

Abstract
The first chemical synthesis of lacto-N-neohexaose (LNnH) has been completed using a convergent synthetic strategy. The reaction conditions and donor–acceptor combinations have been carefully refined to minimize side reactions and achieve high yields in all glycosylation steps. Lacto-N-neotetraose, another common human milk oligosaccharide, was also synthesized en route to the target LNnH.
Human milk oligosaccharides (HMO) are complex glycans that are highly abundant in human milk and have been associated with many beneficial health effects via various mechanisms. HMO composition can vary between different mothers and has also been found to vary geographically.1 All HMO sequences are variations of a specific structural blueprint, but all glycans are composed of lactose (Galβ1 → 4Glc) at their reducing end. The lactose unit can be elongated at the galactose moiety at the C-3 position with lacto-N-biose (Galβ1 → 3GlcNAc, type 1 chain), which prevents further elongation in this direction. The galactose moiety of the reducing-end lactose can also be elongated by N-acetyllactosamine (Galβ1 → 4GlcNAc, type 2 chain) units. And this elongation, which can take place either at the C-3 or C-6 position of galactose, is open for further oligosaccharide chain extension. In addition, the HMO core structures can be fucosylated and/or sialylated.2 Structures of many HMO are known,3,4 and many have already been prepared enzymatically and/or chemically.5 However, the exact biological mechanisms of action of a majority of individual HMO remain unknown6 beyond prebiotic effects7 due to the lack of well-defined HMO in sufficient quantiles.8
With the expectation that developing reliable synthetic methods for obtaining individual HMO will boost further innovations in this area, we have reported the total syntheses of a number of glycans. Our initial efforts included syntheses of two linear tetrasaccharide structures (para-HMO): lacto-N-neotetraose (LNnT)9 and lacto-N-tetraose (LNT).10 Very recently, we reported the first synthesis of a branched hexasaccharide (iso-HMO), lacto-N-hexaose (LNH).11 Reported herein is our further investigation in the field of HMO dedicated to the synthesis of another branched hexasaccharide, lacto-N-neohexaose (LNnH). LNnH 1 is structurally derived from LNnT 2, and represents one of the most common HMO core structures.12 In LNnH lactose disaccharide at the reducing end (AB, Fig. 1), common for all HMO, is branched at the galactose residue (B). Both at C-3 and C-6 it is elongated with N-acetyllactosamine disaccharide (CD and EF).
Fig. 1.
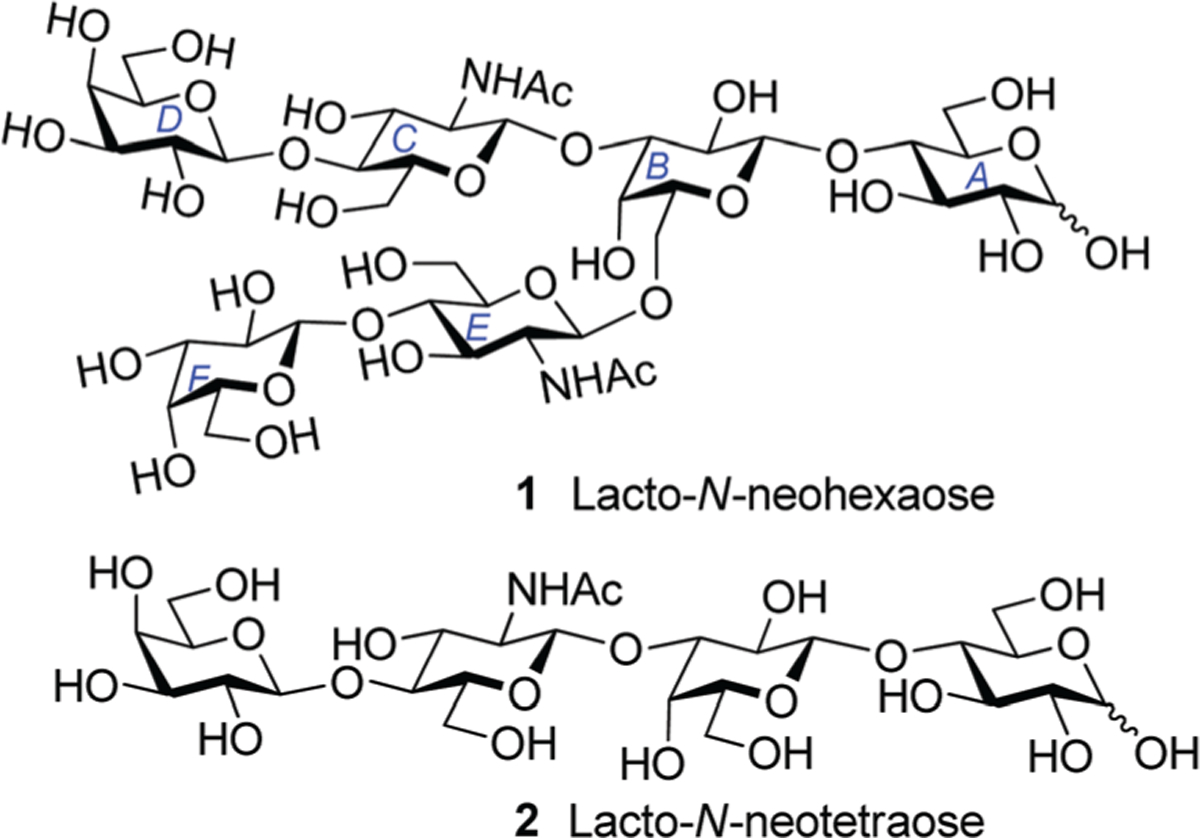
Chemical structures of LNnH and LNnT.
Many chemical9,13,14 and enzymatic syntheses8,15,16 of LNnT are known. LNnH has been synthesized chemoenzymatically,8,17 and some spacer-containing, protected LNnH derivatives have been synthesized chemically using polymer supports.18 However, to the best of our knowledge, no chemical synthesis of LNnH 1 has yet emerged. Human milk isolates of LNnH are available from a variety of commercial vendors, but this common HMO is not yet available in large quantities and at accessible cost for mainstream research and application. Reported herein as an efficient method for the scalable chemical synthesis of LNnH 1 and, by extension, LNnT 2.
Recently we have synthesized LNnT 2 using convergent and linear assembly strategies.9 The protecting groups in tetrasaccharide 3 obtained over the course of our previous work9 were strategically placed to allow for the synthesis of other derivatives (Scheme 1). We envisaged that selective removal of 6′-O-picoloyl (Pico) temporary protecting group in 3 would provide acceptor 4 suitable for subsequent branching to obtain LNnH. To follow this lead, 6′-O-Pico in 3 was removed in the presence of copper(II) acetate monohydrate in MeOH/CH2Cl2, and tetrasaccharide 4 was synthesized in 97% yield. To our disappointment, all attempts to glycosylate acceptor 4 with lactosamine thioglycoside donor 59 were largely unsuccessful and only small amounts (<25%) of the desired hexasaccharide were formed. Instead, many side products derived from glycosyl donor 5 have been observed, whereas a majority of glycosyl acceptor 4 remained intact. In further attempts to enhance the efficiency of this coupling we applied less bulky glucosamine thioglycoside donor 6. Although the formation of minor amounts of the desired pentasaccharide were observed by mass spectroscopy, these glycosylations were largely unsuccessful.
Scheme 1.
