemically administered OVs given the high seroprevalence in the human population.36 We have demonstrated maintenance of therapeutic activity of systemically delivered R-123 in combination with α-PD1 in mice immunized with HSV-1 virus showing anti-HSV neutralizing antibodies. By using a protocol of active immunization, we have likely induced also cellular immunity against HSV that may be beneficial for the anti-tumor effect, consistent with the observations by Ricca et al.37 While the current study suggests that i.v. delivery is feasible and can be effective to control tumor growth, it still represents a challenge because of several components impeding the optimal systemic delivery efficiency of OVs.7,33 Among them, the sequestration and subsequent clearance of the infused viruses by the host’s mononuclear phagocyte system and the inactivation by the host’s defense systems.38 In order to enable the virus to evade the host’s defense machinery and efficiently reach the tumor site, different strategies are being considered, such as virus capsid engineering, chemical modifications of virus capsid (e.g., PEGylation),39 use of carrier cells,40,41 and potentiation of the virus by choice of optimal immunomodulators to strengthen the immune-mediated effect of oHSV. Our findings open the possibility to explore the clinical use of retargeted-armed oHSV for local and systemic treatment of cancer diseases and support combined immunotherapy of checkpoint blockade and endovaccines based on OVs.
Materials and Methods
Mice
Tolerant hHER2-transgenic C57BL/6 mice (B6.Cg-Pds5bTg(Wap ERBB2)229Wzw/J) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Mouse colony management and all day-to-day care were performed by trained mouse house staff at Plaisant (Castel Romano, Italy). Female mice (6–8 weeks of age) were used for in vivo experiments.
Cell Cultures
The SK-OV-3 (human ovarian carcinoma) cell line was purchased from ATCC (Manassas, VA, USA). The HER2-LLC1 cell line was generated by Campadelli’s laboratory.10 The SK-OV-3 cells were cultured in RPMI 1640 medium-GlutaMAX (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with heat-inactivated 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) and 1% (v/v) penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA). HER2-LLC1 cells were maintained in DMEM (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% FBS, 1% (v/v) penicillin/streptomycin, 2 mM l-glutamine (Thermo Fisher Scientific, Waltham, MA, USA), and 2 mM puromycin (Sigma-Aldrich/Merck, St. Louis, MO, USA) for selection. Both cell lines were maintained at 37°C in 5% CO2.
Viruses
R-113 corresponds to the previously described virus named R-LM113.21 R-115, described by Menotti et al.,18 and R-121 are derivatives of R-113. They are single-armed THVs, expressing mIL-12 or murine GM-CSF (mGM-CSF), respectively. R-123 is a double-armed THV, derivative of R-115, expressing both cytokines. The viral sites of insertion are UL26–UL27 for mGM-CSF-expressing cassette and US1–US2 for mIL-12-expressing cassette. All viruses were cultivated and titrated by plaque assay in SK-OV-3 cells.
A WT HSV1, R-LM5,21 was used to induce the development of a pre-existing anti-HSV immunity in mice.
ELISA Assay
SK-OV-3 cells were seeded in 12-well plates at a density of 5 × 105 cells/well 24 h before virus infection. Cells were infected with R-113, R-115, R-121, or R-123 at a multiplicity of infection (MOI) of 0.1 PFU/cell. 24 and 48 h after infection, supernatants were collected to check cytokine production. mIL-12 and mGM-CSF were quantified by means of an IL-12p70 mouse ELISA kit or GM-CSF mouse ELISA kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions, respectively.
In Vivo Studies
For the metastatic tumor setting, 7.5 × 105 HER2-LLC1 cells were injected i.v. into the mouse tail vein. After 3 days, viral and α-PD1 treatments started (day 0). THVs were administered five times at 108 PFU/injection every 2–3 days starting from day 0. α-murine (α-m)PD1, clone RMP1-14 (Bio X Cell, Lebanon, NH, USA), was administered intraperitoneally (i.p.) at a dosage of 200 μg twice a week from day 0 to day 10. On day 14, lungs were perfused with India ink (15%), harvested, and fixed in Fekete’s solution. Lung metastatic colonies were counted using a dissecting microscope.
