

Abstract
We investigated whether microRNA-150 (miR-150)-based RNA interference (RNAi) ameliorates tubular injury and tubulointerstitial fibrosis. Mice injected with folic acid developed tubulointerstitial fibrosis at day 30. miR-150 levels were increased at day 7 and peaked at day 30. At day 30, protein levels of α-smooth muscle actin, fibronectin (FN), and collagen 1 (COL-1) were increased, while suppressor of cytokine signal 1 (SOCS1) was decreased. Kidneys manifested increased macrophage numbers and increased expression of potential mediators: interferon-γ, interleukin-6, and tumor necrosis factor-α. Locked nucleic acid-anti-miR-150, started prior to or after tubular injury and administered twice weekly for 4 weeks, reversed renal inflammation and fibrosis. In HK-2 cells, co-culture with macrophages increased miR-150 expression and decreased SOCS1. Janus kinase (JAK) and signal transducer and activators of transcription (STAT) pathway-related proteins p-JAK1, p-JAK2, p-STAT1, p-STAT3, and pro-fibrotic genes encoding α-smooth muscle actin, FN, and COL-1 were all upregulated. The miR-150 antagonist reversed these transcriptional changes. Lastly, in renal biopsies from patients with chronic interstitial fibrosis, renal miR-150, and pro-fibrotic gene expression and macrophage numbers were increased, while SOCS1 expression was decreased. In conclusion, miR-150-based RNAi is as a potential novel therapeutic agent for tubulointerstitial fibrosis, suppressing the SOCS1/JAK/STAT pathway and reducing macrophage influx.
Keywords: RNAi, renal fibrosis, macrophages, SOCS1, folic acid, HK-2 cells, therapeutics
Graphical Abstract
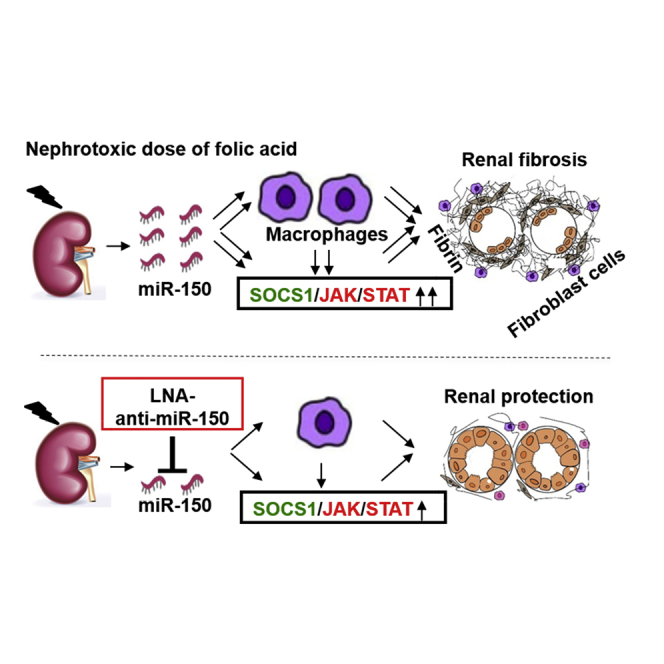
Zhou and colleagues found increased miR-150 expression and macrophage infiltrates in mouse kidneys with folic acid-induced fibrosis. Locked-nucleic acid anti-miR-150 alleviated fibrosis. In HK-2 kidney cell-macrophage cocultures, the miR-150 inhibitor reversed overexpression of profibrotic genes, acting via suppressor of cytokine signal 1/Janus kinase/signal transducer and activator of transcription pathway.
Introduction
The prevalence of chronic kidney disease (CKD) is approximately 8%–16% worldwide and CKD is the 16th most common cause of death.1 Renal tubulointerstitial fibrosis (TIF) is common pathologic feature of all CKD.2 To date, there are few effective therapeutics to prevent or retard progression of TIF. We may reduce the prevalence of CKD through a deeper understanding the pathogenesis of TIF and by identifying novel therapeutic agents that target key mediators of renal fibrosis.
MicroRNAs (miRNAs or miRs) are recognized as critical contributors to kidney diseases.3, 4, 5 We have identified miR-150 as the most overexpressed miRNA in renal biopsies from patients with lupus nephritis and have reported that miR-150 promotes renal fibrosis by downregulating suppressor of cytokine signal 1 (SOCS1) in kidney resident cells in vitro.6 Subsequently, Guan et al.7 reported that in renal ischemia/reperfusion model, miR-150 promotes kidney fibrosis by activating fibroblasts as a signal from injured tubular epithelial cells. Transfection of miR-150 into young mesangial cells promotes cell aging.8 Acute kidney injury (AKI) induced by ischemia/reperfusion is attenuated in miR-150 knockout mice.9 Renal miR-150 levels are increased in minimal change disease cases with poor prognosis compared, to those with good outcome; as these cases often represent unsampled focal segmental glomerulosclerosis, this suggests an association of miR-150 with renal fibrosis.10 In addition, plasma and urinary miR-150 have emerged as potential progression biomarkers in CKD patients, including those with diabetic kidney disease.11, 12, 13 Taken together, these findings suggest that miR-150 contributes to the pathogenesis of various scarring kidney diseases and is an intriguing and novel therapeutic target.
Recently RNA interference (RNAi) has opened a new avenue to develop novel drugs for human diseases.14 In terms of identifying miR-targeted RNAi for treating kidney diseases, many miRs have been reported to be altered in animal models. The inhibitors of miR-21, miR-192, miR-27a, miR-215, miR-34a, miR-29a, or miR-25 ameliorate diabetic nephropathy in mice.9,15, 16, 17, 18, 19, 20 In other animal models, the miR-132 antagomir reduces interstitial myofibroblast numbers in mice with unilateral ureteral obstruction,21 miR-30b inhibitor ameliorates lipopolysaccharide-induced AKI,22 miR-709 antagonist attenuates cisplatin-induced AKI in mice,23 and miR-34a inhibitor alleviates multi-organ injury after ischemia/reperfusion injury in pregnant rats.24 Recently, we reported that locked nucleic acid (LNA)-anti-miR-150 ameliorates lupus nephritis and focal segmental glomerulosclerosis arising from glomerular injury in mouse models.25,26 However, it remains unclear whether miR-150-based RNAi can attenuate TIF initiated from renal tubular damage.