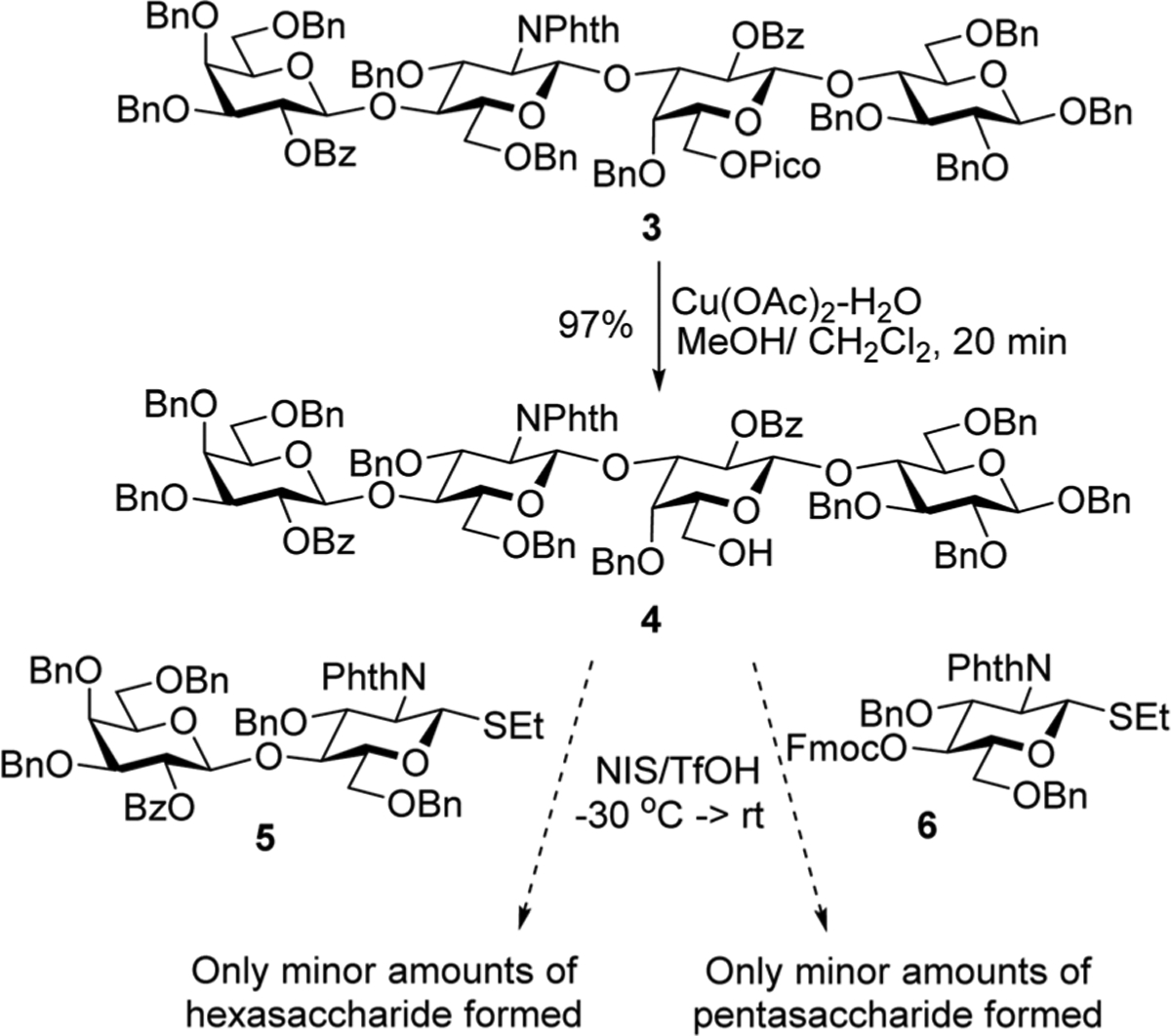
First attempted assembly of LNnH.
Over the course of our previous synthetic studies of HMO, we emphasized that glycosyl donor and acceptor combination was essential to ensure successful glycosylations. However, even upon extended investigation of lactosamine phosphate and trichloroacetimidate donors instead of thioglycosides 5 and 6, we failed to synthesize the backbone of LNnH. Having explored all possibilities available to us, we anticipated that it is axial 4′-O-benzyl group that hinders glycosylation at 6′-OH of acceptor 4. A similar hurdle was encountered during our previous synthesis of LNH.11 In order to decrease the steric hin-drance, we decided to follow the same strategy as that developed for the modified synthesis of LNH, in which we employed 4,6-O-benzylidene protected lactose building block 7.11 Over the course of our previous work with LNH, we observed that if the benzylidene acetal is removed at the tetrasaccharide stage, the resulting 4,6-diol would offer a far more accessible glycosyl acceptor site for glycosylation. This would be essential for our route employing the glycosylation with bulky disaccharide donor 5.
With this strategic adjustment, we attempted to glycosylate acceptor 7 with thioglycoside donor 5. This reaction was slug-gish and inefficient, and despite all attempts to push the reaction to completion, significant amounts of the acceptor remained. After a quick screening of other leaving groups, we discovered that trichloroacetimidate 9 is an effective donor to glycosylate acceptor 7. Glycosyl donor 9 was prepared from thioglycoside precursor 5 that was first converted into the corresponding hemiacetal 8 in 83% over two steps involving the anomeric bromination with Br2 in CH2Cl2 followed by hydrolysis of the intermediate bromide in the presence of Ag2CO3 in wet acetone (Scheme 2). Next, installation of the tri-chloroacetimidoyl group was performed using trichloroaceto-nitrile (CCl3CN) in the presence of 1,8-diazabicylco[5.4.0] undec-7-ene (DBU) to afford lacto-N-biose donor 9 in 87% yield. Glycosylation of acceptor 7 with trichloroacetimidate donor 9 was conducted in the presence of catalytic TMSOTf and the desired tetrasaccharide 10 was obtained in a good yield of 83% with complete β-stereoselectivity. It is noteworthy that the corresponding glycosyl phosphate donor9 gave a somewhat inferior result in this application. Its activation demanded 2.0 equiv. of TMSOTf that often provoked competing cleavage of benzylidene acetal. As a result, product 10 was contaminated with tetrasaccharide 11, which was the result of benzylidene cleavage. While this offers a possibility of obtaining 11 directly from the disaccharide building blocks in one pot, we have not explored this potential advantage. The deprotection of the 4′,6′-O-benzylidene acetal in intermediate 10 was somewhat low-yielding in the presence of TFA in wet DCM, hence we did the reaction in the presence of TsOH and EtSH in MeOH/CH2Cl2, an approach adapted in our previous work.11 As a result, tetrasaccharide diol 11 was obtained in 87% yield (Scheme 2).
Scheme 2.