In the condition of pre-existing immunity against HSV, 12 weeks before HER2-LLC1 implantation, mice were systemically (i.p.) injected with R-LM5 in a prime-boost regimen (week 0, week 3) at a dose of 3 × 106 PFU. Serum neutralization titers were measured over time by a plaque reduction neutralization test.
For the established tumor setting, 5 × 105 HER2-LLC1 cells were s.c. injected into the mouse right flank. At day 0 treatments started in animals previously selected based on the tumor volume (tumor size average per group 110 mm3), according to the doses and the regimen described above for the metastatic setting. α-PD1 antibody was administered in this setting until day 17. Tumor growth was measured using a digital caliper every 3–4 days. Tumor volume was calculated using the formula 0.5 × length × width2, where the length was the longer dimension. Mice were sacrificed as soon as signs of distress or a tumor volume above 1,500 mm3 occurred. 5 × 105 HER2-LLC1 cells were also used for the contralateral tumor challenge, performed at day 40 in cured tumor-free mice.
To deplete immune cells subsets, α-mCD8, α-mCD4, or α-mNK (Bio X Cell, Lebanon, NH, USA) was used. In vivo depletion of IFN-γ was performed with α-mIFN-γ (Bio X Cell, Lebanon, NH, USA). Each antibody was administered five times by i.p. injection at a dosage of 200 μg, every 3–4 days from day 1 to day 15.
Ex Vivo Immune Analysis
Immune cell analysis was performed on blood 10 days post treatments start. Blood collected in heparin tubes was processed to remove erythrocytes; cells were then stained for viability with a Live/Dead fixable near-infrared (IR) dead cell stain kit (Thermo Fisher Scientific, Waltham, MA, USA). Surface staining was then performed to detect CD3+ T cells and both CD4+ and CD8+ T cell subsets, with the following surface antibodies: allophycocyanin anti-mouse CD3e, phycoerythrin anti-mouse CD4, and peridinin chlorophyll protein (PerCP) anti-mouse CD8a (BD Biosciences, San Jose, CA, USA). Stained cells were acquired on a FACSCanto flow cytometer and analyzed using DIVA software (BD Biosciences). The same antibodies were used to check CD8+ and CD4+ T cell depletion. Fluorescein isothiocyanate anti-mouse CD335 (BioLegend, San Diego, CA, USA) was added for the NK cell depletion check.
Plaque Reduction Neutralization Test
SK-OV-3 cells were seeded in 12-well plates at a density of 5 × 105 cells/well and cultured overnight at 37°C in 5% CO2 until the cells became a monolayer. Heat-inactivated mouse sera were first diluted 20-fold in DMEM containing 1% heat-inactivated FBS and 1% (v/v) penicillin/streptomycin, followed by 3-fold serial dilutions from 1:60 to 1:131,220. An equal volume of HSV-1 viral dilution, containing 100 PFU of viruses, was mixed with the diluted samples and incubated at 37°C in 5% CO2 for 30 min. Then, 0.35 mL of the total mixture was added to the SK-OV-3 cells and incubated for 1.5 h at 37°C on a shaker. The infection mixture was then removed and 1.5 mL of fresh culture medium (RPMI 1640 medium-GlutaMAX, supplemented with heat-inactivated 2.5% FBS and 1% [v/v] penicillin/streptomycin) was added to facilitate the viral entry. After 48h incubation, the cells were fixed with 96% EtOH (0.5 mL/well) for 10 min and then stained with 0.8 mL of Giemsa (Sigma-Aldrich/Merck, St. Louis, MO, USA) for 15 min. Plates were then washed and left to dry. The plaques were counted by use of a dissecting microscope. Virus control wells were infected with the same amount of virus as the testing wells mixed with serially diluted samples. Each test was carried out in triplicate.