Here, we aimed to determine whether LNA-anti-miR-150 attenuates TIF induced by folic acid in mice. We investigated the underlying mechanisms by silencing overexpression of miR-150 in HK-2 cells co-cultured with macrophages. We show for the first time the effect of miR-150 based RNAi to reduced tubulointerstitial fibrosis and suggest a possible mechanism of action.
Results
Renal Expression of miR-150 from AKI to TIF following Folic Acid Administration to Mice
AKI developed 2 days after folic acid injection, as indicated by the elevated ratio of kidney weight to heart weight, serum creatinine (Scr), and blood urea nitrogen (BUN). These parameters improved by day 7 and returned to the baseline by day 30 (Figures 1A–1C). At day 2, features of AKI were present: loss of proximal tubular cells brush border, detachment of tubular epithelial cells, and modest inflammatory cells infiltrate were seen on periodic acid-Schiff (PAS) stained sections. At day 7, cortical histological tubular dilation remained and interstitial inflammatory cell numbers were increased, and at day 30, medullary rays displayed patchy fibrosis and severe infiltration of inflammatory cells (Figure 1D). The severe histological fibrosis suggests transition of AKI to chronic interstitial fibrosis. Importantly, miR-150 levels were increased at day 7 and remained increased through day 30, as assessed by fluorescence in situ hybridization (FISH) and qPCR (Figures 1E and 1F).
Figure 1.

Time Course of Folic-Acid-Induced Kidney Injury and miR-150 Expression in Mice
(A–C) Evaluation of kidney injury after the injection of folic acid (FA). Shown are (A) the ratio of kidney weight to heart weight, (B) serum creatinine (Scr), (C) blood urea nitrogen (BUN), and (D) PAS staining. Magnification 400×, scale bar, 10 μm. Black asterisks indicate tubular ectasia, loss of brush border, flattened or detached epithelial cells; yellow arrows indicate infiltration of inflammatory cells in tubulointerstitium; white arrows indicate patchy fibrosis. (E and F) miR-150 expression assessed by (E) fluorescence in situ hybridization (FISH) and (F) qPCR. Magnification 200×, scale bar, 400 μm; n = 6, # p < 0.05, day 2, day 7, day 30 versus day 0; ∗p < 0.05, day 7, day 30 versus day 2. Data represent mean ± SD. See also Figure S4A.
Efficacy and Safety of LNA-Anti-miR-150 in TIF Mice
We have previously demonstrated that LNA-anti-miR-150 is delivered to mouse kidneys following systemic administration.25,26 Therefore, we investigated the efficacy and safety of LNA-anti-miR-150 in folic acid-induced TIF on day 30. Mice received eight LNA anti-miR-150 doses, either prophylactically (on day 0 before folic acid) or therapeutically (on day 2 after folic acid injection). Review of Masson and PAS stained sections, as well as semiquantitative analysis (Figures 2A–2D), revealed that both courses of LNA-anti-miR-150 therapy decreased renal fibrosis, as indicated by reductions in the area of scarred cortex, regions of patchy fibrosis in the medulla, and the severity of inflammatory cell infiltrates. Further, both prophylactic exposure to LNA-anti-miR-150 and therapeutic use (starting after AKI) reduced renal overexpression of miR-150 on day 30 (Figure 2E). LNA therapy appeared to lack toxicity, as judged by stable body weight and stable kidney and liver function (Figures S1A–1C).
Figure 2.

Effect of LNA-Anti-miR-150 on Kidney Injury in TIF Mice
(A and B) Masson (A) and PAS (B) staining. (C and D) Semiquantitative analysis on (C) fibrosis on Masson staining and (D) tubular damage on PAS staining. (E) Renal miR-150 expression measured by qPCR analysis. (n = 6, # p < 0.05, each group versus normal control. ∗p < 0.05, LNA-anti-miR-150 group versus scrambled LNA group. Data represent mean ± SD. For Masson staining, magnification 1.5× and scale bar, 2,000 μm. For PAS staining, magnification 200×, scale bar, 200 μm. Black arrowheads indicate the scarred kidney surface. Black arrows indicate patchy fibrosis. Yellow arrows indicate the infiltration of inflammatory cells into the tubulointerstitium. See also Figures S1 and S4B.
Effect of LNA-Anti-miR-150 on Renal Pro-Fibrotic Genes in TIF Mice
To investigate the underlying anti-fibrotic mechanisms LNA-anti-miR-150 in mice with TIF, we examined the expression of selected pro-fibrotic gene mRNAs and proteins, including α-smooth muscle actin (α-SMA), fibronectin (FN), and collagen 1 (COL-1) in kidney tissues. Levels of α-SMA, FN, and COL-1 mRNA were all increased in TIF mice compared to control mice. These increased mRNA levels were reduced by LNA-anti-miR-150, compared to the scrambled LNA, with both prophylactic treatment and therapeutic use (Figure 3A). The same effect of LNA-anti-miR-150 was seen on the renal protein levels of these pro-fibrotic genes in TIF mice on western blotting and immunofluorescence staining, compared to the scrambled LNA mice (Figures 3B and 3C).
Figure 3.

Effect of LNA-Anti-miR-150 on Renal Expression of Pro-Fibrotic Genes in TIF Mice
(A) Quantification of mRNA encoding α-SMA, FN, and COL-1. (B and C) Renal fibrosis-related proteins α-smooth muscle actin, FN, and COL-1 were assessed on (B) western blotting and by (C) double immunofluorescence staining of α-SMA and FN, as well as COL-1. Magnification 200×. Scale bar, 400 μm; n = 6. # p < 0.05, each experimental group versus normal group. ∗p < 0.05, LNA-anti-miR-150 versus Scrambled LNA respectively. Data are presented as mean ± SD. Red indicates FN and COL-1; green indicate α-SMA, asterisks indicate blood vessels.
Effect of LNA-Anti-miR-150 on Renal SOCS1 in TIF Mice
Our previous study reported that miR-150 promotes renal fibrosis by downregulating anti-fibrotic SOCS1 as assessed by SOCS1 luciferase reporter activity in human proximal tubular cells.6 In present study, we further characterized renal SOCS1 expression in TIF mice. Renal SOCS1 protein expression was downregulated up to 44% in folic-acid-induced TIF compared to control mice. Both early and delayed treatment with LNA-anti-miR-150 restored SOCS1 levels to normal, as assessed by western blotting and immunofluorescence staining, while scrambled LNA had no effect (Figures 4A and 4B).