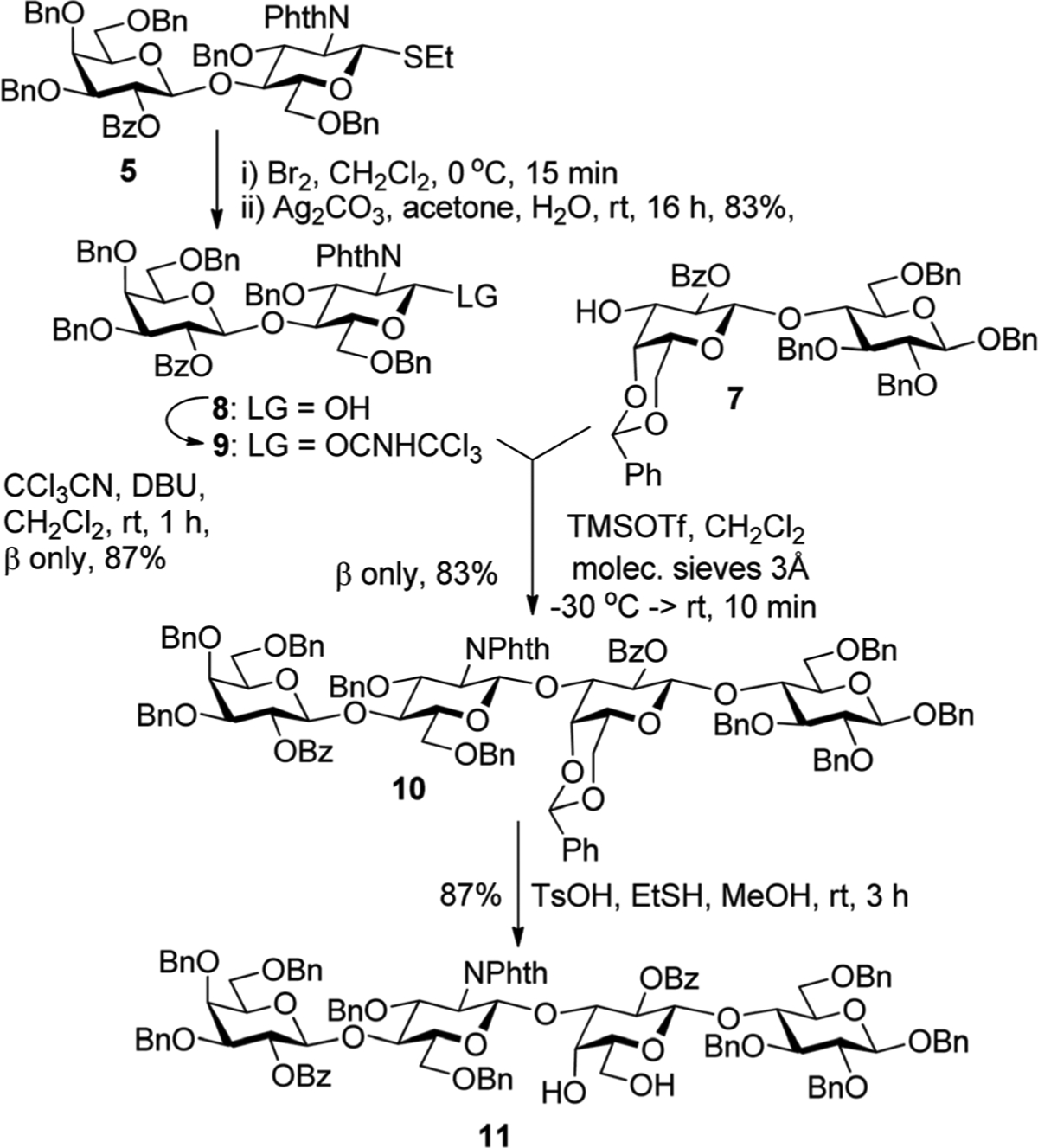
Convergent synthesis of intermediate 11.
Next, we attempted glycosylation of tetrasaccharide diol 11 with lactosamine thioglycoside donor 5. Even glycosylation of this more accessible glycosyl acceptor was not as straight-forward as we had hoped for. Standard reaction condition employing NIS/TfOH as promoters produced the desired hexasaccharide 12 in an unremarkable yield of 57%. Although this result was a significant improvement in respect to our initial attempts to glycosylate acceptor 4, wherein the yields rarely exceeded 25%, we endeavored to pursue further improvement the outcome of this challenging reaction. With this intention, we identified the major side reactions hampering the yield of the product: fairly rapid hydrolysis of lactosamine donor 5 and the formation of the 1 → 4-linked hexasaccharide alongside the desired 1 → 6-linked counterpart 12. The formation of somewhat unexpected 1 → 4-linked hexasaccharide was the main reason we were unable to introduce lactosamine units at C-3 and C-6 positions concomitantly. To suppress these side reactions, we investigated milder promoters, dimethyl(thio-methyl)sulfonium triflate (DMTST)19,20 and NIS/AgOTf.21 The former promoter provided a comparable result that achieved with NIS/TfOH. With the latter promoter, known for providing a slower release of the iodonium ion, minimal side products were observed. After careful refinement of the reaction conditions, we achieved the desired protected LNnH precursor 12 in a commendable yield of 82% (Scheme 3). This result was obtained by conducting the reaction at −40 °C for 30 min, and then allowing the reaction temperature gradually increase (to about −18 °C) over the course of the additional 30 min period.
Scheme 3.
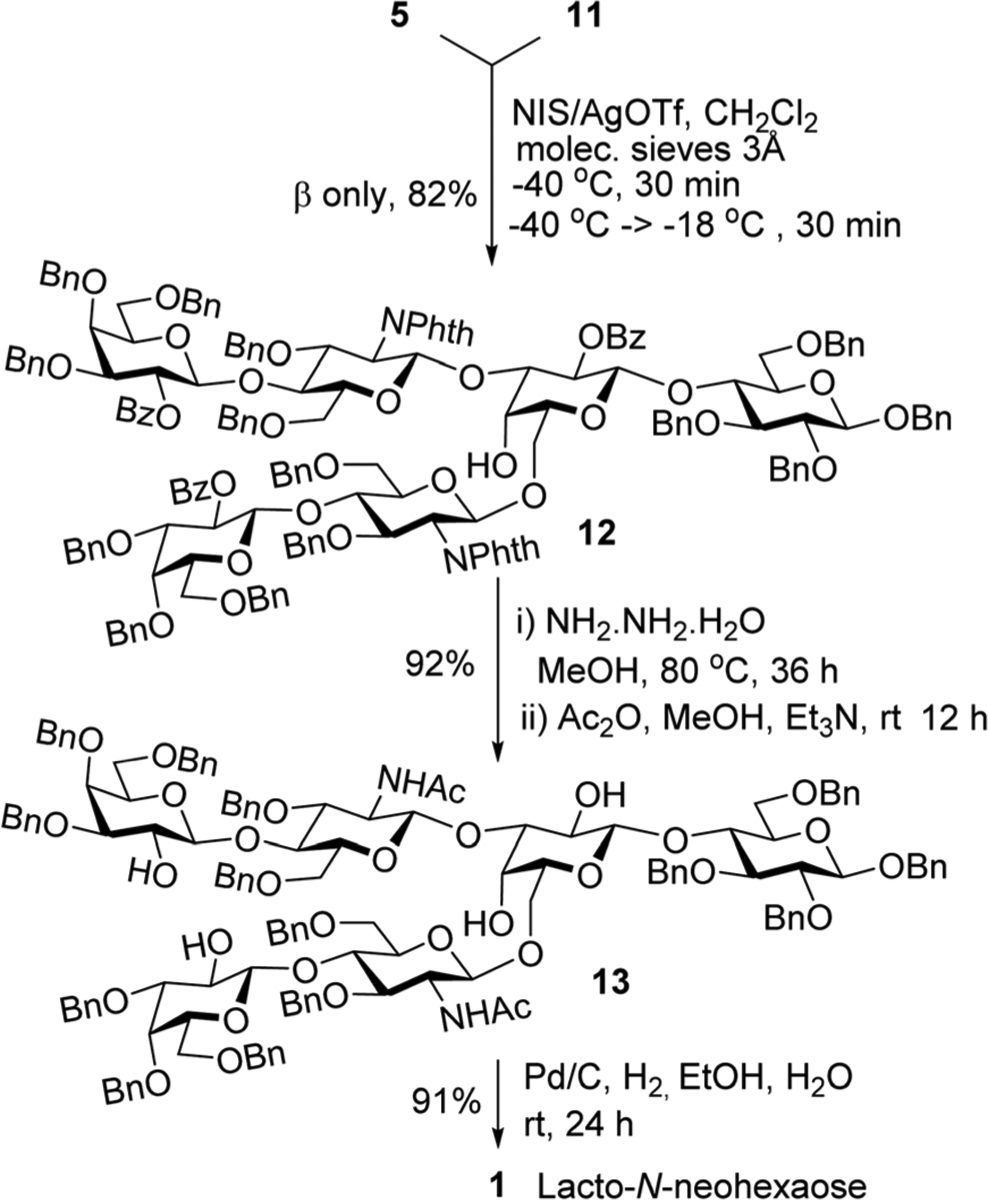
Convergent synthesis of hexasaccharide 12 and its deprotection to obtain LNnH.