The neutralizing titer was expressed as the dilution to which the number of plaques reduces by 50% compared to the control sample (virus alone).
Statistical Analysis
Statistical significance was calculated using the Mann-Whitney test, using Prism 6.0 software. The analysis of contingency data was performed using the mid-p exact test. The significant p values were assigned as ∗p <0.05, ∗∗p <0.01, ∗∗∗p <0.001, and ∗∗∗∗p <0.0001 and designated on the plots.
Study Approval
All experimental procedures were approved by the local animal Ethics Council and were performed in accordance with national and international laws and policies (EEC Council Directive 86/609; Italian Legislative Decree 26/14). The Ethics Committee of the Italian Ministry of Health approved this research.
Author Contributions
A.M.D, E.Scarselli, and A.N. conceived the experiments and supervised the study. A.M.D. and E.Scarselli designed the experiments. M.D.L., G.C., V.B, I.G., L.N., F.L., B.P., E.Sasso, S.P., G.F., C.G., and G.C.-F. conducted the research. M.D.L., A.M.D., and G.C. analyzed the data. G.C.F. and N.Z contributed to data interpretation. M.D.L. and A.M.D. wrote the manuscript.
Conflicts of Interest
E. Scarselli and A.N. are founders of Nouscom S.R.L. G.C.-F. owns shares in Nouscom S.r.l. A.M.D, M.D.L., G.C., V.B., I.G., L.N., F.L., E. Sasso, and B.P. are employees of Nouscom S.r.l. The remaining authors declare no competing interests.
Acknowledgments
We thank Marina Udier for critical reading of the manuscript and helpful discussion. This work was supported by the Grant SATIN, Regione Campania. We acknowledge the animal facility of Plaisant in Castel Romano (Rome) for the maintenance and care of the mice used in this study. In particular, we thank Domenico Salvatori for support with tumor calibrations for in vivo studies.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.10.006.
Supplemental Information
References
- 1.Bartlett D.L., Liu Z., Sathaiah M., Ravindranathan R., Guo Z., He Y., Guo Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer. 2013;12:103. doi: 10.1186/1476-4598-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2016;15:660. doi: 10.1038/nrd.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufman H.L., Bines S.D. OPTIM trial: a phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol. 2010;6:941–949. doi: 10.2217/fon.10.66. [DOI] [PubMed] [Google Scholar]
- 4.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 5.Liu B.L., Robinson M., Han Z.Q., Branston R.H., English C., Reay P., McGrath Y., Thomas S.K., Thornton M., Bullock P. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. doi: 10.1038/sj.gt.3301885. [DOI] [PubMed] [Google Scholar]
- 6.Kohlhapp F.J., Kaufman H.L. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin. Cancer Res. 2016;22:1048–1054. doi: 10.1158/1078-0432.CCR-15-2667. [DOI] [PubMed] [Google Scholar]
- 7.Campadelli-Fiume G., Petrovic B., Leoni V., Gianni T., Avitabile E., Casiraghi C., Gatta V. Retargeting strategies for oncolytic herpes simplex viruses. Viruses. 2016;8:63. doi: 10.3390/v8030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchida H., Hamada H., Nakano K., Kwon H., Tahara H., Cohen J.B., Glorioso J.C. Oncolytic herpes simplex virus vectors fully retargeted to tumor-associated antigens. Curr. Cancer Drug Targets. 2018;18:162–170. doi: 10.2174/1568009617666170206105855. [DOI] [PubMed] [Google Scholar]
- 9.Menotti L., Nicoletti G., Gatta V., Croci S., Landuzzi L., De Giovanni C., Nanni P., Lollini P.L., Campadelli-Fiume G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA. 2009;106:9039–9044. doi: 10.1073/pnas.0812268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leoni V., Vannini A., Gatta V., Rambaldi J., Sanapo M., Barboni C., Zaghini A., Nanni P., Lollini P.L., Casiraghi C., Campadelli-Fiume G. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018;14:e1007209. doi: 10.1371/journal.ppat.1007209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Graaf J.F., de Vor L., Fouchier R.A.M., van den Hoogen B.G. Armed oncolytic viruses: a kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018;41:28–39. doi: 10.1016/j.cytogfr.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi K.J., Zhang S.N., Choi I.K., Kim J.S., Yun C.O. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012;19:711–723. doi: 10.1038/gt.2011.125. [DOI] [PubMed] [Google Scholar]