Figure 4.

Effect of LNA-Anti-miR-150 on Renal SOCS1 in TIF Mice
(A and B) Renal anti-fibrotic SOCS1 protein on (A) western blotting and (B) immunofluorescence staining. Magnification 200×, scale bar, 400 μm; n = 6, #p < 0.05, each group versus normal group. ∗p < 0.05, LNA-anti-miR-150 versus Scrambled LNA, respectively. Data represent mean ± SD; red staining indicates SOCS1 protein expression.
Effect of LNA-Anti-miR-150 on Renal Inflammation in TIF Mice
Macrophages are key contributors to the pathogenesis of tissue inflammation and fibrosis.27 In the present study, we examined the effects of LNA-anti-miR-150 on macrophages. We measured renal RNA expression of cytokines interferon-γ (IFN-γ), interleukin-6 (IL-6), and tumor necrosis factor (TNF-α), all of which are produced by inflammatory cells including macrophages in mice with interstitial fibrosis. Both early and delayed treatment with LNA-anti-miR-150 abolished the increases in renal IFN-γ, IL-6, and TNF-α mRNAs (Figures 5A–5C). We further examined the expression of two macrophage proteins, CD68 and F4/80, in mouse kidneys manifesting interstitial fibrosis. Renal CD68 expression increased on immunofluorescence staining (Figure 5D) and F4/80 protein levels were upregulated on western blotting of tissue from TIF mice. LNA-anti-miR-150 reversed these changes, while scrambled LNA had no effect (Figure 5E). T lymphocytes also secrete cytokines and we found that both early and delayed treatment of LNA-anti-miR-150 reduced infiltration of CD3+ T cells induced by folic acid administration (Figure S2A).
Figure 5.

The Effect of LNA-Anti-miR-150 on Renal Inflammation in TIF Mice
(A–C) Renal mRNA levels of cytokine (A) IFN-γ, (B) IL-6, and (C) TNF. (D and E) Renal infiltration of macrophages expressed by (D) CD68 immunostaining and (E) western blotting of F4/80 (magnification 200×, scale bar, 400 μm; n = 6, # p < 0.05), each group versus normal group. ∗p < 0.05, LNA-anti-miR-150 versus Scrambled LNA respectively, data represent mean ± SD, red dots indicate positive CD68 staining. See also Figure S2A.
Effect of miR-150 Antagonist on SOCS1/JAK/STAT Pathway in HK-2 Cells
To further investigate the mechanism by which LNA-anti-miR-150 attenuated folic acid-induced tubular damage and initiated renal fibrosis, we performed an in vitro study. Previously we found that LNA-anti-miR-150 reduced macrophage infiltration to reach anti-fibrosis effects in lupus nephritis mice with glomerular damage leading to renal fibrosis.25 Therefore, we co-cultured immortalized human kidney proximal tubular cells (HK-2 cells) with macrophages. As we have demonstrated by luciferase reporter assay that miR-150 decreases SOCS1 gene expression,6 therefore we next studied expression of the Janus kinase (JAK)/signal transducer and activators of transcription (STAT) genes, which are mechanistically downstream of SOCS1. We transfected miR-150 antagonist into HK-2 cells. We found that miR-150 and SOCS1 were co-expressed in untreated HK-2 cells (Figure 6A). We further found that miR-150 levels were increased HK-2 cells following co-culture with macrophages, compared to the HK-2 cells cultured alone. The miR-150 antagonist reversed the increment of miR-150 induced by macrophages, compared to scrambled negative control oligonucleotides (Figure 6B). The mRNA levels of fibrosis-related genes including SOCS1 was increased, while α-SMA, FN, and COL-1 were all reduced toward to normal by the miR-150 antagonist (Figures 6C and 6D). With regard to protein expression of SOCS1 and phosphorylation of its downstream target JAK/STAT in HK-2 cells co-cultured with macrophages, we found the following changes: decreased SOCS1; increased p-JAK1, p-JAK2, p-STAT1, and p-STAT3; and increased pro-fibrotic proteins α-SMA, FN, and COL-1. Following transfection of miR-150 into HK-2 cells, the mRNA and protein expression levels of these genes were similar to the normal control (Figure 6E).
Figure 6.

The Effect of miR-150 Inhibitor on SOCS1/JAK/STAT Pathway in HK-2 Cells
(A) miR-150 (green), SOCS1 protein (red), and co-expression (yellow) in untreated HK-2 cells assessed by double staining using FISH and immunofluorescence (magnification 400×, scale bar, 20 μm). (B) qPCR of miR-150 in HK-2 cells co-cultured with macrophages. (C) SOCS1 mRNA expression. (D) mRNA levels of pro-fibrotic genes including α-SMA, FN, and COL-1. (E) Western blotting for SOCS1, p-JAK1, p-JAK2, p-STAT1, p-STAT3, α-SMA, FN, and COL-1 (n = 3, # p < 0.05, HK-2+ macrophages versus HK-2 cells. ∗p < 0.05, siRNA of miR-150 versus scrambled negative control (NC) oligonucleotides. Data represent mean ± SD. See also Figure S4C.
Renal Expression of miR-150 and Its Regulated Proteins in Human Subjects
To extend our findings to the clinical setting, we established biopsies from ten patients with chronic tubulointerstitial nephritis (cTIN). We selected ten kidney tissue samples obtained from age- and sex-matched controls (Table S1). Renal fibrosis was present in renal biopsies from cTIN patients on Masson staining (Figure 7A). FISH showed increased expression of miR-150 in tubular epithelial cells (Figure 7B). In addition, double staining of miR-150 and aquaporin-1 (AQP1), a proximal tubule marker, confirmed localization of miR-150 to proximal tubules (Figure S3). Increased expression of pro-fibrotic proteins α-SMA, FN, and collagen-1 was present (Figure 7C). Infiltration of CD68+ macrophages and CD3+ T cells was notable (Figures 7D and S2B). Expression of anti-fibrotic protein SOCS1 was decreased in cTIN renal biopsies compared to normal control kidneys (Figure 7E).