With the protected intermediate 12 in hand, we then endeavored a series of deprotection steps to obtain the target LNnH hexasaccharide 1. Deprotection of the phthalimido and benzoyl ester groups was achieved by refluxing compound 12 with NH2NH2-H2O in MeOH, followed by N-acetylation with acetic anhydride in the presence of Et3N in MeOH to furnish hexasaccharide 13 in 92% yield (Scheme 3). Finally, the benzyl groups were removed by hydrogenation the presence of 10% palladium on charcoal in wet ethanol to obtain target LNnH 1 in 91% yield.
We also performed deprotection of tetrasaccharide 11 to obtain LNnT 2. This was achieved by concomitant deprotection of the phthalimido and ester groups in the presence of NH2NH2-H2O in refluxing MeOH. Subsequent N-acetylation with acetic anhydride in MeOH afforded intermediate 14 in 94% yield (Scheme 4). Finally, benzyl ethers were removed in the presence of 10% Pd/C in wet ethanol to afford LNnT 2 in 82% yield.
Scheme 4.
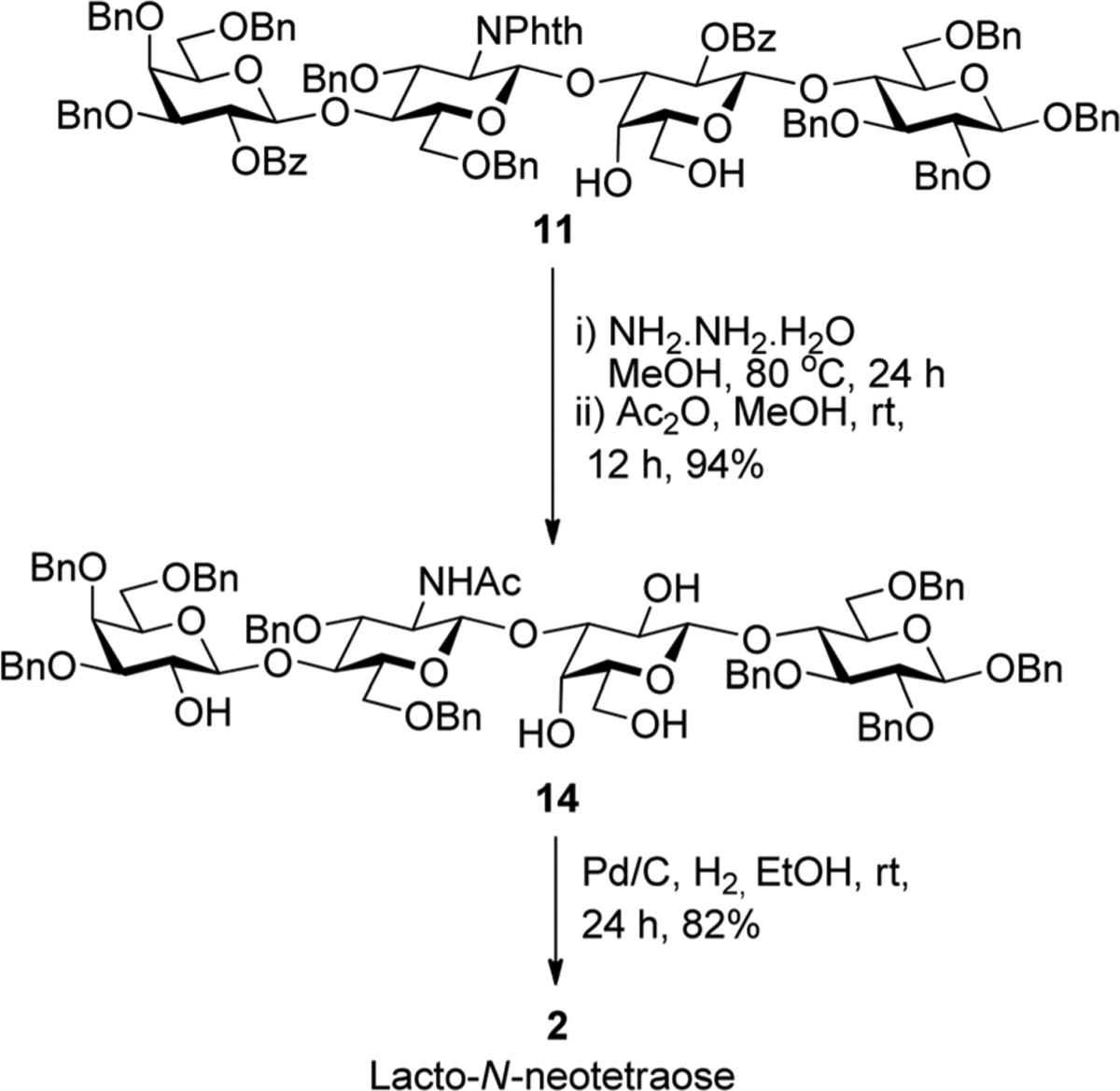
Deprotection of 11 to obtain LNT.
In summary, the first chemical synthesis of lacto-N-neohexaose has been completed using a convergent 2 + 2 + 2 strategy. This approach employed preassembled lactose and lactosamine building blocks. Along the way, we have also obtained lacto-N-neotetraose core sequence. With expectation that new methods for reliable chemical synthesis of individual HMO will boost practical applications of these important bio-molecules, further synthetic studies of other classes of HMO are underway in our laboratory.
Experimental
General methods
Reactions were performed using commercial reagents and the ACS grade solvents were purified and dried according to standard procedures. Column chromatography was performed on silica gel 60 (70–230 mesh) and Sephadex G-25 size exclusion resin, reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 was distilled from CaH2 directly prior to application. Molecular sieves (3 Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. AgOTf was co-evaporated with toluene (3 × 10 mL) and dried in vacuo for 2–3 h directly prior to application. Optical rotations were measured using a Jasco polarimeter. 1H NMR spectra were recorded at 300 MHz or 600 MHz, and 13C{H} NMR spectra were recorded at 75 MHz or 151 MHz. The 1H chemical shifts are referenced to the signal of the residual TMS (δH = 0.00 ppm) for solutions in CDCl3 or the signal of the residual D2O (δH = 4.79 ppm) for solutions in D2O. The 13C chemical shifts are referenced to the central signal of CDCl3 (δC = 77.16 ppm) for solutions in CDCl3 or the central signal of added CD3COCD3 (δC = 29.84 ppm) for solutions in D2O. Accurate mass spectrometry determinations were performed using Agilent 6230 ESI TOF LCMS mass spectrometer.
Benzyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-(1 → 3)-O-(2-O-benzoyl-4-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (4).