- 13.Parker J.N., Gillespie G.Y., Love C.E., Randall S., Whitley R.J., Markert J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA. 2000;97:2208–2213. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stephenson K.B., Barra N.G., Davies E., Ashkar A.A., Lichty B.D. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012;19:238–246. doi: 10.1038/cgt.2011.81. [DOI] [PubMed] [Google Scholar]
- 15.van de Laar L., Coffer P.J., Woltman A.M. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. 2012;119:3383–3393. doi: 10.1182/blood-2011-11-370130. [DOI] [PubMed] [Google Scholar]
- 16.Puzanov I., Milhem M.M., Minor D., Hamid O., Li A., Chen L., Chastain M., Gorski K.S., Anderson A., Chou J. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016;34:2619–2626. doi: 10.1200/JCO.2016.67.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ribas A., Dummer R., Puzanov I., VanderWalde A., Andtbacka R.H.I., Michielin O., Olszanski A.J., Malvehy J., Cebon J., Fernandez E. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031–1032. doi: 10.1016/j.cell.2018.07.035. [DOI] [PubMed] [Google Scholar]
- 18.Menotti L., Avitabile E., Gatta V., Malatesta P., Petrovic B., Campadelli-Fiume G. HSV as a platform for the generation of retargeted, armed, and reporter-expressing oncolytic viruses. Viruses. 2018;10:352. doi: 10.3390/v10070352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piechocki M.P., Ho Y.S., Pilon S., Wei W.Z. Human ErbB-2 (Her-2) transgenic mice: a model system for testing Her-2 based vaccines. J. Immunol. 2003;171:5787–5794. doi: 10.4049/jimmunol.171.11.5787. [DOI] [PubMed] [Google Scholar]
- 20.D’Alise A.M., Leoni G., Cotugno G., Troise F., Langone F., Fichera I., De Lucia M., Avalle L., Vitale R., Leuzzi A. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat. Commun. 2019;10:2688. doi: 10.1038/s41467-019-10594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menotti L., Cerretani A., Hengel H., Campadelli-Fiume G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008;82:10153–10161. doi: 10.1128/JVI.01133-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bullock B.L., Kimball A.K., Poczobutt J.M., Neuwelt A.J., Li H.Y., Johnson A.M., Kwak J.W., Kleczko E.K., Kaspar R.E., Wagner E.K. Tumor-intrinsic response to IFNγ shapes the tumor microenvironment and anti-PD-1 response in NSCLC. Life Sci. Alliance. 2019;2:e201900328. doi: 10.26508/lsa.201900328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertrand F., Montfort A., Marcheteau E., Imbert C., Gilhodes J., Filleron T., Rochaix P., Andrieu-Abadie N., Levade T., Meyer N. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017;8:2256. doi: 10.1038/s41467-017-02358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez C. Genetics of natural resistance to herpesvirus infections in mice. Nature. 1975;258:152–153. doi: 10.1038/258152a0. [DOI] [PubMed] [Google Scholar]
- 25.Gujar S., Pol J.G., Kroemer G. Heating it up: oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. OncoImmunology. 2018;7:e1442169. doi: 10.1080/2162402X.2018.1442169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senzer N.N., Kaufman H.L., Amatruda T., Nemunaitis M., Reid T., Daniels G., Gonzalez R., Glaspy J., Whitman E., Harrington K. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009;27:5763–5771. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 27.Hellums E.K., Markert J.M., Parker J.N., He B., Perbal B., Roizman B., Whitley R.J., Langford C.P., Bharara S., Gillespie G.Y. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro-oncol. 2005;7:213–224. doi: 10.1215/S1152851705000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markert J.M., Cody J.J., Parker J.N., Coleman J.M., Price K.H., Kern E.R., Quenelle D.C., Lakeman A.D., Schoeb T.R., Palmer C.A. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J. Virol. 2012;86:5304–5313. doi: 10.1128/JVI.06998-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cody J.J., Scaturro P., Cantor A.B., Yancey Gillespie G., Parker J.N., Markert J.M. Preclinical evaluation of oncolytic δγ(1)34.5 herpes simplex virus expressing interleukin-12 for therapy of breast cancer brain metastases. Int. J. Breast Cancer. 2012;2012:628697. doi: 10.1155/2012/628697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varghese S., Rabkin S.D., Liu R., Nielsen P.G., Ipe T., Martuza R.L. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006;13:253–265. doi: 10.1038/sj.cgt.7700900. [DOI] [PubMed] [Google Scholar]
- 31.Wong R.J., Patel S.G., Kim S., DeMatteo R.P., Malhotra S., Bennett J.J., St-Louis M., Shah J.P., Johnson P.A., Fong Y. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum. Gene Ther. 2001;12:253–265. doi: 10.1089/10430340150218396. [DOI] [PubMed] [Google Scholar]
- 32.Thomas S., Kuncheria L., Roulstone V., Kyula J.N., Mansfield D., Bommareddy P.K., Smith H., Kaufman H.L., Harrington K.J., Coffin R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer. 2019;7:214. doi: 10.1186/s40425-019-0682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferguson M.S., Lemoine N.R., Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv. Virol. 2012;2012:805629. doi: 10.1155/2012/805629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Streby K.A., Currier M.A., Triplet M., Ott K., Dishman D.J., Vaughan M.R., Ranalli M.A., Setty B., Skeens M.A., Whiteside S. First-in-human intravenous Seprehvir in young cancer patients: a phase 1 clinical trial. Mol. Ther. 2019;27:1930–1938. doi: 10.1016/j.ymthe.2019.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasso E., Froechlich G., Cotugno G., D’Alise A.M., Gentile C., Bignone V., De Lucia M., Petrovic B., Campadelli-Fiume G., Scarselli E. Replicative conditioning of Herpes simplex type 1 virus by Survivin promoter, combined to ERBB2 retargeting, improves tumour cell-restricted oncolysis. Sci. Rep. 2020;10:4307. doi: 10.1038/s41598-020-61275-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradley H., Markowitz L.E., Gibson T., McQuillan G.M. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J. Infect. Dis. 2014;209:325–333. doi: 10.1093/infdis/jit458. [DOI] [PubMed] [Google Scholar]
- 37.Ricca J.M., Oseledchyk A., Walther T., Liu C., Mangarin L., Merghoub T., Wolchok J.D., Zamarin D. Pre-existing immunity to oncolytic virus potentiates its immunotherapeutic efficacy. Mol. Ther. 2018;26:1008–1019. doi: 10.1016/j.ymthe.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu X., Tao L., Zhang X. Genetically coating oncolytic herpes simplex virus with CD47 allows efficient systemic delivery and prolongs virus persistence at tumor site. Oncotarget. 2018;9:34543–34553. doi: 10.18632/oncotarget.26167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eto Y., Yoshioka Y., Mukai Y., Okada N., Nakagawa S. Development of PEGylated adenovirus vector with targeting ligand. Int. J. Pharm. 2008;354:3–8. doi: 10.1016/j.ijpharm.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 40.Leoni V., Gatta V., Palladini A., Nicoletti G., Ranieri D., Dall’Ora M., Grosso V., Rossi M., Alviano F., Bonsi L. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget. 2015;6:34774–34787. doi: 10.18632/oncotarget.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakashima H., Kaur B., Chiocca E.A. Directing systemic oncolytic viral delivery to tumors via carrier cells. Cytokine Growth Factor Rev. 2010;21:119–126. doi: 10.1016/j.cytogfr.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.