Figure 7.

Renal Expression of miR-150 and Target Proteins in cTIN Patients
(A) Masson staining. (B) miR-150 on FISH. (C–E) immunofluorescence (IF) staining: (C) pro-fibrotic α-SMA, FN, and COL-1 protein; (D) macrophage biomarker CD68; and (E) anti-fibrotic SOCS protein. Magnification 400×, scale bar, 10 μm; magnification 100×, scale bar, 200 μm; n = 10, #p < 0.05, renal biopsies from patients with chronic tubulointerstitial nephritis (cTIN) versus normal control kidneys, Data represent mean ± SD. See also Figures S2B, S3, and S4D.
Discussion
The major findings of this study are as follows. In a mouse model of tubulointerstitial fibrosis induced by folic acid, (1) LNA-anti-miR-150 attenuated renal fibrosis, and (2) the decrease of SOCS1 and increase of α-SMA, FN, and COL-1, and macrophage infiltration were reversed by LNA-anti-miR-150. (3) The addition of macrophages to HK-2 cells increased miR-150 expression and regulated the SOCS1/JAK/STAT pathway, while miR-150 antagonist reversed the changes of those proteins, and (4) the changes of miR-150, fibrosis-related proteins, and macrophages present in mice with tubulointerstitial fibrosis and in cultured HK-2 cells were also observed in renal biopsies from patients with chronic tubulointerstitial nephritis.
We previously found an increased expression of miR-150 in renal biopsies from lupus nephritis patients and showed that miR-150 promotes renal fibrosis by downregulating SOCS1 in resident kidney cells.6 Subsequently, transfection of an miR-150 mimic to young mesangial cells was shown to promote cell aging by Liu et al.8 Renal miR-150 levels are increased in minimal change disease cases with a poor prognosis, suggesting that these may represent biopsies that from cases of focal segmental glomerulosclerosis in which the biopsy did not retrieve any scarred glomeruli.10 In mice with AKI following experimental myocardial infarction, miR-150 deficiency alleviated renal fibrosis occurring 8 weeks later.28 Guan et al.7 reported that overexpression of miR-150 in tubular epithelial cells promotes kidney fibrosis by promoting the activation and proliferation of fibroblasts. Taken together, these data suggest that miR-150 contributes to renal fibrosis associated with glomerular injury or tubular damage. Renal fibrosis is a complex process and many genes participate in the pathogenesis.2 It remains unclear whether miR-150 is a key upstream gene that can target multiple pro-fibrotic genes. Inhibition of miR-150 activity is one approach to gain insight into this issue.
Recent clinical trials have used miRNA-based RNAi to treat human diseases.14 These include LNA-anti-miR-155 for lymphoma and leukemia and LNA-anti-miR-9 for heart failure.14 In terms of miRNA-based RNAi in kidney diseases, anti-miR-21 is being tested in a phase II clinical trial targeting hereditary nephritis.29 However, these clinical trials are all ongoing clinical studies and the results have not been disclosed. These clinical trials do suggest a promising era of miRNA-based RNAi serving as a therapeutic approach and the importance of preclinical studies in CKD. Such studies include miRNA-based RNAi including miR-192, miR-34a, miR-29a, and miR-25 in experimental diabetic nephropathy.9,15, 16, 17, 18, 19, 20 Further, miR-132 inhibitor attenuated kidney injury in mice of unilateral ureteral obstruction,21 anti-miR30a ameliorated lipopolysaccharide-induced AKI, and anti-miR-709 improved cisplatin-induced AKI in mice.22,23 Most of the above studies have used a single miRNA to treat a specific disease. Most recently, we found that LNA-anti-miR-150 ameliorates renal fibrosis initialed from glomerular injury in mouse models of lupus nephritis and focal segmental glomerulosclerosis mice.25,26 These findings encouraged us to determine whether miR-150-based RNAi can attenuate tubulointerstitial fibrosis initiated following renal tubular damage.
We focused here on renal fibrosis, a common feature of all end-stage kidney diseases. We selected folic acid-induced a progressive severe renal fibrosis from AKI in mice for in vivo study. We tested initiating treatment with LNA-anti-miR-150 at day 0 before folic acid injection versus at day 2 after AKI onset, with obvious early renal tubular damage. The former experiment model is a prevention approach that could be used in patients at high risk for AKI, while the latter experiment model is a treatment approach for those patients with established AKI. With regard to folic acid nephrotoxicity, we found that both early treatment and delayed treatment attenuated renal fibrosis (Figure 2). These findings are consistent with our previous work in experimental glomerular diseases.25,26 To our knowledge, this is the first experimental study examining the therapeutic effect of miR-150 inhibitor in renal tubulointerstitial fibrosis using both preventive and therapeutic strategies.
To investigate the mechanisms by miR-150 contributes to renal fibrosis in the setting renal tubular damage, we examined fibrosis-related genes, inflammatory cytokines, and kidney resident macrophages. The proteins α-SMA, FN, and COL-1 are characteristic of progressive renal fibrosis.30 We found that both early and delayed treatment with LNA-anti-miR-150 decreased the expression of fibrotic mRNAs (Figure 3) and increased the renal mRNA levels of anti-fibrotic gene SOCS1 (Figure 4). These data are supported by our previous data showing that SOCS1 is a target of miR-150.6 miR-150 has been demonstrated to play a key regulatory role in many immune cells. For example, miR-150 may control B cell development or function by targeting c-Myb;31 miR-150 regulates the development of NK cells and induced NKT cells;32 and miR-150 promotes differentiation and cytolytic function of CD8+ T cells and suppresses function of CD4+ T cells.33,34 In the present study, LNA-anti-miR-150 reversed the renal infiltration of inflammatory cells (Figure 2B) including CD3+ T cells in folic acid-induced renal fibrosis (Figure S2). We speculate that the different effects of miR-150 on different T cell subsets might be due to its cell-specific features. We also found increased numbers of renal macrophages and tubulointerstitial fibrosis, and LNA-anti-miR-150 reversed the increase of macrophages (Figure 5). Macrophages play diverse roles renal inflammation and fibrosis.27 We have explored mechanisms of miR-150 regulation of macrophages.