Cu (OAc)2-H2O (15.3 mg, 0.084 mmol) was added to a solution of benzyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-d-galactopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthlimido-β-d-glucopyranosyl)-(1 → 3)-O-(2-O-benzoyl-4-O-benzyl-6-O-picoloyl-β-d-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside9 (3, 22.0 mg, 0.011 mmol) in MeOH/CH2Cl2 (3.0 mL, 1/3, v/v) and the resulting mixture was stirred for 20 min at rt. Next, the reaction mixture was diluted with CH2Cl2 (~50 mL) was washed with H2O (10 mL), sat. aq. NaHCO3 (10 mL) and water (2 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound as an off-white amorphous solid in 97% yield (20.9 mg, 0.011 mmol). Analytical data for 4: Rf = 0.60 (ethyl acetate/hexane, 1/1, v/v); +14.9 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 2.88 (m, 1H, J = 9.5 Hz, H-5), 3.09–3.63 (m, 15H, 2, 3, 3′, 3‴, 5′, 5″, 5‴, 6a, 6a′, 6a″, 6a‴, 6b, 6b′, 6b″, 6b‴), 3.74–3.78 (m, 2H, H-4, 4′), 3.89–3.95 (dd, 1H, J4″,5″ = 8.9 Hz, H-4″), 3.98 (d, 1H, J3‴,4‴ = 1.6 Hz, H-4‴), 4.14–4.30 (m, 9H, H-1, 1′, 2″, 3″, 5 × CHPh), 4.42–4.59 (m, 7H, 7 × CHPh), 4.61–4.70 (m, 3H, H-1‴, CH2Ph), 4.79–4.82 (dd, 2H, CH2Ph), 4.84–4.88 (dd, 2H, CH2Ph), 4.98 (dd, 2H, CH2Ph), 5.06 (d, 1H, J1″,2″ = 8.4 Hz, H-1″), 5.24 (dd, 1H, J1′,2′ = 8.2, J2′,3′ = 9.9 Hz, H-2′), 5.64 (dd, 1H, J1‴,2‴ = 8.1, J2‴,3‴ = 9.8 Hz, H-2‴), 6.57–8.12 (m, 64H, aromatic) ppm; 13C{H} NMR (151 MHz, CDCl3): δ, 56.1, 61.9, 67.5, 68.1, 68.5, 71.0, 71.4, 71.9, 72.5, 72.7, 73.4 (×2), 73.5, 73.6, 74.3, 74.5, 74.6, 74.7 (×2), 74.8, 74.9, 75.8, 76.0, 76.3, 77.0, 78.4, 79.8, 80.5, 81.4, 82.7, 99.7, 100.6, 101.2, 102.5, 122.8, 123.2, 126.7, 127.4 (×2), 127.5, 127.6 (×2), 127.7 (×3), 127.8 (×8), 127.9 (×5), 128.0 (×4), 128.1 (×5), 128.2 (×3), 128.3 (×6), 128.4 (×8), 128.5 (×4), 128.6 (×3), 128.9 (×2), 129.4, 129.7, 130.0, 130.1, 130.9, 131.5, 132.9, 133.2, 133.4, 137.6, 137.8, 138.0, 138.1, 138.2, 138.6, 138.8 (×2), 138.9, 164.4, 165.2, 167.3, 167.8 ppm; ESI TOF LCMS [M + Na]+ calcd for C116H113NNaO24 1927.7584, found 1927.7586.
O-(2-O-Benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-D-glucopyranose (8).
A freshly prepared solution of Br2 in DCM (2.5 mL, 1/165, v/v) was added to a pre-chilled solution of ethyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-d-galactopyranosyl)-(1 → 4)-4,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside9 (5, 0.28 g, 0.27 mmol) in CH2Cl2 (4.0 mL) and the resulting mixture was stirred for 15 min at 0 °C. After that, the volatiles were removed under reduced pressure. The residue was co-evaporated with CH2Cl2 (3 × 10 mL) and dried in vacuo for 2 h. The crude residue was dissolved in acetone (5.0 mL), water (0.25 mL) and Ag2CO3 (0.04 g, 0.14 mmol) were added, and the resulting mixture was stirred in the absence of light for 16 h at rt. After that, the solids were filtered-off and rinsed successively with CH2Cl2. The combined filtrate (~150 mL) was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford the title compound as a white foam in 83% yield (0.23 g, 0.22 mmol). Analytical data for β−8: Rf = 0.35 (acetone/toluene, 1/4, v/v); 1H NMR (300 MHz, CDCl3): δ, 3.34 (d, 1H, J = 8.4 Hz, OH), 3.36–3.65 (m, 7H, H-3′, 5, 5′, 6a, 6a′, 6b, 6b′), 3.97–4.08 (m, 3H, H-2, 4, 4′), 4.22–4.63 (m, 10H, J1′,2′ = 7.9 Hz, H-1′, 3, 3 × CH2Ph, 2 × CHPh), 4.91 (d, 1H, 2J = 12.1 Hz, CHPh), 4.97 (d, 1H, 2J = 11.6 Hz, CHPh), 5.19 (dd, 1H, J1,2 = 8.4 Hz, H-1), 5.62 (dd, 1H, J2′,3′ = 10.1 Hz, H-2′), 6.73–7.99 (m, 34H, aromatic) ppm; 13C{H} NMR (75 MHz, CDCl3): δ, 57.6, 67.8, 68.2, 71.4 (×2), 72.5, 72.6, 73.4, 73.6 (×2), 74.6 (×2), 74.7, 77.4, 79.8, 93.0, 100.7, 123.6, 126.8, 127.5, 127.6 (×2), 127.8 (×7), 127.9 (×3), 128.0 (×2), 128.1 (×2), 128.2 (×2), 128.3, 128.4 (×2), 128.5 (×5), 129.1, 129.9 (×2), 130.0 (×2), 131.7, 133.2, 133.9, 137.8, 138.0, 138.1, 138.8, 139.0, 168.2 (×3) ppm; ESI TOF LCMS [M + Na]+ calcd for C62H59NNaO13 1048.3884, found 1048.3875.
O-(2-O-Benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl trichloroacetimidate (9).
CCl3CN (0.86 mL, 8.58 mmol) and DBU (5.63 μL, 0.04 mmol) were added to a solution of compound 8 (0.44 g, 0.43 mmol) in CH2Cl2 (8.0 mL) and the resulting mixture was stirred under argon for 1 h at rt. After that, the volatiles were removed under reduced pressure. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford the title compound as a white foam in 87% yield (0.43 g, 0.37 mmol). Analytical data for 9: Rf = 0.65 (acetone/toluene, 1/4, v/v); +52.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 3.44–3.73 (m, 7H, H-3′, 5, 5′, 6a, 6a′, 6b, 6b′), 4.01 (br d, 1H, J3′,4′ = 2.4 Hz, H-4′), 4.16 (dd, 1H, J3,4 = J4,5 = 8.8 Hz, H-4), 4.27–4.75 (m, 11H, H-1′, 2, 3, 4 × CH2Ph), 4.92 (d, 1H, 2J = 12.2 Hz, CHPh), 4.98 (d, 1H, 2J = 11.6 Hz, CHPh), 5.65 (dd, 1H, J1′,2′ = 8.1 Hz, J2′,3′ = 9.8 Hz, H-2′), 6.28 (d, 1H, J = 8.5 Hz, H-1), 6.73–8.00 (m, 34H, aromatic), 8.46 (s, 1H, NH) ppm; 13C{H} NMR (75 MHz, CDCl3): δ, 54.7, 67.2, 68.2, 71.4, 72.4, 72.7, 73.4, 73.5, 73.6, 74.6, 74.8, 75.4, 76.6, 76.9, 79.8, 90.5, 94.2, 100.6, 123.4 (×2), 125.4, 126.9, 127.4, 127.6 (×2), 127.7, 127.8 (×4), 127.9 (×3), 128.0 (×3), 128.2 (×4), 128.3, 128.4 (×2), 128.5 (×3), 128.6 (×2), 129.1, 129.9 (×3), 131.5, 133.2, 133.9, 137.8, 138.0, 138.2, 138.8 (×2), 160.9, 165.1, 167.6 ppm; ESI TOF LCMS [M + Na]+ calcd for C64H59Cl3N2NaO13 1193.2951, found 1193.2967.
Benzyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-(1 → 3)-O-(2-O-benzoyl-4,6-O-benzylidene-β-D-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (10).