We conducted co-culture studies involving renal tubular HK-2 cells and macrophages, followed by transfection of a miR-150 antagonist. We found co-expression of miR-150 and SOCS1 in HK-2 cells by double staining with FISH for miR-150 and IF for SOCS1. These results suggested the interaction between miR-150 and SOCS1 (Figure 6A). We also found that miR-150 antagonist reversed the macrophages-induced decrease of SOCS1, the increase of α-SMA, FN, and COL-1, as well as overexpression of macrophage CD68. In addition, the miR-150 antagonist also partially suppressed the macrophages-induced upregulation of p-JAK1, p-JAK2, p-STAT1, and p-STAT3 in HK-2 cells (Figure 6). Our findings suggest that the miR-150/SOCS1/pro-fibrotic protein pathway was not the only pathway contributing to the pathogenesis of folic acid-induced renal fibrosis. In this model, SOCS1/JAK/STAT pathway may also act downstream of miR-150. Consistent with the findings from other researchers, a dysregulated SOCS1/JAK/STAT pathway also contributes to the pathogenesis of inflammatory disorders in murine diabetic nephropathy and in rats of cisplatin-induced AKI, as shown from in vivo and in vitro data.35,36
Macrophages are known to activate the SOCS1/JAK/STAT pathway.37 Our findings suggest that macrophages activate the SOCS1/JAK/STAT pathway following kidney injury and thus contribute to the pathogenesis of renal fibrosis. The renal protective role of miR-150 inhibitor in renal fibrosis might be mediated by regulating SOCS1/JAK/STAT pathway in addition to inhibiting macrophage functions, as shown in the graphic abstract. To our knowledge, this is the first time the addition of JAK/STAT has been shown to act downstream genes whose expression is activated by miR-150 in renal fibrosis in folic acid-induced tubular injury in a mouse model and in HK-2 cells.
Lastly, we compared findings obtained from mice with folic-acid-induced interstitial fibrosis and co-cultured HK-2 cells with macrophages in renal biopsies from patients with chronic tubulointerstitial nephritis. In the renal biopsies, we found that renal overexpression of miR-150 and pro-fibrotic proteins including of α-SMA, FN, and COL-1, suppression of SOCS1, and the increased infiltration of macrophages indicated by CD68 positive staining (Figure 7). The findings in renal biopsies from patients with chronic interstitial nephritis were similar to those in murine with tubulointerstitial fibrosis and in HK-2 cells co-cultured with macrophages. It is well known that the injured proximal tubules can also elicit an extensive pro-inflammatory response via multiple mechanisms.38 The limitation of our current study is that we did not investigate the possible differentiation between proximal tubule-specific inflammation and macrophage-induced inflammation. This will be conducted in our future project.
In conclusion, the renal protective mechanism of miR-150-based RNAi may be mediated by regulating pro-fibrotic genes and inflammation via infiltration of macrophages and SOCS1/JAK/STAT pathway. miR-150 based RNAi appears to be a novel promising therapeutic agent for renal tubulointerstitial fibrosis.
Materials and Methods
TIF Mouse Model
Male ICR mice (12–16 weeks old, 30–35 g) were purchased from Beijing Vital River Laboratory Animal Technology (Beijing, China), housed 4 per cage, and allowed free access to standard food and drinking water. Mice were maintained under a 12 h light/dark cycle with fixed temperature at 24°C and humidity (40%–55%). Folic acid (Sigma-Aldrich, MO, USA) was peritoneally injected to male ICR mice to induce AKI on day 2 and TIF on day 30 after one bolus of injection.39 Animal studies were approved in advance by the Animal Care and Use Committee of China Medical University (15052111), and were performed following NIH Animal Care and Use Guidelines.
Animal Experimental Design and Samples Collection
Natural History of Folic-Acid-Induced Kidney Injury
Male ICR mice (n = 24) were used to determine the natural history of folic-acid-induced kidney injury. Folic acid was dissolved in 0.3 mM NaHCO3 solution. Approximately 0.2 mL of folic acid (250 mg/kg body weight) or vehicle were injected intraperitoneally into each mouse on day 0. Mice were anesthetized and blood samples were collected from abdominal aorta and the kidneys were collected after perfusion with PBS to remove intrarenal blood on day 2, day 7, and day 30 (Figure S4A).
The Effect of LNA-Anti-miR-150 Treatment on TIF in Mice on Day 30
We administered LNA-anti-miR-150 or scrambled LNA (Exiqon, MA, USA), each at dose of 2 mg/kg, twice weekly for total of eight doses. ICR mice (n = 36) were divided into 6 groups: normal control, folic acid alone, prophylactic treatment (LNA-anti-miR-150 or scrambled LNA) starting at day 0 before AKI induction and delayed treatment (LNA-anti-miR-150 or scrambled LNA) starting at day 2 after AKI induction respectively for 28 days or 30 days. As described above, blood samples and kidneys were collected on day 30 after folic acid injection (Figure S4B).
For all experimental animals, serum was isolated from blood samples and was stored at −80°C. The kidneys were divided into four parts: one piece was fixed into 4% paraformaldehyde and tissue was embedded in paraffin, one piece was put into optimal cutting temperature compound (OCT) (Sakura, CA, USA) and stored at −80°C, and the remaining two tissue pieces were stored at −80°C for isolation of total protein and RNA.
Cell Lines
HK-2 cells and the human monocytic cell line THP1 were purchased from ATCC (Manassas, VA, USA). HK-2 cells were cultured in DMEM/F 12 medium supplemented with 10% FBS. THP1 cell number was adjusted to 1 × 106 cells/mL and plated in RPMI 1640 Basic supplemented with 10% FBS for 24 h. Thereafter, THP1 cells were transferred to the RPMI 1640 Basic medium with 100 ng/mL phorbol 12-myristate 13-acetate (Sigma-Aldrich, CA, USA) and cultured for 48 h at 37°C. These cells were characterized as macrophage.
Co-culture of HK-2 Cells and Macrophage Cells and Transfection of miR-150 Inhibitor
HK-2 cells (105/mL) were seeded in the isolated upper compartments of a Transwell system plate and were allowed to grow to 70%–80% confluence. Small interfering RNA (siRNA; Ribobio, Guangdong, China) including siRNA against miR-150 and its negative control were transfected to HK-2 cells using Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer’s instructions. The lower compartment of the Transwell plate with cultured macrophages covering 90% disc was set on the lower chamber of the co-culture system. HK-2 cells were harvested 24 h after the transfection and co-culture (Figure S4C).