A mixture of donor 9 (0.15 g, 0.13 mmol), acceptor 711 (0.09 g, 0.01 mmol), and freshly activated molecular sieves (3 Å, 450 mg) in CH2Cl2 (5.0 mL) was stirred under argon for 2 h at rt. The mixture was cooled to −30 °C, TMSOTf (5.50 μL, 0.03 mmol) was added, and the resulting mixture was stirred for 10 min while the reaction temperature was allowed to increase gradually. After that, the solids were filtered-off and rinsed successively with CH2Cl2. The combined filtrate (~50 mL) was washed with sat. aq. NaHCO3 (10 mL) and water (2 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford the title compound as a white foam in 83% yield (0.21 g, 0.11 mmol). Analytical data for 10: Rf = 0.55 (acetone/toluene, 1/4, v/v); +7.9 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 2.92–3.00 (m, 2H, H-5, 5′), 3.28–3.65 (m, 13H, H-2, 3, 3′, 3‴, 4′, 5″, 5‴, 6a, 6a″, 6a‴, 6b, 6b″, 6b‴), 3.78–3.89 (m, 2H, H-4, 4″), 4.01 (br s, 1H, H-4‴), 4.09 (br d, 1H, J = 11.9 Hz, H-6a′), 4.12–4.28 (m, 6H, H-2″, 3″, 6b′, 3 × CHPh), 4.29–4.34 (m, 3H, H-1, CH2Ph), 4.39 (d, 1H, 2J = 11.5 Hz, CHPh), 4.45–4.59 (m, 5H, H-1′, 2 × CH2Ph), 4.63–4.72 (m, 4H, H-1‴′, 3 × CHPh), 4.81–4.89 (m, 3H, 3 × CHPh), 5.01 (dd, 2H, CH2Ph), 5.20 (d, 1H, J = 7.6 Hz, H-1″), 5.26–5.32 (dd, 1H, J2′,3′ = 8.7 Hz, H-2′), 5.39 (s, 1H, >CHPh), 5.64–5.71 (dd, 1H, J2‴,3‴ = 8.7 Hz, H-2‴), 6.72–8.13 (m, 64H, aromatic) ppm; 13C{H} NMR (151 MHz, CDCl3): δ, 55.6, 66.7, 68.0, 68.2, 68.6, 69.2, 71.0, 71.1, 71.5, 72.6, 72.8, 73.3, 73.5, 73.6 (×2), 74.3, 74.6 (×4), 74.9, 75.3, 75.6, 75.7, 77.0, 78.5, 79.0, 80.0, 81.8, 83.1, 99.6, 100.6, 100.8, 101.2, 102.5, 126.4 (×2), 126.7, 127.1, 127.5 (×4), 127.7 (×6), 127.8 (×3), 127.9 (×7), 128.0 (×6), 128.1 (×6), 128.2 (×3), 128.3 (×6), 128.4 (×5), 128.5 (×5), 128.6 (×4), 129.4, 129.5 (×2), 130.0, 130.1 (×2), 132.7, 133.3 (×2), 137.6, 137.8, 138.0, 138.1, 138.2, 138.4, 138.7, 138.8 (×2), 139.2, 164.1, 165.2 ppm; ESI TOF LCMS [M + 2Na]2+ calcd for C116H111NNa2O24 974.3662, found 974.3644.
Benzyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-(1 → 3)-O-(2-O-benzoyl-β-D-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (11).
TsOH (10.8 mg, 0.06 mmol) and EtSH (38 μL, 0.54 mmol) were added to a solution of compound 10 (0.17 g, 0.09 mmol) in MeOH/CH2Cl2 (7.0 mL, 1/1, v/v) and the resulting mixture was stirred for 3 h at rt. The reaction was then quenched with triethylamine (~0.5 mL) and the volatiles were removed under reduced pressure. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford the title compound as a white amorphous solid in 87% yield (0.14 g, 0.08 mmol). Analytical data for 11: Rf = 0.50 (acetone/toluene, 1/4, v/v); +36.1 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 2.85 (br s, 1H, H-5′), 2.95 (br d, 1H, J = 9.6 Hz, H-5), 3.21–3.65 (m, 14H, H-2, 3, 3′, 3‴, 5″, 5‴, 6a, 6a′, 6a″, 6a‴, 6b, 6b′, 6b″, 6b‴), 3.80 (dd, 1H, J4,5 = 9.3 Hz, H-4), 3.85 (dd, 1H, J4″,5″ = 9.0 Hz, H-4″), 3.90 (s, 1H, H-4′), 4.00 (s, 1H, H-4‴), 4.10–4.59 (m, 14H, H-1, 1′, 2″, 3″, 5 × CH2Ph), 4.61–4.71 (m, 4H, H-1‴, 3 × CHPh), 4.80–4.87 (m, 3H, 3 × CHPh), 4.90 (d, 1H, 2J = 10.3 Hz, CHPh), 4.97 (d, 1H, 2J = 11.7 Hz, CHPh), 5.13 (d, 1H, J1″,2″ = 8.4 Hz, H-1″), 5.23 (dd, 1H, J2″,3″ = 10.0 Hz, H-2′), 5.65 (dd, 1H, J2″,3″ = 9.6 Hz, H-2‴), 6.66–8.07 (m, 59H, aromatic) ppm; 13C{H} NMR (151 MHz, CDCl3): δ, 55.5, 62.0, 67.6, 68.2, 68.3, 68.4, 71.0, 71.1, 71.5, 72.6, 72.7, 73.4 (×2), 73.6 (×3), 74.3 (×2), 74.6, 74.7, 74.8, 74.9, 75.7, 76.4, 78.3, 79.8, 81.4, 81.6, 82.7, 99.0, 100.3, 101.2, 102.5, 126.8, 127.5 (×3), 127.6, 127.7 (×3), 127.8 (×8), 127.9 (×6), 128.0 (×2), 128.1 (×3), 128.2 (×7), 128.3 (×6), 128.4 (×6), 128.5 (×3), 128.6 (×7), 129.2, 129.4, 130.0 (×2), 132.7, 133.4, 137.6, 137.7, 138.0 (×2), 138.2, 138.6, 138.7, 138.8, 138.9, 164.2, 165.2 ppm; ESI TOF LCMS [M + 2Na]2+ calcd for C109H107NNa2O24 930.3506, found 930.3489.
Benzyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-(1 → 3)-[O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-(1 → 6)]-O-(2-O-benzoyl-β-D-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (12).