Kidney Tissues from Human Subjects
Renal biopsies were from ten cTIN patients and were obtained between June 2018 and December 2019 in the Department of Nephrology. cTIN was diagnosed by two renal pathologists. The exclusion criteria for cTIN were the patients with age <18 or >75, hypertension, diabetes, possible hepatitis B infection, tumors, and pregnancy. Normal kidney tissues were obtained from the nephrectomy of patients with renal tumor at The Department of Urology. These tissues located at least 5 cm away from the tumors.25 A human subject research protocol was approved in advance by the Institutional Review Board of Shengjing Hospital of China Medical University. All subjects provided the written consent form. Renal function of human subjects was measured by clinical central laboratory of the hospital. Age and gender of human subjects were matched (Figure S4D; Table S1).
Serum Biochemistry Assays
Scr, BUN, alanine aminotransferase (ALT), and aspartate amino transferase (AST) were measured by an Architect c16000 device (Abbott, IL, USA).
Histology and Immunofluorescence Studies
Quantitative scoring of tubular injury was assessed on PAS-stained sections using a semiquantitative scale as described previously.40 Tubulointerstitial fibrosis was assessed on Masson stained sections as the estimated percentage of blue area to total area.41 Frozen OCT fixed kidney tissue sections (5 μm) slides were incubated with antibodies of α-SMA, FN, COL-1, SOCS1, CD68, and CD3 were incubated at 4°C overnight followed by incubation with secondary antibodies conjugated with Alexa 568 or Alexa 488 (Table S2) at room temperature for 1 h. After three washes with PBS, slides were mounted with 4’, 6-diamidino-2-phenylindole (DAPI) medium for 10 min. Images were captured using immunofluorescence microscopy (Nikon, Tokyo, Japan). Staining intensity was semi-quantified by Image-Pro Plus 6.0 (Media Cybernetics, MD, USA) as previously described.25,26
Western Blotting
Equal amounts of total protein from kidney tissues (50 μg) or HK2 cells (30 μg) were separated by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore Immobilon-P, MA, USA). And the membranes were incubated at 4°C overnight with primary antibodies against α-SMA, FN, COL-1, SOCS1, F4/80, and JAK/STAT pathway related proteins, including JAK1, p-JAK1, JAK2, p-JAK2, STAT1, p-STAT1, STAT3, and p-STAT3 (Table S2). After washing, antibodies goat anti-mouse/rabbit immunoglobulin G (IgG) was added for 1 h at room temperature (Table S2). Antibody-antigen binding was detected by high-sig ECL western blotting substrate and visualized by the Tanon 5500 imaging system (Shanghai, China). The protein loading variation was normalized by α-tubulin or GAPDH. Blot density was analyzed by NIH ImageJ software (Bethesda, MD, USA)
FISH of Kidney Tissues
For FISH, sections from paraffin-embedded human kidney biopsies, normal control kidney tissues, and mouse kidney tissues were cut at 4 μm thickness. Sections were deparaffinized, rehydrated, digested with trypsin, and hybridized with a digoxigenin-horseradish peroxidase (DIG-HRP)-labeled oligonucleotide probe complementary to miR-150 (Table S3) at 37°C overnight following by stained with anti-DIG- HRP antibody for 50 min, FITC-tyramine signal amplification for 5 min, and DAPI for 5 min. Images were captured by immunofluorescence microscopy (Nikon, Tokyo, Japan). Positive staining areas were semi-quantified as reported by Huang et al.42
Double Staining of FISH and IF
For double staining of miR-150 and AQP1 in human kidneys, FISH of miR-150 was performed with the protocol described above. After washing three times with PBS, slides were incubated with primary antibody against AQP1at 4°C overnight (Table S2). Following three washes with PBS, the slides were incubated with secondary antibody labeled with Alexa 568 for 1 h in the dark at room temperature, followed by a PBS wash. Slides were mounted with DAPI medium for 5 min. Images were captured by immunofluorescent microscopy (Nikon, Tokyo, Japan).
For double immunofluorescenc staining of miR-150 FISH and SOCS1, HK-2 cells were cultured on the slides placed in the culture dishes with HK-2 medium. At confluence, cells were fixed using in situ hybridization fixative (Servicebio, Wuhan, China) for 15 min at room temperature. FISH to detect miR-150 and IF of SOCS1 was performed with the same protocol as described for kidney tissues.
qPCR
Total RNA was isolated from the frozen kidney tissues and harvested HK-2 cells using TRIzol reagent (Life Technologies, CA, USA) according to instructions. Primers were designed using Primer Express (Applied Biosystems, CA, USA) and synthesized by Life Technologies (Shanghai, China). Real-time fluorescence signal was detected with QuantStudio 6 Flex quantitative real-time PCR system (Applied Biosystems). Actin and sno202/U6 were used as endogenous controls for mRNA and mouse/human miR-150, respectively (Table S3). The relative level of each target gene was calculated using 2−ΔΔCt method (Δ Ct: Ct of endogenous control – Ct of individual target gene)
Statistical Analysis
SPSS 22.0 (SPSS, IL, USA) and Prism 8.0 (GraphPad, CA, USA) software were used for statistical analysis and graphing. Quantitative data were expressed as mean ± SD. Differences between groups were analyzed for statistical significance by one or two way-ANOVA for animal and cell data, and t test for human data. A value p <0.05 was considered as statistically significant.
Author Contributions
J.L. contributed to conducting experiments and manuscript writing. H.Z. designed, supervised the experiments, and finalized the manuscript. D.W., X.C., and D.L. participated in data collection and analysis. C.J., Y.Z., and Y.W. contributed to human kidney collection. J.F., P.S.T.Y., and J.P. participated in advisory roles. J.B.K. provided advice and edited the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors thanks Christine Wang for illustration assistance. This research was supported by Chinese Nature Science Foundation 81770698 and 81370835 (H.Z.); National Key R&D Program of China 2017YFC0907400 (Y.Z.); Key R&D Guidance Plan of Liaoning Province (2019JH8/10300009, H.Z.); and Pandeng Scholar of Liaoning Province 2013222 (H.Z.). J.B.K. is supported by the Intramural Research Program, NIDDK, NIH.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.10.008.
Supplemental Information
References
- 1.Chen T.K., Knicely D.H., Grams M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA. 2019;322:1294–1304. doi: 10.1001/jama.2019.14745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou D., Liu Y. Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat. Rev. Nephrol. 2016;12:68–70. doi: 10.1038/nrneph.2015.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang W., Zhang C., Chen H., Li L., Tu Y., Liu C., Shi S., Zen K., Liu Z. Evaluation of microRNAs miR-196a, miR-30a-5P, and miR-490 as biomarkers of disease activity among patients with FSGS. Clin. J. Am. Soc. Nephrol. 2014;9:1545–1552. doi: 10.2215/CJN.11561113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel V., Williams D., Hajarnis S., Hunter R., Pontoglio M., Somlo S., Igarashi P. miR-17∼92 miRNA cluster promotes kidney cyst growth in polycystic kidney disease. Proc. Natl. Acad. Sci. USA. 2013;110:10765–10770. doi: 10.1073/pnas.1301693110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J., Kwan B.C., Lai F.M., Tam L.S., Li E.K., Chow K.M., Wang G., Li P.K., Szeto C.C. Glomerular and tubulointerstitial miR-638, miR-198 and miR-146a expression in lupus nephritis. Nephrology (Carlton) 2012;17:346–351. doi: 10.1111/j.1440-1797.2012.01573.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhou H., Hasni S.A., Perez P., Tandon M., Jang S.I., Zheng C., Kopp J.B., Austin H., 3rd, Balow J.E., Alevizos I., Illei G.G. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. J. Am. Soc. Nephrol. 2013;24:1073–1087. doi: 10.1681/ASN.2012080849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guan H., Peng R., Mao L., Fang F., Xu B., Chen M. Injured tubular epithelial cells activate fibroblasts to promote kidney fibrosis through miR-150-containing exosomes. Exp. Cell Res. 2020;392:112007. doi: 10.1016/j.yexcr.2020.112007. [DOI] [PubMed] [Google Scholar]
- 8.Liu X., Fu B., Chen D., Hong Q., Cui J., Li J., Bai X., Chen X. miR-184 and miR-150 promote renal glomerular mesangial cell aging by targeting Rab1a and Rab31. Exp. Cell Res. 2015;336:192–203. doi: 10.1016/j.yexcr.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 9.Wang J.Y., Gao Y.B., Zhang N., Zou D.W., Wang P., Zhu Z.Y., Li J.Y., Zhou S.N., Wang S.C., Wang Y.Y., Yang J.K. miR-21 overexpression enhances TGF-β1-induced epithelial-to-mesenchymal transition by target smad7 and aggravates renal damage in diabetic nephropathy. Mol. Cell. Endocrinol. 2014;392:163–172. doi: 10.1016/j.mce.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 10.Lu M., Wang C., Yuan Y., Zhu Y., Yin Z., Xia Z., Zhang C. Differentially expressed microRNAs in kidney biopsies from various subtypes of nephrotic children. Exp. Mol. Pathol. 2015;99:590–595. doi: 10.1016/j.yexmp.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Van Craenenbroeck A.H., Van Craenenbroeck E.M., Van Ackeren K., Hoymans V.Y., Verpooten G.A., Vrints C.J., Couttenye M.M. Impaired vascular function contributes to exercise intolerance in chronic kidney disease. Nephrol. Dial. Transplant. 2016;31:2064–2072. doi: 10.1093/ndt/gfw303. [DOI] [PubMed] [Google Scholar]
- 12.Fujii R., Yamada H., Munetsuna E., Yamazaki M., Ohashi K., Ishikawa H., Maeda K., Hagiwara C., Ando Y., Hashimoto S. Associations of Circulating MicroRNAs (miR-17, miR-21, and miR-150) and Chronic Kidney Disease in a Japanese Population. J. Epidemiol. 2020;30:177–182. doi: 10.2188/jea.JE20180233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie Y., Jia Y., Cuihua X., Hu F., Xue M., Xue Y. Urinary Exosomal MicroRNA Profiling in Incipient Type 2 Diabetic Kidney Disease. J. Diabetes Res. 2017;2017:6978984. doi: 10.1155/2017/6978984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Setten R.L., Rossi J.J., Han S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019;18:421–446. doi: 10.1038/s41573-019-0017-4. [DOI] [PubMed] [Google Scholar]
- 15.Putta S., Lanting L., Sun G., Lawson G., Kato M., Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2012;23:458–469. doi: 10.1681/ASN.2011050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu L., Wang Q., Guo F., Ma X., Ji H., Liu F., Zhao Y., Qin G. MicroRNA-27a Induces Mesangial Cell Injury by Targeting of PPARγ, and its In Vivo Knockdown Prevents Progression of Diabetic Nephropathy. Sci. Rep. 2016;6:26072. doi: 10.1038/srep26072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mu J., Pang Q., Guo Y.H., Chen J.G., Zeng W., Huang Y.J., Zhang J., Feng B. Functional implications of microRNA-215 in TGF-β1-induced phenotypic transition of mesangial cells by targeting CTNNBIP1. PLoS ONE. 2013;8:e58622. doi: 10.1371/journal.pone.0058622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L., He S., Guo S., Xie W., Xin R., Yu H., Yang F., Qiu J., Zhang D., Zhou S., Zhang K. Down-regulation of miR-34a alleviates mesangial proliferation in vitro and glomerular hypertrophy in early diabetic nephropathy mice by targeting GAS1. J. Diabetes Complications. 2014;28:259–264. doi: 10.1016/j.jdiacomp.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu Y.C., Chang P.J., Ho C., Huang Y.T., Shih Y.H., Wang C.J., Lin C.L. Protective effects of miR-29a on diabetic glomerular dysfunction by modulation of DKK1/Wnt/β-catenin signaling. Sci. Rep. 2016;6:30575. doi: 10.1038/srep30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y., Li H., Liu J., Han P., Li X., Bai H., Zhang C., Sun X., Teng Y., Zhang Y. Variations in MicroRNA-25 Expression Influence the Severity of Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017;28:3627–3638. doi: 10.1681/ASN.2015091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bijkerk R., de Bruin R.G., van Solingen C., van Gils J.M., Duijs J.M., van der Veer E.P., Rabelink T.J., Humphreys B.D., van Zonneveld A.J. Silencing of microRNA-132 reduces renal fibrosis by selectively inhibiting myofibroblast proliferation. Kidney Int. 2016;89:1268–1280. doi: 10.1016/j.kint.2016.01.029. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L., Li J., Cui L., Shang J., Tian F., Wang R., Xing G. Retraction. Biomed. Pharmacother. 2018;101:842–851. [Google Scholar]