A mixture of donor 59 (38.3 mg, 0.035 mmol), acceptor 11 (50.0 mg, 0.0275 mmol), and freshly activated molecular sieves (3 Å, 100 mg) in CH2Cl2 (4.0 mL) was stirred under argon for 2 h at rt. The mixture was then cooled to −40 °C, NIS (12.3 mg, 0.055 mmol) and freshly conditioned AgOTf (3.5 mg, 0.013 mmol) were added, and the resulting mixture was stirred for 30 min at −40 °C. After that, the reaction mixture was stirred for additional 30 min during which the reaction temperature was allowed to increase gradually to −18 °C. The solids were then filtered-off and rinsed successively with CH2Cl2. The combined filtrate (~50 mL) was washed with sat. aq. NaHCO3 (10 mL), Na2S2O3 (10 mL), and water (2 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford the title compound as a white foam in 82% yield (63.6 mg, 0.0227 mmol). Analytical data for 12: Rf = 0.60 (acetone/toluene, 1/4 v/v); +34.5 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 1.88 (m, 1H, OH), 2.73 (br s, 1H, H-5B), 2.95 (m, 1H, H-5E), 3.01 (m, 1H, J = 9.3 Hz, H-5C), 3.19–3.67 (m, 22H, H-2A, 3B, 3D, 3F, 4A, 4B, 4E, 5A, 5D, 5F, 6aA, 6aB, 6aC, 6aD, 6aE, 6aF, 6bA, 6bB, 6bC, 6bD, 6bE, 6bF), 3.73 (dd, 1H, J = 8.7, 10.7 Hz, H-3E), 3.86–4.10 (m, 9H, H-2C, 2E, 3A, 3C, 4C, 4D, 4F, CH2Ph), 4.23–4.34 (m, 7H, H-1A, 1F, 5 × CHPh), 4.38–4.56 (m, 14H, 1B, 1D, 1E, 11 × CHPh), 4.61–4.64 (dd, 2H, CH2Ph), 4.69–4.75 (dd, 2H, CH2Ph), 4.78–4.84 (m, 4H, 2 × CH2Ph), 4.91 (d, 1H, J1C,2C = 8.4 Hz, H-1C), 4.94–4.98 (dd, 2H, 2 × CHPh), 5.19 (dd, 1H, J1B,2B = 8.3, J2B,3B = 9.5 Hz, H-2B), 5.61–5.65 (m, 2H, H-2D, 2F), 6.72–8.09 (m, 93H, aromatic) ppm; 13C{H} NMR (151 MHz, CDCl3): δ, 55.2, 55.6, 64.6, 65.9, 67.3, 67.9, 68.1, 68.2, 70.6, 71.0, 71.2, 71.4 (×2), 72.4, 72.5, 72.6, 72.7, 73.3, 73.4, 73.5, 73.6 (×5), 73.7, 73.8, 74.2, 74.3, 74.6 (×2), 74.7 (×3), 75.0, 75.6, 75.9, 76.3, 77.1, 77.3, 79.8, 81.6, 81.9, 82.8, 97.3, 97.9, 100.2, 100.6 (×2), 102.5, 123.2, 123.9, 126.9 (×2), 127.4 (×2), 127.5 (×2), 127.6 (×2), 127.7 (×3), 127.8 (×11), 127.9 (×12), 128.1 (×5), 128.2 (×6), 128.3 (×9), 128.4 (×8), 128.5 (×10), 128.6 (×8), 129.0, 129.2, 129.4, 129.9 (×2), 130.0 (×2), 130.9, 131.2 (×2), 131.9, 132.5, 133.1, 133.4, 133.7, 133.9, 134.6 (×3), 137.7, 137.9, 138.0, 138.1, 138.2 (×2), 138.4, 138.7, 138.8 (×3), 138.9, 139.0, 164.0, 165.2, 165.4, 166.6, 167.0, 168.0, 168.1 ppm; ESI TOF LCMS [M + Na]+ calcd for C171H164N2NaO36 2845.0995, found 2845.0944.
Benzyl O-(3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-O-(2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranosyl)-(1 → 3)-[O-(3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-O-(2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranosyl)-(1 → 6)]-O-(β-D-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (13).
Compound 12 (72.0 mg, 0.025 mmol) was dissolved in NH2NH2-H2O/MeOH (3.0 mL, 1/2, v/v), and the resulting mixture was kept for 36 h at reflux. After that, the volatiles were removed under reduced pressure. The residue was dissolved in MeOH/CH2Cl2 (~5 mL, 1/9, v/v) and filtered through a pad of silica gel eluting with MeOH/CH2Cl2 (1/9, v/v). The combined eluate (~50 mL) was concentrated under reduced pressure and dried in vacuo for 3 h. The crude residue was dissolved in Ac2O/MeOH/Et3N (2.0 mL, 1/1/0.1, v/v/v) and the resulting mixture was stirred for 12 h at rt. The volatiles were then removed under reduced pressure. The residue was diluted with CH2Cl2 (~50 mL), and washed with sat. aq. NaHCO3 (10 mL) and brine (2 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford the title compound as an off-white amorphous solid in 92% yield (54.7 mg, 0.023 mmol). Analytical data for 13: Rf = 0.60 (acetone/toluene, 2/3, v/v); +11.8 (c 1.0, CHCl3); 1.74, 1.78 (2 s, 6H, 2 × COCH3), 2.96–3.97 (m, 36H, H-2A, 2B, 2C, 2D, 2E, 2F, 3A, 3B, 3C, 3D, 3E, 3F, 4A, 4B, 4C, 4D, 4E, 4F, 5A, 5B, 5C, 5D, 5E, 5F, 6aA, 6aB, 6aC, 6aD, 6aE, 6aF, 6bA, 6bB, 6bC, 6bD, 6bE, 6bF), 4.20–4.98 (m, 34H, H-1A, 1B, 1C, 1D, 1E, 1F, 14 × CH2Ph), 5.34, 5.62 (2 d, 2H, 2 × NH), 7.12–7.38 (m, 70H, aromatic) ppm; 13C{H} NMR (151 MHz, CDCl3): δ, 23.6, 23.7, 55.4, 56.0, 67.8, 68.0, 68.2 (×2), 68.6, 68.7, 70.8, 71.2, 71.9, 72.2, 72.3 (×3), 72.9, 73.0, 73.3, 73.4 (×4), 73.5 (×6), 73.9, 74.0, 74.3, 74.6, 74.7, 74.8, 75.0, 75.1, 75.6, 76.8, 77.0, 79.6, 80.3, 81.9, 82.4, 83.4, 101.0, 101.3, 102.7, 102.8, 103.3, 103.4, 127.2, 127.5 (×2) 127.6 (×4), 127.7 (×6), 127.8 (×6), 127.9 (×8), 128.0 (×12), 128.2 (×5), 128.3 (×3), 128.4 (×6), 128.5 (×15), 128.6 (×2), 137.5, 137.8, 137.9, 138.0 (×2), 138.1, 138.2, 138.4 (×2), 138.8 (×2), 139.0, 139.1, 139.2, 170.3, 171.2 ppm; ESI TOF LCMS [M + Na]+ calcd for C138H152N2NaO31 2357.0310, found 2357.0316.
O-(β-D-Galactopyranosyl)-(1 → 4)-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1 → 3)-[O-(β-D-galactopyranosyl)-(1 → 4)-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1 → 6)]-O-(β-D-galactopyranosyl)-(1 → 4)-β-D-glucopyranose (1, LNnH).
10% Pd on charcoal (200 mg) was added to a solution of 13 (70.0 mg, 0.030 mmol) in EtOH/H2O (7.0 mL, 4/1, v/v), and the resulting mixture was stirred under hydrogen atmosphere for 24 h at rt. After that, the solids were filtered off and rinsed successively with methanol and water. The combined filtrate (~50 mL) was concentrated under reduced pressure. The residue was purified by size-exclusion chromatography on Sephadex G-25 (water elution) to afford the title compound as a white amorphous solid in 91% yield (29.2 mg, 0.027 mmol). Selected analytical data for 1:17 Rf = 0.50 (chloroform/methanol/water, 2/1/0.4, v/v/v); 1H NMR (600 MHz, D2O): δ, 1.99, 2.02 (2 s, 6H, COCH3), 3.25 (dd, 1H, J = 8.1, 9.1 Hz), 3.47–3.59 (m, 9H), 3.61–3.64 (m, 2H), 3.65–3.84 (m, 22H), 3.92 (m, 7H), 4.11 (d, 1H, J = 3.3 Hz), 4.39 (d, 1H, J = 7.9 Hz), 4.43 (m, 2H), 4.59 (d, 1H, J = 7.9 Hz), 4.62 (d, 1H, J = 8.0 Hz), 4.66 (d, 1H, J = 8.3 Hz), 5.18 (d, 0.5H, J = 3.7 Hz) ppm; 13C{H} NMR (151 MHz, D2O): δ, 22.5, 22.8, 55.3, 55.5, 60.2 (×2), 60.4, 61.4, 68.7, 68.9, 69.0, 70.2 (×2), 70.3, 71.3, 71.5, 71.7, 72.5, 72.8 (×2), 73.8 (×2), 74.2, 74.7, 74.9, 75.0, 75.1, 75.7, 78.4, 78.6, 79.2, 79.3, 82.1, 92.1, 96.0, 101.3, 103.1, 103.2 (×2), 103.3, 174.9, 175.2 ppm; ESI TOF LCMS [M + H]+ calcd for C40H69N2O31 1073.3884, found 1073.3859.
Benzyl O-(3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1 → 4)-O-(2-acetamido-3,6-di-O-benzyl-2-deoxy-β-D-glucopyranosyl)-(1 → 3)-O-(β-D-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (14).
Compound 11 (47.0 mg, 0.026 mmol) was dissolved in NH2NH2-H2O/MeOH (2.5 mL, 1/4, v/v), and the resulting mixture was kept for 24 h at reflux. After that, the volatiles were removed under reduced pressure. The residue was dissolved in MeOH/CH2Cl2 (~5 mL, 1/9, v/v) and filtered through a pad of silica gel eluting with MeOH/CH2Cl2 (1/9, v/v). The combined eluate (~50 mL) was concentrated under reduced pressure and dried in vacuo for 3 h. The crude residue was dissolved in Ac2O/MeOH (2.0 mL, 1/1, v/v), and the resulting mixture was stirred for 12 h at rt. The volatiles were then removed under reduced pressure. The residue was diluted with CH2Cl2 (~50 mL) and washed with sat. aq. NaHCO3 (10 mL) and brine (2 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford the title compound as an off-white amorphous solid in 94% yield (37.0 mg, 0.024 mmol). Analytical data for 14: Rf = 0.65 (acetone/toluene, 2/3, v/v); +20.4 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 1.73 (s, 3H, COCH3), 2.85–4.01 (m, 24H, H-2, 2′, 2″, 2‴, 3, 3′, 3″, 3‴, 4, 4′, 4″, 4‴, 5, 5′, 5″, 5‴, 6a, 6a′, 6a″, 6a‴, 6b, 6b′, 6b″, 6b‴), 4.30 (dd, 2H, 2J = 11.7 Hz, CH2Ph), 4.43–4.61 (m, 9H, H-1, 1′, 1‴, 3 × CH2Ph), 4.62–4.71 (m, 4H, 2 × CH2Ph), 4.82–4.95 (m, 7H, H-1″, 3 × CH2Ph), 5.64 (d, 1H, J = 7.5 Hz, NH), 7.19–7.37 (m, 45H, aromatic) ppm; 13C{H} NMR (151 MHz, CDCl3): δ, 23.6, 56.7, 62.6, 68.3, 68.6, 68.7, 68.8, 70.9, 71.3, 71.9, 72.4, 72.8, 73.4, 73.6 (×2), 73.7, 74.1, 74.3 (×2), 74.7, 74.8, 75.0, 75.1, 75.3, 77.1, 79.6, 82.0, 82.1, 82.9, 83.1, 101.5, 102.8 (×2), 103.4, 127.5 (×3), 127.6 (×2), 127.8 (×4), 127.9, 128.0 (×15), 128.3 (×6), 128.4 (×5), 128.5 (×7), 128.6 (×2), 137.6, 137.8, 137.9, 138.1 (×2), 138.4, 138.8 (×2), 139.0, 171.3 ppm; ESI TOF LCMS [M + Na]+ calcd for C89H99NNaO21 1540.6607, found 1540.6625.
O-(β-D-Galactopyranosyl)-(1 → 4)-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1 → 3)-O-(β-D-galactopyranosyl)-(1 → 4)-D-glucopyranose (2, LNnT).
10% Pd on charcoal (75 mg) was added to a solution of 14 (30.0 mg, 0.019 mmol) in EtOH/H2O (4.0 mL, 4/1, v/v), and the resulting mixture was stirred under hydrogen atmosphere for 24 h at rt. After that, the solids were filtered off and rinsed successively with methanol and water. The combined filtrate (~50 mL) was concentrated under reduced pressure. The residue was purified by size exclusion chromatography on Sephadex G-25 (water elution) to afford the title compound as a white amorphous solid in 82% yield (11.0 mg, 0.015 mmol). Analytical data for 2 was in agreement with that reported previously:9 Rf = 0.30 (chloroform/methanol/water, 2/1/0.4, v/v/v); ESI TOF LCMS [M + Na]+ calcd for C26H45NNaO21 730.2382, found 730.2381.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of General Medical Sciences (GM120673 and GM111835).
Footnotes
Electronic supplementary information (ESI) available: NMR spectra for all new compounds. See DOI: 10.1039/d0ob00172d
Conflicts of interest
The authors declare no competing financial interests.
References
- 1.Wu X, Jackson RT, Khan SA, Ahuja J and Pehrsson PR, Curr. Dev. Nutr, 2018, 2, nzy025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bode L, Glycobiology, 2012, 22, 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruhaak LR and Lebrilla CB, Adv. Nutr, 2012, 3, 406S–414S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Urashima T, Hirabayashi J, Sato S and Kobata A, Trends Glycosci. Glycotechnol, 2018, 30, SE51–SE65. [Google Scholar]
- 5.Yu H and Chen X, in Synthetic Glycomes, RSC, 2019, p. 254–280. [Google Scholar]
- 6.Craft KM and Townsend SD, ACS Infect. Dis, 2018, 4, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramani S, Stewart CJ, Laucirica DR, Ajami NJ, Robertson B, Autran CA, Shinge D, Rani S, Anandan S, Hu L, Ferreon JC, Kuruvilla KA, Petrosino JF, Prasad BVV, Bode L, Kang G and Estes MK, Nat. Commun, 2018, 9, 5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prudden AR, Liu L, Capicciotti CJ, Wolfert MA, Wang S, Gao Z, Meng L, Moremen KW and Boons GJ, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 6954–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bandara MD, Stine KJ and Demchenko AV, Carbohydr. Res, 2019, 483, 107743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bandara MD, Stine KJ and Demchenko AV, Carbohydr. Res, 2019, 486, 107824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bandara MD, Stine KJ and Demchenko AV, J. Org. Chem, 2019, 84, 16192–16198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dua VK, Goso K, Dube VE and Bush CA, J. Chromatogr, 1985, 328, 259–269. [DOI] [PubMed] [Google Scholar]
- 13.Aly MRE, Ibrahim ESI, El Ashry ESH and Schmidt RR, Carbohydr. Res, 1999, 316, 121–132. [DOI] [PubMed] [Google Scholar]
- 14.Yan F, Wakarchuk WW, Gilbert M, Richards JC and Whitfield DM, Carbohydr. Res, 2000, 328, 3–16. [DOI] [PubMed] [Google Scholar]
- 15.Prudden AR, Chinoy ZS, Wolfert MA and Boons GJ, Chem. Commun, 2014, 50, 7132–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rich JR, Cunningham A-M, Gilbert M and Withers SG, Chem. Commun, 2011, 47, 10806–10808. [DOI] [PubMed] [Google Scholar]
- 17.Xiao Z, Guo Y, Liu Y, Li L, Zhang Q, Wen L, Wang X, Kondengaden SM, Wu Z, Zhou J, Cao X, Li X, Ma C and Wang PG, J. Org. Chem, 2016, 81, 5851–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roussel F, Takhi M and Schmidt RR, J. Org. Chem, 2001, 66, 8540–8548. [DOI] [PubMed] [Google Scholar]
- 19.Ravenscroft M, Roberts RMG and Tillett JG, J. Chem. Soc., Perkin Trans 2, 1982, 1569–1972. [Google Scholar]
- 20.Andersson F, Fugedi P, Garegg PJ and Nashed M, Tetrahedron Lett., 1986, 27, 3919–3922. [Google Scholar]
- 21.Konradsson P, Udodong UE and Fraser-Reid B, Tetrahedron Lett., 1990, 31, 4313–4316. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.