- 23.Guo Y., Ni J., Chen S., Bai M., Lin J., Ding G., Zhang Y., Sun P., Jia Z., Huang S. MicroRNA-709 Mediates Acute Tubular Injury through Effects on Mitochondrial Function. J. Am. Soc. Nephrol. 2018;29:449–461. doi: 10.1681/ASN.2017040381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin L.B., Li Z.Y., Li H., Fan X.Q., Liu H.G., Dong X.M., Jia W.Y. Inhibitive effects of microRNA-34a on protecting against ischemia-reperfusion injury of vital organs in hemorrhagic shock pregnant mice. Eur. Rev. Med. Pharmacol. Sci. 2018;22:1812–1818. doi: 10.26355/eurrev_201803_14600. [DOI] [PubMed] [Google Scholar]
- 25.Luan J., Fu J., Chen C., Jiao C., Kong W., Zhang Y., Chang Q., Wang Y., Li D., Illei G.G. LNA-anti-miR-150 ameliorated kidney injury of lupus nephritis by inhibiting renal fibrosis and macrophage infiltration. Arthritis Res. Ther. 2019;21:276. doi: 10.1186/s13075-019-2044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi H., Fu J., Luan J., Jiao C., Cui X., Cao X., Zhang Y., Wang Y., Kopp J.B., Pi J., Zhou H. miR-150 inhibitor ameliorates adriamycin-induced focal segmental glomerulosclerosis. Biochem. Biophys. Res. Commun. 2020;522:618–625. doi: 10.1016/j.bbrc.2019.11.096. [DOI] [PubMed] [Google Scholar]
- 27.Tang P.M., Nikolic-Paterson D.J., Lan H.Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019;15:144–158. doi: 10.1038/s41581-019-0110-2. [DOI] [PubMed] [Google Scholar]
- 28.Ranganathan P., Jayakumar C., Tang Y., Park K.M., Teoh J.P., Su H., Li J., Kim I.M., Ramesh G. MicroRNA-150 deletion in mice protects kidney from myocardial infarction-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2015;309:F551–F558. doi: 10.1152/ajprenal.00076.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bajan S., Hutvagner G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells. 2020;9:137. doi: 10.3390/cells9010137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Djudjaj S., Boor P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Aspects Med. 2019;65:16–36. doi: 10.1016/j.mam.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Xiao C., Calado D.P., Galler G., Thai T.H., Patterson H.C., Wang J., Rajewsky N., Bender T.P., Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 32.Bezman N.A., Chakraborty T., Bender T., Lanier L.L. miR-150 regulates the development of NK and iNKT cells. J. Exp. Med. 2011;208:2717–2731. doi: 10.1084/jem.20111386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith N.L., Wissink E.M., Grimson A., Rudd B.D. miR-150 Regulates Differentiation and Cytolytic Effector Function in CD8+ T cells. Sci. Rep. 2015;5:16399. doi: 10.1038/srep16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sang W., Sun C., Zhang C., Zhang D., Wang Y., Xu L., Zhang Z., Wei X., Pan B., Yan D. MicroRNA-150 negatively regulates the function of CD4(+) T cells through AKT3/Bim signaling pathway. Cell. Immunol. 2016;306-307:35–40. doi: 10.1016/j.cellimm.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Liu Q., Xing L., Wang L., Yao F., Liu S., Hao J., Liu W., Duan H. Therapeutic effects of suppressors of cytokine signaling in diabetic nephropathy. J. Histochem. Cytochem. 2014;62:119–128. doi: 10.1369/0022155413512493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsogbadrakh B., Ryu H., Ju K.D., Lee J., Yun S., Yu K.S., Kim H.J., Ahn C., Oh K.H. AICAR, an AMPK activator, protects against cisplatin-induced acute kidney injury through the JAK/STAT/SOCS pathway. Biochem. Biophys. Res. Commun. 2019;509:680–686. doi: 10.1016/j.bbrc.2018.12.159. [DOI] [PubMed] [Google Scholar]
- 37.Malemud C.J. Suppression of Autoimmune Arthritis by Small Molecule Inhibitors of the JAK/STAT Pathway. Pharmaceuticals (Basel) 2010;3:1446–1455. doi: 10.3390/ph3051446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato Y., Takahashi M., Yanagita M. Pathophysiology of AKI to CKD progression. Semin. Nephrol. 2020;40:206–215. doi: 10.1016/j.semnephrol.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 39.Doi K., Leelahavanichkul A., Hu X., Sidransky K.L., Zhou H., Qin Y., Eisner C., Schnermann J., Yuen P.S., Star R.A. Pre-existing renal disease promotes sepsis-induced acute kidney injury and worsens outcome. Kidney Int. 2008;74:1017–1025. doi: 10.1038/ki.2008.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong W., Fu J., Liu N., Jiao C., Guo G., Luan J., Wang H., Yao L., Wang L., Yamamoto M. Nrf2 deficiency promotes the progression from acute tubular damage to chronic renal fibrosis following unilateral ureteral obstruction. Nephrol. Dial. Transplant. 2018;33:771–783. doi: 10.1093/ndt/gfx299. [DOI] [PubMed] [Google Scholar]
- 41.Liu F., Wei C.C., Wu S.J., Chenier I., Zhang S.L., Filep J.G., Ingelfinger J.R., Chan J.S. Apocynin attenuates tubular apoptosis and tubulointerstitial fibrosis in transgenic mice independent of hypertension. Kidney Int. 2009;75:156–166. doi: 10.1038/ki.2008.509. [DOI] [PubMed] [Google Scholar]
- 42.Huang X., Li Z., Zhang Q., Wang W., Li B., Wang L., Xu Z., Zeng A., Zhang X., Zhang X. Circular RNA AKT3 upregulates PIK3R1 to enhance cisplatin resistance in gastric cancer via miR-198 suppression. Mol. Cancer. 2019;18:71. doi: 10.1186/s12943-019-0969-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.